
Abstract
Dyslipidemia is a major risk factor for cardiovascular (CV) disease – the primary cause of death, worldwide. Although reducing levels of low-density lipoprotein-cholesterol can significantly reduce CV risk, a high level of residual risk persists, especially in people with obesity-related conditions, such as metabolic syndrome and type 2 diabetes mellitus. Peroxisome proliferator-activated receptor alpha- (PPARα-) agonists (e.g. fibrates), play a central role in the reduction of macro- and microvascular risk in these patients. However, the currently available fibrates are weak (PPARα-agonists) with limited efficacy due to dose-related adverse effects. To address this problem, a new generation of highly potent and selective PPARα-modulators (SPPARMα) is being developed that separate the benefits of the PPARα-agonists from their unwanted side effects. Among these, aleglitazar (a dual PPARα/γ agonist) and GFT505 (a dual PPAR α/δ agonist) have recently entered late-phase development. Although both compounds are more potent PPARα-activators than fenofibrate in vitro, only aleglitezar is more effective in lowering triglycerides and raising high-density lipoprotein-cholesterol (HDL-C) in humans. However, it is also associated with a potential risk of adverse effects. More recently, a highly potent, specific PPARα-agonist (K-877) has emerged with SPPARMα characteristics. Compared to fenofibrate, K-877 has more potent PPARα-activating efficacy in vitro, greater effects on triglycerides- and HDL-C levels in humans, and a reduced risk of adverse effects. If successful, K-877 has the potential to supersede the fibrates as the treatment of choice for patients with residual CV risk associated with metabolic syndrome and type 2 diabetes.
Similar content being viewed by others
Introduction
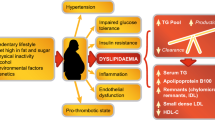
Dyslipidemia is a major risk factor for cardiovascular (CV) disease – the leading cause of morbidity and mortality in the developed world[1]. Lowering low-density lipoprotein-cholesterol (LDL-C) levels using lifestyle change and pharmacotherapy can significantly reduce CV risk in people with and without cardiometabolic diseases, such as metabolic syndrome (MetS) and type 2 diabetes mellitus (T2D)[2–4]. However, the risk of macrovascular events in those attaining the maximum levels of LDL-C reduction is only reduced by around 30%, leaving substantial residual risk[2]. Moreover, the risk of microvascular events in people with MetS or T2D can only be reduced by approximately 50% using the current standard of care (intensive treatment to reduce LDL-C, blood pressure and blood glucose)[5]. Recent studies demonstrate that low levels of high-density lipoprotein-cholesterol (HDL-C) (<1.0 mmol/L; 40 mg/dL) and high levels of triglycerides (TGs) (≥1.7 mmol/L; 150 mg/dL) are independent risk factors for both macro- and microvascular diseases[6–8]. Consequently, treatment guidelines highlight the importance of targeting these risk factors in addition to LDL-C[9]. This is particularly important for patients with cardiometabolic diseases who often have atherogenic dyslipidemia – a triad of lipid abnormalities that includes low levels of HDL-C, high levels of TG and a preponderance of atherogenic small, dense LDL particles[9–11]. In addition, patients with these lifestyle-related conditions typically present with chronic inflammation and other obesity-related risk factors, such as insulin resistance and hyperglycemia. The ideal strategy for reducing CV risk in patients with MetS or T2D should therefore encompass many aspects of cardiometabolic control in addition to lipid homeostasis[9, 11].
