
Abstract
Idiopathic pulmonary fibrosis (IPF) is a prototype of lethal, chronic, progressive interstitial lung disease of unknown etiology. Over the past decade, macrophage has been recognized to play a significant role in IPF pathogenesis. Depending on the local microenvironments, macrophages can be polarized to either classically activated (M1) or alternatively activated (M2) phenotypes. In general, M1 macrophages are responsible for wound healing after alveolar epithelial injury, while M2 macrophages are designated to resolve wound healing processes or terminate inflammatory responses in the lung. IPF is a pathological consequence resulted from altered wound healing in response to persistent lung injury. In this review, we intend to summarize the current state of knowledge regarding the process of macrophage polarization and its mediators in the pathogenesis of pulmonary fibrosis. Our goal is to update the understanding of the mechanisms underlying the initiation and progression of IPF, and by which, we expect to provide help for developing effective therapeutic strategies in clinical settings.
Similar content being viewed by others


Background
Idiopathic pulmonary fibrosis (IPF) is the most common fibrosing lung disease with poor prognosis and no effective treatment. It is characterized by the development of excessive extracellular matrix deposition, leading to decreased static lung compliance, disrupted gas exchange, and ultimately, respiratory failure and death [1,2,3,4]. The median survival time for IPF patients is approximately 3 years after the initial diagnosis [5, 6]. Notably, the incidence of IPF rises dramatically with age and is estimated to be 3–9 cases per 100,000 people in Europe and North America [7, 8], which is slightly higher than that in Asia and South America.
It is believed that IPF is resulted from a complex interaction between multiple genetic and non-genetic risk factors (e.g., cigarette smoking and infection). However, the relative contribution of genetic and non-genetic risk factors is likely to vary among individuals [9]. Past genetic studies in familial and sporadic IPF patients have characterized that the surfactant-associated genes (surfactant protein C, SFTPC; surfactant protein A2, SFTPA2; and ATP binding cassette member A3, ABCA3) [10, 11] and the telomerase-related genes (telomerase reverse transcriptase, TERT; telomerase RNA component, TERC; and regulator of telomere elongation helicase 1, RTEL1) [12] are associated with IPF susceptibility. More recently, two large genome-wide association studies (GWASs) conducted in patients with sporadic and familiar IPF not only confirmed known associations with TERC, TERT, and mucin 5B gene (MUC5B), but also identified novel risk genes such as the toll interacting protein (TOLLIP) and the signal peptide peptidase like 2C (SPPL2C) [13, 14]. Of note, excess matrix accumulation is thought to be an important part in the pathological process of IPF, and its related proteins such as matrix metalloproteinase 1 (MMP1) and matrix metalloproteinase 19 (MMP19) that are strongly upregulated in IPF are proposed to be potential peripheral blood biomarkers [15, 16]. In support of this notion, a frameshift deletion (c.988delG, p.A330fs) in MMP1 and a nonsense mutation (c.T1155A, p.Y385X) in MMP19 were characterized in IPF patients [17]. Although these discoveries are exciting, how non-genetic factors trigger those genetic predisposed individuals to initiate IPF process is unfortunately remained poorly understood, which is not our focus in the current review and will be reviewed elsewhere.
Monocytes/macrophages, which originate from progenitors in the bone marrow, circulate in the peripheral blood or migrate into different tissues and constitute the foremost controllers of both innate and acquired immunity [18, 19]. In general, circulating monocytes leave the blood and migrate into tissues, where they differentiate into macrophages following exposure to different stimuli, including local growth factors, cytokines and microbial debris [20, 21]. Macrophages are critical to antigen removal by phagocytosis and the clearance of microbes, apoptotic cells and neoplastic cells [22, 23]. Furthermore, in activated immune responses, macrophages are an extremely heterogeneous population, exerting a combination of pro-inflammatory and anti-inflammatory functions [20, 24]. Typically, macrophages that mainly produce pro-inflammatory cytokines are called classically activated macrophages (M1), which can be activated either by IFN-γ or lipopolysaccharide (LPS). In contrast, macrophages that attenuate inflammation and encourage wound repair are referred to as alternatively activated macrophages (M2), which can be activated by IL-4 or IL-13 [25]. It is believed that enhanced macrophage M2 program is associated with fibrotic remodeling of internal organs, including the heart, kidneys, liver, gastrointestinal tract and lungs [26]. Therefore, in this review, we mainly discuss the current state of knowledge regarding the process of macrophage polarization and its mediators in the pathogenesis of pulmonary fibrosis. Our goal is to update the understanding of the mechanisms underlying the initiation and progression of IPF, and by which, we expect to provide help for developing effective therapeutic strategies in clinical settings.
