
Abstract
Angiopoietin-like protein 3 (ANGPTL3) is a secretory protein regulating plasma lipid levels via affecting lipoprotein lipase- and endothelial lipase-mediated hydrolysis of triglycerides and phospholipids. ANGPTL3-deficiency due to loss-of-function mutations in the ANGPTL3 gene causes familial combined hypobetalipoproteinemia (FHBL2, OMIM # 605019), a phenotype characterized by low concentration of all major lipoprotein classes in circulation. ANGPTL3 is therefore a potential therapeutic target to treat combined hyperlipidemia, a major risk factor for atherosclerotic coronary heart disease. This review focuses on the mechanisms behind ANGPTL3-deficiency induced FHBL2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dietary fats are transported in the circulation in lipoprotein particles, lipid-apolipoprotein complexes containing a surface of phospholipid monolayer together with free cholesterol and structural apolipoproteins (apo) and a hydrophobic core including cholesterol esters and triglycerides (TG) [1, 2]. In humans, TGs are packed and secreted in the small intestine in apoB-48-containing chylomicrons (CM) and in the liver in apoB-100 containing very low density lipoproteins (VLDL) [1, 2]. TG in CM and VLDL are hydrolyzed in circulation by lipoprotein lipase (LPL) [3]. The resulting free fatty acids are taken up by tissues primarily via the function of CD36 transporter [4]. After deprivation of TG, CM, and VLDL remnants are cleared via specific liver receptors. Some VLDL remnants are converted in circulation, via hepatic lipase (HL) function, into cholesterol-rich low density lipoproteins (LDL). LDL-receptors recognize LDL-bound apoB-100, resulting in the uptake of the circulating LDL, mainly by the liver and by the steroidogenic tissues [3]. Another class of lipoproteins, high density lipoproteins (HDL), are functionally important in reverse cholesterol transport, to clear excess accumulated cholesterol from the periphery, and transport it back to the liver for excretion [5]. Disturbances in lipoprotein metabolism by genetic variants in genes affecting LPL activity (ANGPTL3, APOC3, APOC2, APOA5), remnant clearance (APOE, LIPC, LRP1), LDL receptor activity (PCSK9, LDLR), lipoprotein secretion (APOB, MTP) and HDL (APOA1, ABCA1) have been detected in humans [6, 7].
High blood levels of saturated fat and cholesterol are major risk factors for coronary heart disease (CHD). Lipids within apoB-containing lipoproteins can accumulate in arterial intima and result in plaque formation and the development of atherosclerosis [8]. LDL-Cholesterol is a major cause for the generation of atherosclerotic plaques [8], however, an independent predictive value for elevated circulating TG in coronary heart disease (CHD) risk has been demonstrated in prospective studies [9, 10]. Existing pharmaceuticals, such as statins, fibrates, niacin, and fish oil, which target LDL-cholesterol (LDL-C), TG, and HDL-cholesterol (HDL-C) are prescribed alone or in various combinations to target dyslipidemias [11]. ANGPTL3 is a potential therapeutic target for alternative treatment of combined hyperlipidemia.
Characteristics of ANGPTL3
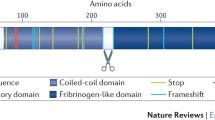
The angiopoietin-like proteins (ANGPTLs) form collectively a specific family of secretory proteins sharing a structural similarity to angiopoietins, the key factors that regulate angiogenesis [12]. ANGPTL3 is a 460-amino-acid (aa) polypeptide with a distinctive signal peptide sequence, a N-terminal helical domain (predicted to form dimeric or trimeric coiled-coil structures) and a C-terminal globular fibrinogen homology domain [13]. The N-terminal coiled—coil region (17–207 aa), specifically the amino—acid domain 61–66, affects plasma triglyceride (TG) levels via reversibly inhibiting catalytic activity of LPL [14, 15]. The fibrinogen-like domain (207–460 aa) binds to integrin αvβ3 receptor and affects angiogenesis [16]. A short linker region (at 221–222 and 224–225) between N- and C-terminal domains functions as a furin cleavage site. The truncated form of cleaved ANGPTL3 displays enhanced inhibitory activity for LPL and endothelial lipase (EL) suggesting that furin-facilitated cleavage of ANGPTL3 is physiologically important [14, 17]. Angiopoietin-like protein 8 (ANGPTL8), an insulin-induced protein sharing sequence homology with ANGPTL3, may also regulate ANGPTL3 proteolytic activation in vivo [18–20].