Peroxisome proliferator-activated receptor alpha (PPARα) agonists
Effects in vitro and in vivo
The PPARα-agonists (e.g. fibrates) play a central role in reducing plasma concentrations of TG-rich lipoproteins, increasing HDL-C levels and reducing vascular inflammation in people with atherogenic dyslipidemia[10, 12, 13]. Furthermore, PPARα-agonists can increase the stability of atherosclerotic plaques, reduce the risk of atherothrombosis, slow the progression of intimal hyperplasia following surgery, and reduce hepatic fat accumulation leading to non-alcoholic steatohepatitis/fatty liver disease (NASH/NAFLD)[14]. The net effect of these actions is a significant reduction in macrovascular events in people with, but not without, atherogenic dyslipidemia[12, 13, 15–18], and significant reductions in T2D-related microvascular events[19–22]. For example, the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study demonstrated a 27% reduction in the risk of major CV events in patients with atherogenic dyslipidemia (P<0.005)[23], a 14% reduction in albuminuria progression over 5 years (P<0.001)[21], a 36% reduction in the risk of first amputation (P=0.02)[19], and a 31% reduction in the need for first laser treatment (P=0.0002)[20]. Although fibrates are the only lipid-lowering drugs to demonstrate clinically significant benefits for both macro- and microvascular disease in people with T2D, they are weak PPARα-agonists whose efficacy is partly limited by dose-dependent side effects. Commonly-reported adverse events (AEs) include elevations in markers for CV disease (e.g. homocysteine), renal disease (e.g. creatinine) and liver dysfunction (e.g. alanine aminotransferase [ALT] and γ-glutamyl transpeptidase)[12, 13, 24]. There is therefore an unmet medical need for a new generation of more potent PPARα-agonists with a lower potential for AEs.
Mechanism of action
Three PPAR isoforms have been identified (PPARα, γ and δ), each encoded by a separate gene[25]. PPARα is abundant in highly active metabolic tissues including the liver, kidney, heart, muscle, brown adipose and macrophages, whereas PPARγ is predominantly found in adipose tissue, macrophages and large intestine. In contrast, PPARδ (also called PPARβ) is ubiquitously expressed.
PPARs are activated when endogenous ligands (e.g. prostaglandins, leukotrienes, free fatty acids) or synthetic PPAR agonists (e.g. glitazones, fibrates) bind to the lipid-binding domain enabling heterodimerisation with a ligand-activated retinoid X-receptor (RXR)[26–30]. This process triggers a conformational change, leading to the transrepression or transactivation of target genes. During transrepression, the activated PPAR binds to cytokine-activated transcription factors, such as nuclear factor kappa B or activator protein-1[31, 32]. Under normal conditions, these transcription factors induce the synthesis of proteins involved in the inflammatory response. PPARs can inhibit this process by blocking the interaction between activated transcription factors and the promoter region of the target gene, thereby preventing transcription and reducing inflammation. During transactivation, the activated PPAR binds to a specific sequence of nucleotides (the PPAR receptor response element) upstream of the target gene. A cofactor (either a coactivator or a corepressor) renders the PPAR complex ‘transcriptionally active’ and gene transcription begins. A large number of genes carry response elements for PPARs. Targets for PPARα include key genes involved in lipid metabolism, such as apo A-I, A-II and A-V, LPL and SR-BI, whereas PPARγ targets include genes involved in obesity and insulin resistance, such as ADIPOQ and ADRP. Consequently, PPARα primarily regulates lipid homeostasis[14], whereas PPARγ largely regulates adipogenesis and glucose homeostasis[33]. However, the precise receptor-mediated response depends on the individual agonist and the tissue in which it is expressed.
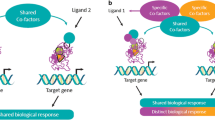
Compared to other nuclear receptors, PPARs have a large lipid-binding pocket capable of encompassing a range of endogenous ligands[34]. This provides a variety of potential contact points that, when occupied, can trigger different conformational changes. Since each PPAR conformation is associated with a unique cofactor recruitment pattern, the same PPAR subtype has the potential to induce a wide range of biological effects depending on its agonist (Figure 1). The number of possible effects is further increased by the fact that PPAR-activation requires both a specific ligand and an activated RXR complex, the concentration and affinity of which can vary between tissues, organs, and individuals. It is therefore possible for a ligand to act as a full agonist in a tissue where sufficient concentrations of a specific coactivator are present, and as a partial agonist in another tissue with higher concentrations of the same coactivator. For example, gemfibrozil and fenofibrate (both PPARα-agonists) have a similar impact on levels of HDL-C, TG and small dense LDL but, whereas fenofibrate (a full PPARα-agonist) has additional benefits on apolipoprotein A-I (apo A-I) and fibrinogen levels[35], gemfibrozil (a partial PPARα-agonist) has little or no effect[36]. Moreover, whereas most fibrates specifically activate PPARα, bezafibrate is a pan PPAR-agonist, activating all three PPAR subtypes (α, γ and δ) at comparable doses[37]. Although this effect can be exploited to increase the number of biological benefits, it can also be associated with an increased risk of unwanted side effects. Consequently, the development of ideal PPAR-agonists involves a series of in vitro and in vivo assays to identify the most potent molecules that differentially induce receptor-mediated beneficial effects in specific tissues whilst avoiding unwanted side effects[34]. This is the pharmacological basis for the selective PPARα modulators (SPPARMs) (Figure 2).