Origin of macrophages
Macrophages are small populations of leukocytes that play an important role in the cross-talk between innate and adaptive immunity [27]. As early as 1893, Metchnikoff first described the term “phagocytes” when he used a rose thorn to challenge a starfish [28]. He classified the phagocytes into macrophages (macro = big, phage = eater) and microphages (now better known as neutrophils), which promoted the development of a theory of phagocytosis [29, 30]. In the 1960s, several studies proposed that macrophages were solely derived from the terminal differentiation of circulating monocytes [31,32,33]. However, this theory has been refuted by the recent discoveries that most adult tissue-resident macrophages are seeded before birth, stem from the embryonal yolk sac and fetal liver, possess a self-renewing capacity and are maintained independent of circulating monocytes [27, 34]. In addition, the function of macrophages varies significantly based on their anatomical location, functional phenotype, morphology, as well as their gene expression profile, such as the alveolar macrophages, adipose tissue macrophages, Kupffer cells in the liver, and microglia cells in the central nervous system [26, 35].
Pulmonary macrophages
Pulmonary macrophage populations are classified into two main categories: alveolar macrophages (AMs), which strategically reside in the alveoli, and interstitial macrophages (IMs), which are located within the lung parenchymal tissue [36, 37]. Given the fact that previous studies in macrophages were conducted in animals or human mononuclear phagocytes from blood and small tissue resections around tumors, Desch and colleagues recently collected pulmonary mononuclear phagocytes from fully intact non-diseased human lungs and identified five extravascular macrophage phagocytes based on phenotype, location, and gene expression, supporting the growing approval of myeloid cell diversity in tissues [38]. It is now clear that AMs colonize the lung tissues shortly after birth and display remarkable self-renewal properties independent of blood monocyte input in the steady-state [39,40,41]. In contrast, IMs are less well studied. Scott et al. demonstrated that the IM compartment contains cells derived from both yolk-sac macrophages and bone marrow-derived monocytes (BMDMs) [42]. However, the contribution of BMDMs to this population is still under investigation. Functionally, AMs are the chief effector cells of immune responses and have both proinflammatory and anti-inflammatory properties, whereas IMs play a major role in maintaining immune homeostasis in the respiratory tract and inducing immune tolerance to harmless antigens [43, 44]. Currently, a growing body of evidence supports a role for both AMs and IMs in the pathogenesis of pulmonary fibrosis.
Macrophage polarization in IPF
Macrophages are remarkable plastic cells that can transform from one phenotype to another [42, 45]. Macrophage polarization is a dynamic process whereby macrophages manifest different functional phenotypes in response to micro-environmental stimuli and signals [25]. As the most abundant immune cells in the lungs (approximately 70% of the immune cells), macrophages play a vital role in airway remodeling in pulmonary fibrosis [43]. Beyond the AM/IM macrophage subtype delineation, pulmonary macrophages as aforementioned, can be characterized as classically activated macrophages (also termed M1) or alternatively activated macrophages (M2) [46] (Fig. 1). These macrophage subtypes differ in the expression of their cell surface markers, production of specific factors, and biological activities. Both M1 and M2 macrophages have been noted to be involved in the pathogenesis of pulmonary fibrosis, which will be discussed in detail in the following sections.

Schematic diagram of macrophage subtypes. The M1 subtype is generally considered to be proinflammatory. The M2a subtype is induced by IL-4 and IL-13, which are critical mediators of allergic inflammation. The M2b and M2c subtypes predominately participate in tissue remodeling and fibrosis. BV, blood vessel; M0, monocyte; LPS, lipopolysaccharide; IL, interleukin
Classically activated macrophages (M1) and IPF
M1 macrophages contribute to the host defense against pathogens by generating reactive nitric oxide (NO) via inducible nitric oxide synthase (iNOS) and releasing proinflammatory cytokines and chemokines such as IL-1β, IL-12, IL-23, CCL2 and TNF-α [47, 48]. Phenotypically, this cell type is characterized by the expression of high levels of CD80, TLR4, MHCII and CD86 [25, 49]. Therefore, M1 macrophages have robust anti-microbial and anti-tumor activities, mediate tissue damage and initiate inflammatory responses. Potent inducers such as LPS, IFN-γ, and granulocyte-macrophage colony-stimulating factor (GM-CSF), stimulate the generation of M1 macrophages, from either naïve M0 or polarized M2 macrophages [50]. Other endogenous alarmins such as oxidized low-density lipoprotein, fatty acids, caveolin-1 (Cav-1), and high-mobility group box 1 (HMGB1), are also found to induce M1 biased macrophage polarization [51,52,53,54]. Nevertheless, it still remains unknown whether they exert similar effects on pulmonary fibrosis. Of importantly note, there is evidence that the polarized M1 macrophages can be switched back to the M0 state in a cytokine-deficient medium for 12 days or re-differentiated into another cell phenotype after culture in an alternative polarizing medium [55].