The association of ANGPTL3 with lipoprotein metabolism was discovered in hypertriglyceridemic and hyperglycemic obese KK mice, which spontaneously inherited a recessive trait causing low levels of triglyceride in plasma [21] and elevated post-heparin LPL activity [22]. The hypolipidemic mice carried a 4-base pair insertion in exon 6 introducing a stop codon in the Angptl3 gene. Low lipid levels in the mutant mice were normalized by an intravenous injection of ANGPTL3 [21]. When the region including Angptl3 LOF was introduced to atherogenic apoE-knock out mice, the prevalence of atherosclerotic lesions significantly declined [23] indicating that ANGPTL3-deficiency and the resulting hypolipidemia in these mice, was protective against the development of atherosclerosis.
Population studies and clinical characterization of ANGPTL3 sequence variants in humans
In humans, both common and rare genetic variants in ANGPTL3 gene have been reported to associate with plasma lipids. Three genome-wide scans (n = 8800) from Finnish and Italian subjects showed a strong association between ANGPTL3 and plasma TG levels [6]. Since then, more data have been published on the association between ANGPTL3 variants and plasma TG, LDL, HDL, and total cholesterol levels [24]. As much as 4 % of the Dallas Heart Study population (n = 3,551) with plasma TG in the lowest quartile carried rare LOF mutations in ANGPTL3, ANGPTL4, or ANGPTL5 [25].
Genetic LOF variants in ANGPTL3 cause a rare recessive disorder known as familial combined hypobetalipoproteinemia (FHBL2, OMIM # 605019). FHBL2 subjects display a distinct plasma lipid phenotype including very low VLDL, LDL, and HDL levels, and consequently low total TG and cholesterol levels ([26], see Table 1). Involvement of ANGPTL3 LOF mutation in FHBL2 was discovered by genome sequencing of two siblings of European descent who were compound heterozygotes for two distinct nonsense mutations in ANGPTL3 (p.E129* and p.S17*) [27]. p.S17* LOF mutation is common among the residents of a town Campodimele (Latina, Italy), of which 9.4 % carry the mutant variant [26]. Only the homozygous carriers of p.S17*, who have undetectable levels of ANGPTL3 in circulation, manifest low lipid and lipoprotein levels whereas heterozygote carriers, with a 50 % reduction in circulatory ANGPTL3, are not affected by FHBL2 (26, 34, see Table 1). In addition to p.S17* and p.E129* LOF mutations, more subjects with rare LOF variants in ANGPTL3 are detected in Spanish and Italian FHBL2 families [28–30]. Among 78 sequenced American and Italian FHBL2 subjects, 8 subjects carried 9 different nonsense mutations in ANGPTL3, with no mutations in APOB, PCSK9, or MTP. The prevalence for ANGPTL3 LOF mutations in all of the FHBL2 cases was therefore 10 % [31].
ANGPTL3 function
An increasing number of evidence indicates that the hypolipidemic phenotype in ANGPTL3-deficiency is driven by accelerated turnover of lipoproteins and the resulting altered energy substrate distribution among tissues (see Fig. 1).

Function of ANGPTL3 in lipoprotein metabolism. ANGPTL3-deficiency causes enhanced activity of lipoprotein lipase (LPL) in the muscle and adipose tissue and accelerated clearance of TG-rich lipoproteins. Decreased release of FFA, from adipose tissue (lipolysis), hypothetically results in scarcity of FFA substrates for hepatic de novo synthesis of TG and cholesterol, and consequently decreased lipidation of VLDL. There are no reports on whether ANGPTL3-deficiency would affect the lipidation and secretion of intestinal TG-enriched chylomicron (CM)-particles
ANGPTL3 and lipoprotein clearance
Lipoprotein lipase (LPL) is located on the luminal side of the vascular endothelium of the capillaries in extra hepatic tissues [3]. LPL plays a critical role in hydrolyzing TG carried by VLDL and chylomicron (CM) particles in the circulation. Activity of LPL in white adipose tissue (WAT) is elevated in fed state and reduced in fasted state [3]. Therefore, in the fed state, the flow of dietary fat is primarily targeted into WAT, not into skeletal muscle, which would rely on glucose in energy production [3]. During fasting, skeletal muscle is believed to be the main site for LPL activity [3]. Endothelial lipase (EL) is located on the luminal side of the vascular endothelial cells (like LPL) and shares 44 % homology with LPL and 41 % with hepatic lipase (HL) [32]. Unlike LPL, EL is more specific in hydrolyzing lipoprotein phospholipids, especially in HDL-particles, rather than TG [33].