PPAR-agonists have the potential to trigger different biological responses via the same receptor[34].

SPPARMα characteristics. ALT alanine aminotransferase; Apo apolipoprotein; FGF-21 fibroblast growth factor 21; γGT γ-glutamyl transpeptidase; HDL high-density lipoprotein; NASH/NASFL non-alcoholic steatohepatitis/fatty liver disease; NR nuclear receptor; TG triglyceride.
The next generation of selective PPARα modulators
The concept of SPPARMs (and other selective nuclear receptor modulators) was initially based on the paradigm of tamoxifen, a pioneering selective estrogen receptor modulator that exhibits anti-estrogenic activity in the mammary gland and partial pro-estrogenic activity in bone and uterus[38]. The observed increase in the incidence of uterine cancer with prolonged tamoxifen use led to the development of raloxifene, a second-generation estrogen receptor modulator with highly selective, tissue-specific activity that avoids uterotrophic effects. Since then, selective modulators have been identified for most classes of ligand-modulated nuclear receptor[39]. For example, a number of PPARγ-agonists with SPPARM properties (e.g. INT131 and MK0533) have been developed for the treatment of T2D[40, 41]. Preclinical studies show that these molecules have comparable or more potent antidiabetic benefits to the gold-standard treatment, pioglitazone, with fewer AEs[41]. More recently, the SPPARM concept has been extended to other PPAR subtypes, including PPARα. If successful, these molecules have the potential to become superior therapeutics for the treatment of CV risk associated with MetS and T2D.
Dual PPAR-agonists with SPPARMα properties
Whereas PPARα-agonists can improve lipid control and PPARγ-agonists can improve glucose homeostasis, dual PPARα/γ-or α/δ agonists can potentially be used to treat a range of cardiometabolic imbalances at the same time. Moreover, these molecules can be designed to offset each other’s side effects[26, 42]. For example, weight increases due to the adipogenic effects of PPARγ-agonists can potentially be negated by PPARα-mediated increases in lipid catabolism. Although many dual PPAR-agonists have undergone clinical trials, none have progressed past Phase III due to unresolved safety concerns[42]. Muraglitazar, tesaglitazar, ragaglitazar, TAK559 and KRP292, for example, were discontinued due to an increased risk of CV events, renal dysfunction, weight gain and edema. However, two dual agonists with possible SPPARM properties (aleglitazar and GFT505) have recently entered late-phase development.
Aleglitazar (Figure 3) is a dual PPARα/γ-agonist developed by Roche Holding for the treatment of residual CV risk in people with T2D[26, 43, 44]. Compared to pioglitazone, it is a more potent PPARγ-agonist with greater effects on glucose homeostasis and a reduced risk of AEs[26]. In addition to its PPARγ agonist properties, aleglitazar is a more potent PPARα-agonist than fenofibrate, both in vitro (Table 1)[26] and in vivo (Table 2)[44]. For example, a Phase II study in 332 people with T2D showed that 16 weeks treatment with aleglitazar 150 μg once daily (QD) was associated with significant placebo-adjusted changes in both TG (−43.4%) and HDL-C (+20.7%) (Table 2)[44]. In comparison, 16 weeks treatment with fenofibrate 200mg QD in the FIELD study reduced TG levels by 28.6% vs. placebo and increased HDL-C by 5.1% (Table 2)[12]. Although aleglitazar has greater PPARα-mediated effects on the lipid profile than fenofibrate, it is associated with several potential safety concerns, including weight gain, peripheral edema and increased creatinine kinase levels with corresponding decreases in estimated glomerular filtration rate[43–46]. Since it is unable to completely separate the beneficial effects of the PPARα-agonists from their unwanted AEs, aleglitazar is unlikely to be classified as a true SPPARMα.