In the current paradigm of IPF pathogenesis, pulmonary fibrosis progresses as a final pathological outcome of aberrant wound healing responses to persistent lung injury. Pulmonary cellular damage caused by environmental particulates, infection or mechanical damage, often result in the disruption of normal lung architecture, initiating a wound healing response [56]. At the early inflammatory stages, acute lung injury promotes an M1 phenotype under the control of transcription factor interferon regulatory factor 5 (IRF-5) with expression of high levels of iNOS and proinflammatory cytokines,which is associated with Th1 immune responses and responds to IFN-γ and toll-like receptor (TLR) ligands to maximize cytotoxic activity [57,58,59]. However, sustained inflammatory responses would serve as a trigger to initiate fibrotic responses in the lung. Indeed, during the course of aberrant wound healing processes, there is an early and sustained enrichment in the numbers of exudate macrophages (ExM) and their precursors, the Ly-6Chigh monocytes [60, 61]. For example, targeted type II alveolar epithelial cell (AEC II) injury induces a modest inflammatory response that is nonetheless significantly enriched for alternatively activated and profibrotic ExM and Ly-6Chigh monocytes. The accumulation of these nonresident cells and the development of pulmonary fibrosis in response to targeted alveolar injury is CCR2 dependent, thereby implicating ExM and Ly-6Chigh monocytes and the CCR2/CCR2-ligand axis as potential therapeutic targets to treat or prevent fibrotic lung disease [62].
Generally, the LPS/TLR4 pathway has been considered one of the major pathways in M1 macrophage polarization [63]. For example, the LPS/TLR4 pathway activates NF-kB and IRF3 and promotes the secretion of proinflammatory cytokines (e.g., IL-6 and TNF-α) to exacerbate macrophage M1 polarization [64]. This was further supported by studies showing that the LPS/TLR4 activates STAT1 to induce M1 polarization in an MyD88-independent fashion [65]. Interestingly, deletion of both TLR2 and TLR4 increased the susceptibility to bleomycin- and radiation-induced pulmonary fibrosis [66, 67]. Similarly, IL-1R–associated kinase-M (IRAK-M), an MyD88-dependent inhibitor of TLR signaling, suppresses deleterious inflammation but may paradoxically promote fibrogenesis, where IRAK-M was noted to promote macrophage M2 program in the setting of bleomycin-induced lung injury, which drives lung fibrogenesis in an IL-13–dependent fashion [68]. Furthermore, during the evolution of radiation-induced pulmonary fibrosis, patients manifested enhanced mRNA and protein expression of iNOS within the radiation pneumonic stage, whereas the high levels of arginase 1 (Arg 1) expression occurred within the radiation-induced fibrotic phase [69]. Intriguingly, macrophage inflammatory protein-1α (MIP-1α, also known as CCL3) arises mainly from M1, while its chemotactic affinity toward M2 is stronger than it is for M1. The interaction between MIP-1α and macrophages in different activated states may play a crucial role in regulating the transition from radiation pneumonitis to radiation pulmonary fibrosis [70]. It was also noted that TLR2 was elevated in IPF patients [71], and pharmacologic inhibition of TLR2 protected mice from bleomycin induced lung injury and fibrosis [72]. Similarly, mice with macrophage specific deletion of IRF-5 manifested fibrotic responses in adipose tissue following high-fat diet induction [73]. Collectively, those data indicate that M1 macrophage plays an essential role for wound repair, but it may also serve as a double edged sword to either trigger or prevent fibrotic responses. Nevertheless, the exact extracellular microenvironments relevant to its functional direction (e.g., in favor or against fibrotic responses) are yet to be elucidated.
Alternatively activated macrophages (M2) and IPF
M2 macrophages could be induced by a broad array of mediators such as IL-4, IL-13, TGF-β, and IL-10, and are implicated in the aberrant wound-healing cascade during fibrosis [74]. Depending upon the specific stimulators, M2 macrophages are further subdivided into 3 subgroups, namely, M2a, M2b, and M2c [23, 45]. The M2a subtype is induced by IL-4, IL-13, fungal and helminthic infections. M2b is elicited by IL-1 receptor ligands, immune complexes, and LPS, whereas M2c is activated by IL-10, TGF-β1, and glucocorticoids. More recently, an additional M2 subset, the M2d macrophage, was identified designated by the reduced secretion of IL-12 and enhanced release of IL-10 [75] (Fig. 1). However, there is no unique surface marker to distinguish each subset of M2 macrophages, and they are characterized by low levels of MHCII, CD86, and iNOS2 but high levels of arginase-1, the family proteins chitinase-like Ym1/2 and Fizz1/RELM-α (found in inflammatory zone 1) [76, 77], and cell surface receptors such as macrophage mannose receptor, also called CD206. Analysis of over 72 non-diseased human lungs consistently indicated that CD206 is exclusively expressed on alveolar macrophages rather than on cells found in blood or in the intravascular compartment of the lung [38], which exerts an important function in the phagocytosis of M2 cells via increasing efferocytosis of invading pathogens and apoptotic cells [78]. In addition, recent studies have indicated that transcription factors and other intracellular proteins, such as tuberous sclerosis complex 1 (TSC1) [79], stress-responsive activating transcription factor 7 (ATF 7) [80], STIP1 homology and U-Box containing protein 1 (STUB1) [81], ten eleven translocation (Tet) methylcytosine dioxygenase (Tet2) [82], microRNA (MiR-511) [83], interferon regulatory factor 4 (IRF4) [84], peroxisome proliferator-activated receptor gamma (PPARγ) [85], and Krueppel-like factor 4 (KLF-4) [86] are also involved in the polarization of M2 macrophages.