Both LPL and EL activities are increased in humans and mice with Angptl3 deficiency [34–36]. There are no reports on enhanced HL activity in ANGPTL3-deficiency. In Angptl3-knockout mice, TGs in postprandial lipoproteins were directed in brown adipose tissue (BAT) and muscle instead of WAT [37]. The study indicates that ANGPTL3-deficiency might induce LPL activity in the oxidative tissues during feeding and accelerate the clearance of TG-rich particles. Since LPL or EL do not hydrolyze cholesterol esters, the mechanism for low LDL-C levels in ANGPTL3-deficiency, has remained elusive. Wang et al. reported that LDL-cholesterol levels were equally reduced in wildtype mice and LDLR, LRP1, or APOE knockout mice treated with ANGPTL3 inactivating antibody [38]. These results suggest that reduced LDL-cholesterol in ANGPTL3-deficiency is not caused by enhanced clearance of cholesterol via LDL-receptors. Alternatively, reduced LDL-C levels may be a result of lower secretion rates of LDL precursors, hepatic VLDL particles.
ANGPTL3-deficiency in FHBL2 causes a reduction in HDL-particles, the facilitators of reverse cholesterol transport. As increased activity of EL is associated with reduced plasma HDL-cholesterol (HDL-C) levels [39], higher activity of EL might contribute to low HDL levels demonstrated in ANGPTL3-deficiency. Even though low HDL is an established risk factor for atherosclerosis [40], decreased HDL levels did not result in accelerated atherosclerosis in ANGPTL3-deficient subjects, probably because of the lifelong exposure to low levels of VLDL and LDL [30]. The lipolysis of TG-rich lipoproteins and HDL, via LPL and EL, respectively, may result in increased turnover of lipoprotein remnants and therefore low plasma apoB and apoA-I levels.
The role of ANGPTL3 in lipoprotein production
Hepatic VLDL synthesis relies on the availability of triglycerides which are synthesized from substrates derived from the supply of free fatty acids from adipocytes [41], from VLDL and chylomicron remnants [1, 2] and from simple sugars [42] via the portal vein. Insulin signaling reduces hepatic VLDL synthesis and secretion which is manifested by reduced lipidation of TG-rich VLDL particles [43]. Mice treated with monoclonal ANGPTL3 inactivating antibody did not show lower production rates for apoB-100 [38], the major structural protein of VLDL, indicating that there are no changes within the amount of secreted VLDL particles. However, the amount of TG in each VLDL particle might be declined [44]. Reduced lipidation of VLDL in ANGPTL3-deficiency may be caused by decreased supply of free fatty acids from the circulation into the liver. These observations are in agreement with the reported low FFA levels in ANGPTL3-deficient subjects [34]. A metabolic study with ANGPTL3-silenced hepatocytes suggests that silencing of ANGPTL3 causes a shift in substrate utilization to favor glucose, instead of FFA [44], which supports the theory of attenuated FFA supply into the liver.
Cholesterol is either ingested from nutrition or synthesized de novo by the liver. Reduced availability of substrates may decrease hepatic de novo synthesis of cholesterol resulting in the secretion of cholesterol-poor VLDL in ANGPTL3-deficiency. Interestingly, ANGPTL3 is a downstream target for liver X receptors (LXR’s), transcription factors with sterol binding ability [45]. Oxysterols, cholesterol, and other cholesterol metabolites are natural ligands for LXRs, and LXR activation protects the liver from cholesterol overload by stimulating bile acid formation and excretion, lipogenesis, HDL-mediated reverse cholesterol transport, and glucose metabolism [46, 47]. Synthetic LXR ligands and a high cholesterol diet induce Angptl3 expression in the liver [48, 49]. On the contrary, low LXR, and low Angptl3 expression (in ANGPTL3-deficiency) may therefore be linked to hypocholesterolemic and antilipogenic status in the liver.