Chemical structures of PPARα agonists.
Another dual agonist with SPPARM characteristics is GFT505 (Figure 3)[47, 48]. Developed by GenFit, GFT505 is a dual PPARα/δ-agonist with preferential activity on PPARα (Table 1)[48]. Phase II studies in people with combined dyslipidemia and abdominal obesity (N = 97) or prediabetes (N = 47) showed that 28 days treatment with GFT505 80 mg QD was associated with significant reductions in ALT and γ-glutamyl transpeptidase levels (Table 3)[48], suggesting potential benefits for GFT505 in patients with NASH/NAFLD. This is particularly important because, although the obesity-related incidence of fatty liver disease continues to rise, no effective treatments are currently available[49]. However, although GFT505 was associated with significant placebo-adjusted changes in the levels of TGs (−16.7% to −24.8%) and HDL-C (+7.8% to +9.3%)[48], the magnitude of the effects was similar to those obtained in clinical studies with fenofibrate (Table 2)[12]. GFT505 is, therefore, unlikely to be classified as a true SPPARMα due to its lack of superior lipid-modifying efficacy vs. fenofibrate.
K-877: The first of the SPPARMαs
K-877 (Kowa Co. Ltd.) is a highly potent and selective PPARα-agonist with SPPARM properties (Figure 2)[50–52]. It is currently undergoing Phase I trials in Europe and the USA and Phase III trials in Japan for the treatment of atherogenic dyslipidemia. K-877 contains an acidic region similar to that found in other PPARα-agonists but, to enhance PPARα activity and selectivity, unique benzoxazole and phenoxyalkyl side-chains have been added (Figure 3)[53]. Cell-based transactivation assays using hPPAR-GAL-4 chimeric receptors confirmed that, compared to fenofibrate, K-877 is a more potent PPARα-agonist, with a high degree of PPAR subtype-selectivity (Table 1)[52].
Pre-clinical studies in animal models for obesity demonstrated that low doses of K-877 (0.3-3.0 mg/kg) had a greater TG-lowering efficacy than 1,000-fold higher doses (300 mg/kg) of fenofibrate, an effect that was accompanied by higher levels of plasma FGF-21[52]. Furthermore, K-877 0.01-0.1 mg/kg significantly reduced atherosclerotic lesion area in LDL receptor-null mice fed a Western diet, and significantly reduced the expression of TNF-α and MCP-1 genes. Although there were slight reductions in all three of these parameters with fenofibrate 100 mg/kg, the benefit was not significant.
More recently, a Phase II 12-week dose-finding study (N = 224) showed that K-877 100 μg twice daily (BID) was well tolerated and had a greater lipid modifying efficacy than fenofibrate 100mg QD in patients with atherogenic dyslipidemia (13% with T2D)[50]. The incidences of AEs (47.4% with K-877, 47.2% with placebo and 56.8% with fenofibrate) and adverse drug reactions (5.3% with K-877, 8.3% with placebo and 10.8% with fenofibrate) were similar for K-877 and placebo and slightly higher for fenofibrate. No serious AEs were reported for K-877. As expected, fenofibrate was associated with significant increases vs. baseline in serum creatinine and homocysteine levels and little or no effect on ALT or γ-glutamyl transpeptidase levels (Table 3). In contrast, K-877 100 μg BID had little or no effect on CV or renal markers, and hepatic markers were improved. Together, these results suggest that K-877 has a better safety and tolerability profile than fenofibrate and might be useful for people with NASH/NAFLD.
As suggested by pre-clinical studies, the Phase II study demonstrated greater changes from baseline in fasting plasma TG and HDL-C levels with K-877 100 μg BID than with fenofibrate in people with atherogenic dyslipidemia (Table 2). Compared to placebo, significant changes in TG, HDL-C, non-HDL-C and very low-density lipoprotein were observed for K-877. Although not directly compared, all changes were more pronounced with K-877 than with fenofibrate. In addition, K-877 was associated with greater beneficial changes in the size of atherogenic lipoproteins than fenofibrate[50], suggesting that K-877 has the potential to improve lipoprotein quality as well as quantity.