During the course of IPF development and progression, the predominant infiltration of M2 macrophages in the areas of lung fibrosis acts as a vital regulator of fibrogenesis [58, 87, 88]. Generally, excessive M2-associated and Th2-driven responses are an important part of many fibrotic diseases [58, 89]. For example, alveolar M2 macrophages, which are located close in proximity to sites of pulmonary injury undergoing repair, release CCL18 to stimulate fibroblast producing collagen. On the other hand, the proximate M2 macrophages bind to collagen type I via β2-integrins and scavenger receptors, thereby further increasing their CCL18 production, which creates a self-perpetuating vicious cycle of augmented, continuous M2 macrophage activation and excessive collagen production by lung fibroblasts [90]. Indeed, upon activation, M2 macrophages can produce profibrotic mediators such as TGF-β and PDGF to induce continuous fibroblast activation and to promote myofibroblast proliferation. Similarly, IL-10 generates a Th2 microenvironment in favor of bleomycin- or helminth-induced lung fibrosis [91,92,93], which involves fibrocyte recruitment and M2 macrophage activation likely through the CCL2/CCR2 axis [94], leading to excessive extracellular matrix (ECM) deposition along with distorted lung tissue architecture, and ultimately resulting in pulmonary fibrosis and respiratory failure [95], although the underlying mechanisms are yet to be fully established.
In addition to Th2 cells, eosinophils, innate lymphoid type 2 cells (ILC2s), CD4+CD25+ regulatory T cells (Tregs), and mesenchymal stromal cells (MSCs) were recently reported to drive the polarization of M2 macrophages [96,97,98,99]. For example, mice infused with syngeneic CD4+CD25+ Treg cells have an increased population of CD206+ peritoneal macrophages, with low levels of CD80 and MHCII [100]. Interestingly, Tregs lack of Tim-3 are noted to repress the expression of p-STAT3, thereby attenuating the induction of M2 macrophages in acute respiratory distress syndrome (ARDS)-associated pulmonary fibroproliferation [101]. Similarly, natural killer T cells (NKTs), a subset of T lymphocytes expressing membrane receptors from both T and NK lineages [102], can mediate a protective effect against fibrosis by inhibiting a Th2 response and preventing M2 macrophage polarization [103] in the bleomycin animal model.
It has been well known that IL-4 is the major inducer for macrophage M2 program by activating its two major downstream signals, JAK1/STAT6 and PI3K/AKT [88, 104]. Indeed, Guo et al. reported that elevated Gab1 and Gab2 regulate IL-4-induced macrophage polarization in bleomycin-induced fibrotic lungs, with Gab1 positively regulating AKT signaling and Gab2 positively regulating STAT6 signaling [105]. Similarly, Kral et al. noted that myeloid PTEN deficiency mice following bleomycin induction manifest sustained PI3K activation to enhance macrophage M2 program, leading to increased morbidity and decreased survival, although these mice exhibit impaired recruitment function [106]. Furthermore, deficiency of shp2, a ubiquitously expressed cytoplasmic tyrosine phosphatase, increases the sensitivity to chitin-induced M2 macrophage polarization and bleomycin-induced pulmonary fibrosis by enhancing IL-4–induced JAK1/STAT6 signaling [107]. Other than cytokines, altered adenosine metabolism was also noted to regulate macrophage M2 program. Adenosine is generated in response to cellular stress and damage, and extracellular accumulation of adenosine and subsequent activation of its receptor, ADORA2B, polarizes macrophages to a fibrotic phenotype. Therefore, mice with macrophage specific deletion of ADORA2B are protected from bleomycin-induced lung injury and fibrosis [108]. It was further noted that hypoxia-inducible factor 1-α (HIF-1α) enhances ADORA2B expression to regulates M2 macrophage differentiation and production of profibrotic mediators [109].