Adipose tissue
White adipose tissue (WAT) serves as the primary tissue for storage of triglycerides. Postprandial TGs are primarily directed to WAT via LPL-mediated hydrolysis [3]. In Angptl3-knockout mice, TG-derived FFA uptake was decreased in WAT while elevated in BAT and muscle. Such a shift in substrate distribution between tissues, however, did not result in differences in TG-content of the liver, adipose tissue, and heart, possibly because reduced lipid uptake in WAT was balanced by elevated glucose uptake [37].
TG-deposits in adipose tissue are dismantled by lipolysis, a catabolic breakdown of TG mediated by concerted action of adipose TG lipase (ATGL), and hormone sensitive lipase (HSL) [41]. Lipolysis is inhibited in the postprandial state by elevated insulin concentrations [50]. During fasting, lipolytic activity increases the release of FFA and free glycerol into circulation, to provide substrates for energy production in the oxidative tissues [41]. In Angptl3-knockout mice, both FFA and glycerol were reduced in plasma and were preserved by administration of ANGPTL3 [51]. Therefore, absence of circulatory ANGPTL3 (in ANGPTL3-deficiency) may decrease FFA lipolysis in the adipose tissue and consequently reduce plasma FFA and glycerol levels.
Conclusions
Several mutations in the APOB, PCSK9, and MTP genes result in familial hypobetalipoproteinemia (FHBL) and abetalipoproteinemia (ABL) defined as low, or absent levels of apoB-100, and LDL-C in plasma [52, 53]. LOF mutations in the ANGPTL3 results in familial combined hypobetalipoproteinemia (FHBL2) with reduced levels of all major lipoprotein classes (VLDL, LDL, HDL) in plasma. The hypolipidemic phenotype caused by ANGPTL3 inactivation is likely an outcome of increased activity of two lipolytic enzymes, LPL, and EL, which account for increased turnover of lipoproteins, and reduced TG and HDL levels in the plasma. Low plasma FFA levels in ANGPTL3-deficiency may be due to decreased lipolysis in the adipose tissue which may also contribute to reduced lipidation of hepatic VLDL, and LDL, and consequently low TG and LDL-C. The impact of ANGPTL3-deficiency on intestinal chylomicron production remains unknown. However, there are no reports on steatorrhea among ANGPTL3-deficient subjects.
No adverse health effects or developmental alterations during early or later life-time are reported in ANGPTL3-deficient p.s17* LOF mutation carriers [26, 34]. Perhaps ANGPTL3 may have been functionally important during long periods of fasting to primarily ensure FFA release from TG-deposits, via controlling lipolytic activity, and by guiding energy substrates into either oxidative tissues or WAT by selective LPL inhibition. Such tight regulation between energy storage and substrate release might be important during times with less frequent access to energy rich diet.
The striking hypolipidemic phenotype in ANGPTL3 deficiency shows the potential of ANGPTL3 inactivation as a treatment for correcting hyperlipidemia. Therefore, ANGPTL3 inactivation may have important therapeutic implications for treatment of metabolic syndrome, type 2 diabetes, and patients in high risk of heart disease. The first reports on ANGPTL3 inhibitors have shown promising results. Administration of ANGPTL3 inactivating antibody to monkeys and dyslipidemic mice reduced circulating plasma levels of TG, LDL-cholesterol, and HDL-cholesterol significantly [54]. Another strategy for ANGPTL3 inactivation, and perhaps a better one to avoid immune response associated with administration of antibodies, could be selective antisense inhibition of ANGPTL3 expression in the liver. Despite favorable reduction in LDL-C and TG, it is still uncertain whether decreased HDL-C would have an influence on overall CVD risk in ANGPTL3-inhibitor treated patients.