Results from a ‘Cookie test’[54] subanalysis (N = 143) showed that 12 weeks treatment with K-877 had a significantly greater potential than fenofibrate to improve postprandial hyperlipidemia, a major risk factor for CV disease in people with MetS or T2D[51]. For example, K-877 suppressed the postprandial increase in TG, apo B-48 and remnant-like particle cholesterol, a major risk factor for ischemic heart disease[55]. Although no clear between-group differences in glucose or insulin levels were observed in the substudy, results from the Phase II trial showed that K-877 was associated with significant increases in plasma levels of FGF-21. Similarly, K-877 was associated with increased levels of FGF21 gene expression in the livers of LDL receptor-null mice and increased levels of plasma FGF-21 in ZF rats[52]. These observations are important because FGF21 expression in white adipose tissue increases in response to feeding and is generally associated with weight loss, antidiabetic and hypolipidemic effects in animal models of T2D and obesity[56, 57]. Further studies are required to fully understand the implications of these results and to examine the effects of K-877 on vascular inflammation in humans.
Conclusions
Fibrates play a central role in the reduction of macro- and microvascular risk associated with MetS and T2D[12, 13, 20–23]. However, they are weak PPARα agonists with limited efficacy due to dose-related AEs. To address this problem, a new generation of tissue-specific PPARα agonists — the SPPARMαs — is being developed that separates the receptor-mediated beneficial effects of the PPARα agonists from their unwanted side effects. Although a number of dual PPARα/γ and α/δ-agonists have been developed with SPPARMα characteristics, most are associated with unresolved safety issues[26, 42–44] or fail to provide a superior efficacy vs. standard treatment[47, 48].
Recently, a highly specific PPARα-agonist (K-877) has emerged with SPPARM properties. Although K-877 has not been compared with fenofibrate in head-to-head studies, it is approximately 10,000-fold more potent than fenofibrate in vitro[52], has a greater lipid-modifying efficacy at considerably lower doses both in animal models for obesity/T2D and in humans, and is associated with an improved safety/tolerability profile[50–52]. In addition, K-877 100 μg BID is associated with beneficial changes in markers for liver disease. This suggests that, in addition to improving lipid parameters in people with cardiometabolic diseases, K-877 might also be useful for the prevention of NASH/NAFLD. Further studies are required to characterize the effects of K-877 on the recruitment of cofactors and gene expression to clarify the precise molecular mechanism, and to investigate the long-term safety and clinical efficacy of K-877 in people with atherogenic dyslipidemia. However, results to date suggest that K-877 is the first true member of the SPPARMα family. If successful, this next generation of PPARα-agonists has the potential to supersede fibrates as the treatment of choice in patients with atherogenic dyslipidemia and could have a major impact on the management of residual macro- and microvascular risk associated with MetS and T2D.
Authors’ information
JCF is Past-President of the International Atherosclerosis Society. He is currently President of the Residual Risk Reduction Initiative (http://www.r3i.org/pg/1/), an international, academic, multidisciplinary non-profit organization, which is focused on addressing the high residual risk of macro- and microvascular complications in patients with atherogenic dyslipidemia.
Abbreviations
- AE:
-
Adverse event
- ALT:
-
Alanine aminotransferase
- Apo:
-
Apolipoprotein
- BID:
-
Twice daily
- CV:
-
Cardiovascular
- FIELD:
-
Fenofibrate intervention and event lowering in diabetes
- HDL-C:
-
High-density lipoprotein-cholesterol
- LDL-C:
-
Low-density lipoprotein-cholesterol
- MetS:
-
Metabolic syndrome
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- PPAR:
-
Peroxisome proliferator-activated receptor
- QD:
-
Once daily
- SPPARM:
-
Selective peroxisome proliferator-activated modulator
- T2D:
-
Type 2 diabetes
- TG:
-
Triglyceride.
References
World Health Organisation cardiovascular disease statistics. Fact sheet No. 317.http://www.who.int/mediacentre/factsheets/fs317/en/index.html.
Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R, Cholesterol Treatment Trialists’ (CTT) Collaboration: Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010, 376: 1670-1681. 10.1016/S0140-6736(10)61350-5.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R, Cholesterol Treatment Trialists' (CTT) Collaborators: Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet. 2005, 366: 1267-1278.
Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Peto R, Armitage J, Baigent C, Cholesterol Treatment Trialists' (CTT) Collaborators: Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008, 371: 117-125. 10.1016/S0140-6736(08)60104-X.
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O: Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003, 348: 383-393. 10.1056/NEJMoa021778.
Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J, Emerging Risk Factors Collaboration: Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009, 302: 1993-2000.
Millán Núñez-Cortés J, Montoya JP, Salas XP, Hernández Mijares A, Carey VJ, Hermans MP, Sacks FM, Fruchart JC: The REALIST (REsiduAl risk, LIpids and Standard Therapies) study: an analysis of residual risk attributable to lipid profile in acute coronary syndrome. Endocrinol Nutr. 2011, 58: 38-47. 10.1016/j.endonu.2010.10.004.
Carey VJ, Bishop L, Laranjo N, Harshfield BJ, Kwiat C, Sacks FM: Contribution of high plasma triglycerides and low high-density lipoprotein cholesterol to residual risk of coronary heart disease after establishment of low-density lipoprotein cholesterol control. Am J Cardiol. 2010, 106: 757-763. 10.1016/j.amjcard.2010.05.002.
Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvänne M, op Reimer WJ S, Vrints C, Wood D, Zamorano JL, Zannad F, European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG): European guidelines on cardiovascular disease prevention in clinical practice 2012: The Fifth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice. Eur Heart J. 2012, 33: 1635-1701.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A, Watts GF, European Atherosclerosis Society Consensus Panel: Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011, 32: 1345-1361. 10.1093/eurheartj/ehr112.
International Diabetes Federation Global guidelines for Type 2 diabetes 2012.http://www.idf.org/global-guideline-type-2-diabetes-2012.
Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, Forder P, Pillai A, Davis T, Glasziou P, Drury P, Kesäniemi YA, Sullivan D, Hunt D, Colman P, d'Emden M, Whiting M, Ehnholm C, Laakso M, FIELD study investigators: Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005, 366: 1849-1861.
Ginsberg HN, Elam MB, Lovato LC, Crouse JR, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Cushman WC, Simons-Morton DG, Byington RP, ACCORD Study Group: Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010, 362: 1563-1574.
Fruchart JC: Peroxisome proliferator-activated receptor-alpha (PPARalpha): at the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis. 2009, 205: 1-8. 10.1016/j.atherosclerosis.2009.03.008.
Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P, Manninen V: Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987, 317: 1237-1245. 10.1056/NEJM198711123172001.
The BIP Study Group: Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000, 102: 21-27.
Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J: Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999, 341: 410-418. 10.1056/NEJM199908053410604.
Sacks FM, Carey VJ, Fruchart JC: Combination lipid therapy in type 2 diabetes. N Engl J Med. 2010, 363: 692-694.
Rajamani K, Colman PG, Li LP, Best JD, Voysey M, D'Emden MC, Laakso M, Baker JR, Keech AC, FIELD study investigators: Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): A prespecified analysis of a randomised controlled trial. Lancet. 2009, 373: 1780-1788. 10.1016/S0140-6736(09)60698-X.
Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d'Emden MC, Crimet DC, O'Connell RL, Colman PG, FIELD study investigators: Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007, 370: 1687-1697. 10.1016/S0140-6736(07)61607-9.
Davis TM, Ting R, Best JD, Donoghoe MW, Drury PL, Sullivan DR, Jenkins AJ, O'Connell RL, Whiting MJ, Glasziou PP, Simes RJ, Kesäniemi YA, Gebski VJ, Scott RS, Keech AC, Fenofibrate Intervention and Event Lowering in Diabetes Study investigators: Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia. 2011, 54: 280-290. 10.1007/s00125-010-1951-1.
Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, Hubbard L, Esser BA, Lovato JF, Perdue LH, Goff DC, Cushman WC, Ginsberg HN, Elam MB, Genuth S, Gerstein HC, Schubart U, Fine LJ, ACCORD Study Group; ACCORD Eye Study Group: Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010, 363: 233-244.
Scott R, O'Brien R, Fulcher G, Pardy C, D'Emden M, Tse D, Taskinen MR, Ehnholm C, Keech A, Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators: Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care. 2009, 32: 493-498. 10.2337/dc08-1543.