It is noteworthy that mitochondrial oxidative stress and mitochondrial turnover in alveolar macrophages are directly linked to pulmonary fibrosis [110,111,112], although the exact molecular mechanisms that modulate mitochondrial dynamics are currently not known. The mitochondrial calcium uniporter (MCU) is a highly selective ion channel to transport Ca2+ into the mitochondrial matrix for modulation of cellular metabolism [113, 114]. MCU was recently found to polarize macrophages to a profibrotic phenotype after exposure to asbestos by regulating ATP production [115]. Similarly, mitochondrial Cu,Zn-SOD accelerates the development of pulmonary fibrosis by inducing early and sustained alternative activation of macrophages through redox regulation of the Jumonji domain-containing protein 3 (Jmjd3) [116, 117]. Collectively, these data provided convincing evidence that mitochondrial stress also regulates macrophage M2 program implicated in the pathogenesis of pulmonary fibrosis.
Interestingly, there is compelling evidence that endoplasmic reticulum (ER) stress modulates the activation of M2 macrophages [118, 119]. During the past few years, our laboratory has been focused on the effect of ER stress on fibrogenesis. We first demonstrated evidence that pulmonary fibrosis manifests altered C/EBP homologous protein (CHOP) expression and ER stress in both IPF patients and animals with bleomycin-induced pulmonary fibrosis. In consistent with these observations, mice deficient in Chop were protected from bleomycin-induced lung injury and fibrosis. Specifically, the loss of Chop significantly attenuated TGF-β production along with reduced M2 macrophage infiltration in the lung following bleomycin induction. Mechanistic studies revealed that Chop deficiency suppressed the M2 program in macrophages, which then attenuated TGF-β secretion. The loss of Chop enhanced the expression of SOCS1 and SOCS3, thereby inhibiting STAT6/PPARγ signaling that is essential for macrophage M2 program [87]. Similarly, an ovalbumin (OVA)-induced allergic airway inflammatory model revealed that Chop regulates STAT6 phosphorylation, thereby enhancing the expression of mouse transcription factor EC (Tfec), which then transcribes IL-4 receptor a (IL-4Ra) expression to promote M2 program in macrophages [120]. Taken together, those data provide novel insights into the role of ER stress in modulating macrophage M2 program implicated in the pathogenesis of pulmonary fibrosis.
Conclusion
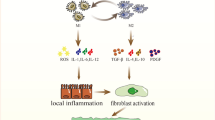
Lung M1 and M2 macrophages are distinct cell subtypes and are both involved in the pathogenesis of pulmonary fibrosis. M1 macrophages express high levels of proinflammatory cytokines, while M2 macrophages express high levels of Th2-type cytokines. Owing to their different cytokine expression profiles, M1 and M2 macrophages play different roles in the pathogenesis of pulmonary fibrosis. Generally, M1 macrophages are responsible for wound healing after alveolar epithelial injury, while M2 macrophages are designated to resolve wound healing processes or terminate inflammatory responses in the lung. IPF is a pathological consequence resulted from altered wound healing in response to persistent lung injury. A variety of regulatory cytokines, chemokines, mediators, and immune-regulatory cells affect polarization and chemotaxis of the lung macrophages (Fig. 2). These mediators interplay and influence disease duration and severity through altered polarization of M1 and M2 cells. Therefore, strategies aimed at modulation of lung macrophage phenotypes may have great potential for prevention and treatment of pulmonary fibrosis in clinical settings.

Macrophages in the pathogenesis of pulmonary fibrosis. This schematic diagram demonstrates the mediators for modulation of macrophage subtypes and how macrophages contribute to IPF initiation and progression. M1 macrophages play an essential role for wound healing after alveolar epithelial injury, while M2 macrophages are require to resolve inflammatory responses in the lung, and IPF is a pathological outcome resulted from altered wound healing in response to persistent lung injury. Ly-6C (H), Ly-6Chigh monocytes; MCS, mesenchymal stem cells; ILC2s, lymphoid type 2 cells; NKTs, natural killer T cells; MCU, mitochondrial calcium uniporter; CHOP, C/EBP homologous protein; IRAK-M, IL-1R–associated kinase-M; Arg 1, arginase 1; iNOS, inducible nitric oxide synthase
As noted above, IPF is a hard-to-diagnose fatal interstitial lung disease with a poor response to traditional therapies. Therefore, the development of reliable and fast diagnostic techniques and effective therapies is highly desirable, while targeting the activation and recruitment of M2 macrophages could be a viable therapeutic strategy. Indeed, pirfenidone, one of the two currently US Food and Drug Administration (FDA) approved drugs (pirfenidone and nintedanib) for treatment of human IPF, exerts its anti-fibrotic property in part by suppressing TGF-β expression relevant to macrophage M2 polarization and fibroblast activation [121]. Moreover, neotuberostemonine (NTS), one of the traditional Chinese medicines included in all versions of the Chinese Pharmacopoeia, can effectively attenuate BLM-induced lung fibrosis via suppressing the recruitment and polarization of M2 macrophages [122]. Intriguingly, Ji et al. demonstrated that mineralocorticoid receptor (MR) antagonism by liposome-encapsulated spironolactone (Lipo-SP) could alleviate bleomycin-induced acute pulmonary injury and fibrosis, partially by reducing circulating inflammatory Ly6Chigh monocyte expansion and repressing alternatively activated mononuclear phagocytes in the alveolar compartment [123, 124].