References
T.G. Redgrave, Chylomicrons, in Lipoproteins in Health and Disease, ed. by D.J. Betteridge, D.R. Illingworth, J. Shepherd (Arnold, London, 1999), pp. 31–54
H.N. Ginsberg, J.L. Dixon, I.J. Goldberg, Illingworth, J. Shepherd, VLDL/LDL cascade system: assembly, secretion and intravascular metabolism of apoprotein B-containing lipoproteins, in Lipoproteins in Health and Disease, ed. by D.J. Betteridge (Arnold, London, 1999), pp. 55–70
C.J. Packard, J. Shepher, Physiology of the lipoprotein transport system: an overview of lipoprotein metabolism, in Lipoproteins in Health and Disease, ed. by D.J. Betteridge, D.R. Illingworth, J. Shepherd (Arnold, London, 1999), pp. 17–30
I.J. Goldberg, R.H. Eckel, N.A. Abumrad, Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 50, S86–S90 (2009)
A.R. Tall, L. Yvan-Charvet, N. Terasaka, T. Pagler, N. Wang, HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 7(5), 365–375 (2008)
C. Willer et al., Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 40(2), 161–169 (2008)
S. Kathiresan, O. Melander, C. Guiducci, A. Surti, NP. Burtt, MJ. Rieder, GM. Cooper, C. Roos, BF. Voight, AS. Havulinna, B. Wahlstrand, T. Hedner, D. Corella, ES. Tai, JM. Ordovas, G. Berglund, E. Vartiainen, P. Jousilahti, B. Hedblad, MR. Taskinen, C. Newton-Cheh, V. Salomaa, L. Peltonen, L. Groop, DM. Altshuler, M. Orho-Melander, Sixnew loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 40(2),189–97 (2008). Erratum in: Nat. Genet. 40(11):1384 (2008)
N. Woolf, Pathology of atherosclerosis, in Lipoproteins in Health and Disease, ed. by D.J. Betteridge, D.R. Illingworth, J. Shepherd (Arnold, London, 1999), pp. 533–540
B.G. Nordestgaard, M. Benn, P. Schnohr, A. Tybjaerg-Hansen, Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA 298(3), 299–308 (2007)
H.L. Staniak, W. Salgado Filho, M.H. Miname, I.M. Benseñor, P.A. Lotufo, R. Sharovsky, C.E. Rochitte, R.D. Bittencourt, R.D. Santos, Association between postprandialtriglycerides and coronary artery disease detected by coronary computed tomography angiography. Atherosclerosis 233(2), 381–386 (2014)
R.C. Oh, J.B. Lanier, Management of hypertriglyceridemia. Am Fam. Physician. 75(9), 1365–1371 (2007)
T. Hato, M. Tabata, Y. Oike, The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 18, 6–14 (2008)
D. Conklin, D. Gilbertson, D.W. Taft, M.F. Maurer, T.E. Whitmore, D.L. Smith, K.M. Walker, L.H. Chen, S. Wattler, M. Nehls, K.B. Lewis, Identification of a mammalian angiopoietin-related protein expressed specifically in liver. Genomics 62(3), 477–482 (1999)
M. Ono, T. Shimizugawa, M. Shimamura, K. Yoshida, C. Noji-Sakikawa, Y. Ando, R. Koishi, H. Furukawa, Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J Biol. Chem. 278(43), 41804–41809 (2003)
L. Shan, X.C. Yu, Z. Liu, Y. Hu, L.T. Sturgis, M.L. Miranda, Q. Liu, The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J Biol. Chem. 284(3), 1419–1424 (2009)
G. Camenisch, M.T. Pisabarro, D. Sherman, J. Kowalski, M. Nagel, P. Hass, M.H. Xie, A. Gurney, S. Bodary, X.H. Liang, K. Clark, M. Beresini, N. Ferrara, H.P. Gerber, ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. J Biol. Chem. 277(19), 17281–17290 (2002)
W. Jin, X. Wang, J.S. Millar, T. Quertermous, G.H. Rothblat, J.M. Glick, D.J. Rader, Hepatic proprotein convertases modulate HDL metabolism. Cell Metab. 6(2), 129–136 (2007)
F. Quagliarini, Y. Wang, J. Kozlitina, N.V. Grishin, R. Hyde, E. Boerwinkle, D.M. Valenzuela, A.J. Murphy, J.C. Cohen, H.H. Hobbs, Atypical angiopoietin-like protein that regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 109(48), 19751–19756 (2012)