Hottelart C, el Esper N, Achard JM, Pruna A, Fournier A: Fenofibrate increases blood creatinine, but does not change the glomerular filtration rate in patients with mild renal insufficiency. Nephrologie. 1999, 20: 41-44.
Issemann I, Green S: Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990, 347: 645-650. 10.1038/347645a0.
Dietz M, Mohr P, Kuhn B, Maerki HP, Hartman P, Ruf A, Benz J, Grether U, Wright MB: Comparative molecular profiling of the PPARalpha/gamma activator aleglitazar: PPAR selectivity, activity and interaction with cofactors. Chem Med Chem. 2012, 7: 1101-1111.
Fruchart JC, Duriez P, Staels B: Peroxisome proliferator-activated receptor-alpha activators regulate genes governing lipoprotein metabolism, vascular inflammation and atherosclerosis. Curr Opin Lipidol. 1999, 10: 245-257. 10.1097/00041433-199906000-00007.
Fruchart JC, Staels B, Duriez P: PPARS, metabolic disease and atherosclerosis. Pharmacol Res. 2001, 44: 345-352. 10.1006/phrs.2001.0871.
Fruchart JC, Duriez P, Staels B: Molecular mechanism of action of the fibrates. J Soc Biol. 1999, 193: 67-75.
Fruchart JC, Duriez P: Mode of action of fibrates in the regulation of triglyceride and HDL-cholesterol metabolism. Drugs Today (Barc). 2006, 42: 39-64. 10.1358/dot.2006.42.1.963528.
Neve BP, Corseaux D, Chinetti G, Zawadzki C, Fruchart JC, Duriez P, Staels B, Jude B: PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation. 2001, 103: 207-212. 10.1161/01.CIR.103.2.207.
Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B: Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999, 274: 32048-32054. 10.1074/jbc.274.45.32048.
Roberts LD, Murray AJ, Menassa D, Ashmore T, Nicholls AW, Griffin JL: The contrasting roles of PPARdelta and PPARgamma in regulating the metabolic switch between oxidation and storage of fats in white adipose tissue. Genome Biol. 2011, 12: R75-10.1186/gb-2011-12-8-r75.
Gervois P, Fruchart JC, Staels B: Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab. 2007, 3: 145-156. 10.1038/ncpendmet0397.
Fruchart JC, Davignon J, Bard JM, Grothe AM, Richard A, Fievet C: Effect of fenofibrate treatment on type III hyperlipoproteinemia. Am J Med. 1987, 83: 71-74. 10.1016/0002-9343(87)90874-6.
Duez H, Lefebvre B, Poulain P, Torra IP, Percevault F, Luc G, Peters JM, Gonzalez FJ, Gineste R, Helleboid S, Dzavik V, Fruchart JC, Fiévet C, Lefebvre P, Staels B: Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arterioscler Thromb Vasc Biol. 2005, 25: 585-591. 10.1161/01.ATV.0000154140.73570.00.
Tenenbaum A, Fisman EZ: Balanced pan-PPAR activator bezafibrate in combination with statin: comprehensive lipids control and diabetes prevention?. Cardiovasc Diabetol. 2012, 11: 140-10.1186/1475-2840-11-140.
Lewis JS, Jordan VC: Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005, 591: 247-263. 10.1016/j.mrfmmm.2005.02.028.
Balint BL, Nagy L: Selective modulators of PPAR activity as new therapeutic tools in metabolic diseases. Endocr Metab Immune Disord Drug Targets. 2006, 6: 33-43. 10.2174/187153006776056620.
Higgins SL, dePaoli AM: Selective peroxisome proliferator-activated receptor gamma (PPARγ) modulation as a strategy for safer therapeutic PPARγ activation. Am J Clin Nutr. 2010, 91: 267S-272S. 10.3945/ajcn.2009.28449E.
Doshi LS, Brahma MK, Bahirat UA, Dixit AV, Nemmani KV: Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin Investig Drugs. 2010, 19: 489-512. 10.1517/13543781003640169.