Although therapies for IPF include a variety of drugs and non-pharmacological interventions, there is still a pressing need for new therapeutic approaches as current therapies are unable to effectively attenuate disease progression or reverse lung fibrosis. Despite past extensive studies, many questions remain unsolved regarding the exact mechanisms of manipulating the balance of M1/M2 phenotype in IPF pathogenesis. Future studies aimed at dissecting the interplay between macrophages and fibroblasts and how macrophages create an extracellular milieu in favor of fibroblast activation and myofibroblast proliferation, would be crucial for developing effective therapies against IPF and other fibrotic diseases.
Abbreviations
- ABCA3:
-
ATP binding cassette member A3
- AEC II:
-
Type II alveolar epithelial cell
- AMs:
-
Alveolar macrophages
- ARDS:
-
Acute respiratory distress syndrome
- Arg 1:
-
Arginase 1
- ATF 7:
-
Activating transcription factor 7
- BMDMs:
-
Bone marrow-derived monocytes
- Cav-1:
-
Caveolin-1
- CHOP:
-
C/EBP homologous protein
- ECM:
-
Excessive extracellular matrix
- ER:
-
Endoplasmic reticulum
- ExM:
-
Exudate macrophages
- FDA:
-
Food and drug administration
- Fizz1/RELM-α:
-
Found in inflammatory zone 1
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- GWASs:
-
Genome-wide association studies
- HIF-1α:
-
Hypoxia-inducible factor 1-α
- HMGB1:
-
High-mobility group box 1
- IL-4Ra:
-
IL-4 receptor a
- ILC2s:
-
Innate lymphoid type 2 cells
- IMs:
-
Interstitial macrophages
- iNOS:
-
Inducible nitric oxide synthase
- IPF:
-
Idiopathic pulmonary fibrosis
- IRAK-M:
-
IL-1R–associated kinase-M
- IRF4:
-
Interferon regulatory factor 4
- IRF-5:
-
Interferon regulatory factor 5
- Jmjd3:
-
Jumonji domain-containing protein 3
- KLF-4:
-
Krueppel-like factor 4
- Lipo-SP:
-
liposome-encapsulated spironolactone
- LPS:
-
Lipopolysaccharide
- M1:
-
Classically activated macrophages
- M2:
-
Alternatively activated macrophages
- MCU:
-
Mitochondrial calcium uniporter
- MIP-1α:
-
Macrophage inflammatory protein-1α
- MMP1:
-
Matrix metalloproteinase 1
- MMP19:
-
Matrix metalloproteinase 19
- MR:
-
mineralocorticoid receptor
- MSCs:
-
Mesenchymal stem cells
- MUC5B:
-
Mucin 5B gene
- NKTs:
-
Natural killer T cells
- NO:
-
Nitric oxide
- NTS:
-
Neotuberostemonine
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- RTEL1:
-
Regulator of telomere elongation helicase 1
- SFTPA2:
-
Surfactant protein A2
- SFTPC:
-
Surfactant protein C
- SPPL2C:
-
Signal peptide peptidase like 2C
- STUB1:
-
STIP1 homology and U-Box containing protein 1
- TERC:
-
Telomerase RNA component
- TERT:
-
Telomerase reverse transcriptase
- Tet:
-
Ten eleven translocation
- Tet2:
-
Ten eleven translocation methylcytosine dioxygenase
- Tfec:
-
Transcription factor EC
- TLR:
-
Toll-like receptor
- TOLLIP:
-
Toll interacting protein
- Tregs:
-
Regulatory T cells
- TSC1:
-
Tuberous sclerosis complex 1
References
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–21.
Helling BA, Yang IV. Epigenetics in lung fibrosis: from pathobiology to treatment perspective. Curr Opin Pulm Med. 2015;21:454–62.
Hewlett JC, Kropski JA, Blackwell TS. Idiopathic pulmonary fibrosis: epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018.
Zhang L, Wang Y, Pandupuspitasari NS, Wu G, Xiang X, Gong Q, Xiong W, Wang CY, Yang P, Ren B. Endoplasmic reticulum stress, a new wrestler, in the pathogenesis of idiopathic pulmonary fibrosis. Am J Transl Res. 2017;9:722–35.
Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. 2013;6:9–17.
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795–806.