Z. Fu, F. Yao, A.B. Abou-Samra, R. Zhang, Lipasin, thermoregulated in brown fat, is a novel but atypical member of the angiopoietin-like protein family. Biochem. Biophys. Res. Commun. 430(3), 1126–1131 (2013)
R. Zhang, A.B. Abou-Samra, A dual role of lipasin (betatrophin) in lipid metabolism and glucose homeostasis: consensus and controversy. Cardiovasc. Diabetol. 13, 133 (2014)
R. Koishi, Y. Ando, M. Ono, M. Shimamura, H. Yasumo, T. Fujiwara, H. Horikoshi, H. Furukawa, Angptl3 regulates lipid metabolism in mice. Nat. Genet. 30(2), 151–157 (2002)
T. Shimizugawa, M. Ono, M. Shimamura, K. Yoshida, Y. Ando, R. Koishi, K. Ueda, T. Inaba, H. Minekura, T. Kohama, H. Furukawa, ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J. Biol. Chem. 277(37), 33742–33748 (2002)
Y. Ando, T. Shimizugawa, S. Takeshita, M. Ono, M. Shimamura, R. Koishi, H. Furukawa, A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. J. Lipid Res. 44(6), 1216–1223 (2003)
T.M. Teslovich, K. Musunuru et al., Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466(7307), 707–713 (2010)
S. Romeo, W. Yin, J. Kozlitina, L.A. Pennacchio, E. Boerwinkle, H.H. Hobbs, J.C. Cohen, Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin. Invest. 119(1), 70–79 (2009)
I. Minicocci, A. Montali, M.R. Robciuc, F. Quagliarini, V. Censi, G. Labbadia, C. Gabiati, G. Pigna, M.L. Sepe, F. Pannozzo, D. Lütjohann, S. Fazio, M. Jauhiainen, C. Ehnholm, M. Arca, Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J. Clin. Endocrinol. Metab. 97(7), E1266–E1275 (2012)
K. Musunuru et al., Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 363, 2220–2227 (2010)
M. Arca, I. Minicocci, M. Maranghi, The angiopoietin-like protein 3: a hepatokine with expanding role in metabolism. Curr. Opin. Lipidol. 24(4), 313–320 (2013)
J.M. Martín-Campos, R. Roig, C. Mayoral, S. Martinez, G. Marti, J.A. Arroyo, J. Julve, F. Blano-Vaca, Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clin. Chim. Acta. 413, 552–555 (2012)
L. Pisciotta et al., Characterization of three kindred with familial combined hypolipidemia due to loss of function mutations of ANGPTL3. Cardiovasc. Genet. 5, 42–50 (2012)
D. Noto, A.B. Cefalù, V. Valenti, F. Fayer, E. Pinotti, M. Ditta, R. Spina, G. Vigna, P. Yue, S. Kathiresan, P. Tarugi, M.R. Averna, Prevalence of ANGPTL3 and APOB gene mutations in subjects with combined hypolipidemia. Arterioscler. Thromb. Vasc. Biol. 32(3), 805–809 (2012)
K. Hirata, H.L. Dichek, J.A. Cioffi, S.Y. Choi, N.J. Leeper, L. Quintana, G.S. Kronmal, A.D. Cooper, T. Quertermous, Cloning of a unique lipase from endothelial cells extends the lipase gene family. J. Biol. Chem. 274(20), 14170–14175 (1999)
M.G. McCoy, G.S. Sun, D. Marchadier, C. Maugeais, J.M. Glick, D.J. Rader, Characterization of the lipolytic activity of endothelial lipase. J. Lipid Res. 43, 921–929 (2002)
M.R. Robciuc, M. Maranghi, A. Lahikainen, D. Rader, A. Bensadoun, K. Öörni, J. Metso, I. Minicocci, E. Ciociola, F. Ceci, A. Montali, M. Arca, C. Ehnholm, M. Jauhiainen, Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler. Thromb. VascBiol. 33(7), 1706–1713 (2013)
J. Liu, H. Afroza, D.J. Rader, W. Jin, Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J. Biol. Chem. 285(36), 27561–27570 (2010)
M. Shimamura, M. Matsuda, H. Yasumo, M. Okazaki, K. Fujimoto, K. Kono, T. Shimizugawa, Y. Ando, R. Koishi, T. Kohama, N. Sakai, K. Kotani, R. Komuro, T. Ishida, K. Hirata, S. Yamashita, H. Furukawa, I. Shimomura, Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler. Thromb. Vasc. Biol. 27(2), 366–372 (2007)