Rosenson RS, Wright RS, Farkouh M, Plutzky J: Modulating peroxisome proliferator-activated receptors for therapeutic benefit? Biology, clinical experience, and future prospects. Am Heart J. 2012, 164: 672-680. 10.1016/j.ahj.2012.06.023.
Younk LM, Uhl L, Davis SN: Pharmacokinetics, efficacy and safety of aleglitazar for the treatment of type 2 diabetes with high cardiovascular risk. Expert Opin Drug Metab Toxicol. 2011, 7: 753-763. 10.1517/17425255.2011.579561.
Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M: Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2009, 374: 126-135. 10.1016/S0140-6736(09)60870-9.
Herz M, Gaspari F, Perico N, Viberti G, Urbanowska T, Rabbia M, Wieczorek Kirk D: Effects of high dose aleglitazar on renal function in patients with type 2 diabetes. Int J Cardiol. 2011, 151: 136-142. 10.1016/j.ijcard.2010.08.037.
Investor update 2012: Roche's aleglitazar renal safety study AleNephro meets primary endpoint.http://www.roche.com/investors/ir_update/inv-update-2012-11-05.htm.
Fruchart JC: Novel peroxisome proliferator activated receptor-alpha agonists. Am J Cardiol. 2007, 100 (Suppl 11A): n41-n46.
Cariou B, Zair Y, Staels B, Bruckert E: Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011, 34: 2008-2014. 10.2337/dc11-0093.
Tailleux A, Wouters K, Staels B: Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 1821, 2012: 809-818.
Ishibashi S, Arai H, Yamashita S, Araki E, Yamada N, Ishibashi S, Arai H, Yamashita S, Araki E, Yamada N: Benefical effects of K-877, a potent and highly selective PPARα agonist, on plasma lipoprotein profile in patients with atherogenic dyslipidemia. Abstract 525.http://www.kenes.com/eas2012/abstracts/pdf/525.pdf.
Ishibashi S, Yamashita S, Arai H, Araki E, Yamada N: Suppression of postprandial triglyceride, Remnant-like Particles-Cholesterol (RLP-C) and ApoB48 surge by K-877, a potent and highly selective PPARα agonist. Abstract 618.http://www.kenes.com/eas2012/abstracts/pdf/618.pdf.
Takizawa T: K-877, a highly potent and selective PPARα agonist, improves dyslipidemia and atherosclerosis in experimental animal models. Abstract 787.http://www.kenes.com/eas2012/abstracts/pdf/787.pdf.
Yamazaki Y, Abe K, Toma T, Nishikawa M, Ozawa H, Okuda A, Araki T, Oda S, Inoue K, Shibuya K, Staels B, Fruchart JC: Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorg Med Chem Lett. 2007, 17: 4689-4693. 10.1016/j.bmcl.2007.05.066.
Harano Y, Miyawaki T, Nabiki J, Shibachi M, Adachi T, Ikeda M, Ueda F, Nakano T: Development of cookie test for the simultaneous determination of glucose intolerance, hyperinsulinemia, insulin resistance and postprandial dyslipidemia. Endocr J. 2006, 53: 173-180. 10.1507/endocrj.53.173.
Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG: Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013, 61: 427-436. 10.1016/j.jacc.2012.08.1026.
Canto C, Auwerx J: Cell biology: FGF21 takes a fat bite. Science. 2012, 336: 675-676. 10.1126/science.1222646.
Ong KL, Rye KA, O'Connell R, Jenkins AJ, Brown C, Xu A, Sullivan DR, Barter PJ, Keech AC, FIELD study investigators: Long-term fenofibrate therapy increases Fibroblast Growth Factor 21 and Retinol-Binding Protein 4 in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2012, 97: 4701-4708. 10.1210/jc.2012-2267.
Acknowledgements
There are no sources of funding for this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
J-C Fruchart has received honoraria as a consultant for SMB laboratories, McCain and Kowa Co. Ltd.
Authors’ contribution
J-C Fruchart confirms he is the sole author of this review.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fruchart, JC. Selective peroxisome proliferator-activated receptorα modulators (SPPARMα): The next generation of peroxisome proliferator-activated receptor α-agonists. Cardiovasc Diabetol 12, 82 (2013). https://doi.org/10.1186/1475-2840-12-82
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2840-12-82