Y. Wang, M.C. McNutt, S. Banfi, M.G. Levin, W.L. Holland, V. Gusarova, J. Gromada, J.C. Cohen, H.H. Hobbs, Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc. Natl. Acad. Sci. USA 112(37), 11630–11635 (2015)
Y. Wang, V. Gusarova, S. Banfi, J. Gromada, J.C. Cohen, H.H. Hobbs, Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J. Lipid Res. 56(7), 1296–1307 (2015)
M. Jaye, K.J. Lynch, J. Krawiec, D. Marchadier, C. Maugeais, K. Doan, V. South, D. Amin, M. Perrone, D.J. Rader, A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 21(4), 424–428 (1999)
J.W. Gofman, W. Young, R. Tandy, Ischemic heart disease, atherosclerosis, and longevity. Circulation 34(4), 679–697 (1966)
R.E. Duncan, M. Ahmadian, K. Jaworski, E. Sarkadi-Nagy, H.S. Sul, Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27, 79–101 (2007)
M.C. Moore, K.C. Coate, J.J. Winnick, Z. An, A.D. Cherrington, Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. 3(3), 286–294 (2012)
M. Adiels, J. Westerbacka, A. Soro-Paavonen, A.M. Häkkinen, S. Vehkavaara, M.J. Caslake, C. Packard, S.O. Olofsson, H. Yki-Järvinen, M.R. Taskinen, J. Borén, Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 50(11), 2356–2365 (2007)
A. Tikka, J. Soronen, P.P. Laurila, J. Metso, C. Ehnholm, M. Jauhiainen, Silencing of ANGPTL 3 (angiopoietin-like protein 3) in human hepatocytes results in decreased expression of gluconeogenic genes and reduced triacylglycerol-rich VLDL secretion upon insulin stimulation. Biosci. Rep. 34(6), e00160 (2014)
M. Baranowski, Biological role of liver X receptors. J. Physiol. Pharmacol. 59(Suppl 7), 31–55 (2008)
S.D. Lee, P. Tontonoz, Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis 242(1), 29–36 (2015)
B.A. Laffitte, L.C. Chao, J. Li, R. Walczak, S. Hummasti, S.B. Josephm, A. Castrillo, D.C. Wilpitz, D.J. Mangelsdorf, J.L. Collins, E. Saez, P. Tontonoz, Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. USA 100(9), 5419–5424 (2003)
R. Kaplan, T. Zhang, M. Hernandez, R.X. Gan, S.D. Wright, M.G. Waters, T.Q. Cai, Regulation of the angiopoietin-like protein 3 gene by LXR. J. Lipid Res. 44(1), 136–143 (2003)
T. Inaba, M. Matsuda, M. Shimamura, N. Takei, N. Terasaka, Y. Ando, H. Yasumo, R. Koishi, M. Makishima, I. Shimomura, Angiopoietin-like protein 3 mediates hypertriglyceridemia induced by the liver X receptor. J. Biol. Chem. 278(24), 21344–21351 (2003)
S.M. Choi, D.F. Tucker, D.N. Gross, R.M. Easton, L.M. DiPilato, A.S. Dean, B.R. Monks, M.J. Birnbaum, Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell Biol. 30(21), 5009–5020 (2010)
M. Shimamura, M. Matsuda, S. Kobayashi, Y. Ando, M. Ono, R. Koishi, H. Furukawa, M. Makishima, I. Shimomura, Angiopoietin-like protein 3, a hepatic secretory factor, activates lipolysis in adipocytes. Biochem. Biophys. Res. Commun. 301(2), 604–609 (2003)
F.K. Welty, Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol. 25(3), 161–168 (2014)
J.I. Pulai, R.J. Neuman, A.W. Groenewegen, J. Wu, G. Schonfeld, Genetic heterogeneity in familial hypobetalipoproteinemia: linkage and non-linkage to the apoB gene in Caucasian families. Am. J. Med. Genet. 76, 79–86 (1998)
V. Gusarova, C.A. Alexa, Y. Wang, A. Rafique, J.H. Kim, D. Buckler, I.J. Mintah, L.M. Shihanian, J.C. Cohen, H.H. Hobbs, Y. Xin, D.M. Valenzuela, A.J. Murphy, G.D. Yancopoulos, J. Gromada, ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J. Lipid Res. 56(7), 1308–1317 (2015)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tikka, A., Jauhiainen, M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine 52, 187–193 (2016). https://doi.org/10.1007/s12020-015-0838-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0838-9