Hopkins RB, Burke N, Fell C, Dion G, Kolb M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur Respir J. 2016;48:187–95.
Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, Yang IV, Schwartz DA. Idiopathic pulmonary fibrosis: a genetic disease that involves Mucociliary dysfunction of the peripheral airways. Physiol Rev. 2016;96:1567–91.
Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, Markin C, Renzoni E, Lympany P, Thomas AQ, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59:977–80.
Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–9.
Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS, Rosenblatt RL, Girod CE, Garrity ER, Xing C, Garcia CK. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5:e10680.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20.
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–17.
Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93.
Yu G, Kovkarova-Naumovski E, Jara P, Parwani A, Kass D, Ruiz V, Lopez-Otin C, Rosas IO, Gibson KF, Cabrera S, et al. Matrix metalloproteinase-19 is a key regulator of lung fibrosis in mice and humans. Am J Respir Crit Care Med. 2012;186:752–62.
Deng Y, Li Z, Liu J, Wang Z, Cao Y, Mou Y, Fu B, Mo B, Wei J, Cheng Z, et al. Targeted resequencing reveals genetic risks in patients with sporadic idiopathic pulmonary fibrosis. Hum Mutat. 2018;39:1238–1245.
Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev. 2017;280:102–11.
Ginhoux F, Guilliams M, Naik SH. Editorial: dendritic cell and macrophage nomenclature and classification. Front Immunol. 2016;7:168.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95.
Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383–96.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79:541–66.
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804.
Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35.
Gordon S. Elie Metchnikoff: father of natural immunity. Eur J Immunol. 2008;38:3257–64.
Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol. 2015;36:307–14.
Stefater JA 3rd, Ren S, Lang RA, Duffield JS. Metchnikoff's policemen: macrophages in development, homeostasis and regeneration. Trends Mol Med. 2011;17:743–52.
van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. 1972;46:845–52.
Marchesi VT, Florey HW. Electron micrographic observations on the emigration of leucocytes. Q J Exp Physiol Cogn Med Sci. 1960;45:343–8.
Volkman A, Gowans JL. The origin of macrophages from bone marrow in the rat. Br J Exp Pathol. 1965;46:62–70.
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91.
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55.
Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–92.
Bedoret D, Wallemacq H, Marichal T, Desmet C, Quesada Calvo F, Henry E, Closset R, Dewals B, Thielen C, Gustin P, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest. 2009;119:3723–38.
Desch AN, Gibbings SL, Goyal R, Kolde R, Bednarek J, Bruno T, Slansky JE, Jacobelli J, Mason R, Ito Y, et al. Flow cytometric analysis of mononuclear phagocytes in nondiseased human lung and lung-draining lymph nodes. Am J Respir Crit Care Med. 2016;193:614–26.
Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in Fibrosing lung disease? Trends Mol Med. 2016;22:303–16.
Van den Bossche J, Malissen B, Mantovani A, De Baetselier P, Van Ginderachter JA. Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood. 2012;119:1623–33.
Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387–404.
Scott CL, Henri S, Guilliams M. Mononuclear phagocytes of the intestine, the skin, and the lung. Immunol Rev. 2014;262:9–24.
Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda MJ. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol. 2014;192:2821–9.
Ji WJ, Ma YQ, Zhou X, Zhang YD, Lu RY, Sun HY, Guo ZZ, Zhang Z, Li YM, Wei LQ. Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J Immunol Methods. 2014;403:7–16.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86.
Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. 2013;120:163–84.
Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res. 2018;191:1–14.
Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–63.
Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614.
Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215:788–95.
van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–9.
Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. 2015;308:F69–75.
Shivshankar P, Halade GV, Calhoun C, Escobar GP, Mehr AJ, Jimenez F, Martinez C, Bhatnagar H, Mjaatvedt CH, Lindsey ML, Le Saux CJ. Caveolin-1 deletion exacerbates cardiac interstitial fibrosis by promoting M2 macrophage activation in mice after myocardial infarction. J Mol Cell Cardiol. 2014;76:84–93.
Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17:216–7.
Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. 2015;53:676–88.
Burgy O, Konigshoff M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018;68-69:67–80.
Richards CD. Innate immune cytokines, fibroblast phenotypes, and regulation of extracellular matrix in lung. J Interf Cytokine Res. 2017;37:52–61.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62.
Weiss M, Byrne AJ, Blazek K, Saliba DG, Pease JE, Perocheau D, Feldmann M, Udalova IA. IRF5 controls both acute and chronic inflammation. Proc Natl Acad Sci U S A. 2015;112:11001–6.
Xiang GA, Zhang YD, Su CC, Ma YQ, Li YM, Zhou X, Wei LQ, Ji WJ. Dynamic changes of mononuclear phagocytes in circulating, pulmonary alveolar and interstitial compartments in a mouse model of experimental silicosis. Inhal Toxicol. 2016;28:393–402.
Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–81.
Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N, Browning K, Lin Y, Morey RE, Dayrit JK, et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190:3447–57.
Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci. 2015;72:4111–26.
Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. 2014;6:716–26.
Toshchakov V, Jones BW, Lentschat A, Silva A, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, et al. TLR2 and TLR4 agonists stimulate unique repertoires of host resistance genes in murine macrophages: interferon-beta-dependent signaling in TLR4-mediated responses. J Endotoxin Res. 2003;9:169–75.
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9.
Paun A, Fox J, Balloy V, Chignard M, Qureshi ST, Haston CK. Combined Tlr2 and Tlr4 deficiency increases radiation-induced pulmonary fibrosis in mice. Int J Radiat Oncol Biol Phys. 2010;77:1198–205.
Ballinger MN, Newstead MW, Zeng X, Bhan U, Mo XM, Kunkel SL, Moore BB, Flavell R, Christman JW, Standiford TJ. IRAK-M promotes alternative macrophage activation and fibroproliferation in bleomycin-induced lung injury. J Immunol. 2015;194:1894–904.
Zhang H, Han G, Liu H, Chen J, Ji X, Zhou F, Zhou Y, Xie C. The development of classically and alternatively activated macrophages has different effects on the varied stages of radiation-induced pulmonary injury in mice. J Radiat Res. 2011;52:717–26.
He Z, Zhang H, Yang C, Zhou Y, Zhou Y, Han G, Xia L, Ouyang W, Zhou F, Zhou Y, Xie C. The interaction between different types of activated RAW 264.7 cells and macrophage inflammatory protein-1 alpha. Radiat Oncol. 2011;6:86.
Samara KD, Antoniou KM, Karagiannis K, Margaritopoulos G, Lasithiotaki I, Koutala E, Siafakas NM. Expression profiles of toll-like receptors in non-small cell lung cancer and idiopathic pulmonary fibrosis. Int J Oncol. 2012;40:1397–404.
Liu HZ, Yang HZ, Mi S, Cui B, Hua F, Hu ZW. Toll like receptor 2 mediates bleomycin-induced acute lung injury, inflammation and fibrosis in mice. Yao Xue Xue Bao. 2010;45:976–86.
Dalmas E, Toubal A, Alzaid F, Blazek K, Eames HL, Lebozec K, Pini M, Hainault I, Montastier E, Denis RG, et al. Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat Med. 2015;21:610–8.
Zhong B, Yang X, Sun Q, Liu L, Lan X, Tian J, He Q, Hou W, Liu H, Jiang C, et al. Pdcd4 modulates markers of macrophage alternative activation and airway remodeling in antigen-induced pulmonary inflammation. J Leukoc Biol. 2014;96:1065–75.
Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20:701–12.
Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602.
Raes G, Noel W, Beschin A, Brys L, de Baetselier P, Hassanzadeh GH. FIZZ1 and Ym as tools to discriminate between differentially activated macrophages. Dev Immunol. 2002;9:151–9.
Hong JY, Chung Y, Steenrod J, Chen Q, Lei J, Comstock AT, Goldsmith AM, Bentley JK, Sajjan US, Hershenson MB. Macrophage activation state determines the response to rhinovirus infection in a mouse model of allergic asthma. Respir Res. 2014;15:63.
Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, Zhang L, Tian H, Zhao Q, Peng J, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. 2014;5:4696.
Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, Uchiyama T, Ishibashi K, Yamada T, Ohno N, et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol. 2015;16:1034–43.
Wei Q, Sha Y, Bhattacharya A, Abdel Fattah E, Bonilla D, Jyothula SS, Pandit L, Khurana Hershey GK, Eissa NT. Regulation of IL-4 receptor signaling by STUB1 in lung inflammation. Am J Respir Crit Care Med. 2014;189:16–29.
Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, Zhao D, Liu Y, Wang C, Zhang X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–93.
Karo-Atar D, Itan M, Pasmanik-Chor M, Munitz A. MicroRNA profiling reveals opposing expression patterns for miR-511 in alternatively and classically activated macrophages. J Asthma. 2015;52:545–53.
Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–44.
Chang HY, Lee HN, Kim W, Surh YJ. Docosahexaenoic acid induces M2 macrophage polarization through peroxisome proliferator-activated receptor gamma activation. Life Sci. 2015;120:39–47.
Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–49.
Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24:915–25.
Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–43.
Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–76.
Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, Vollmer E, Muller-Quernheim J, Zissel G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–92.
Joshi AD, Raymond T, Coelho AL, Kunkel SL, Hogaboam CM. A systemic granulomatous response to Schistosoma mansoni eggs alters responsiveness of bone-marrow-derived macrophages to toll-like receptor agonists. J Leukoc Biol. 2008;83:314–24.
Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, McKenzie A, Lukacs NW, Phan SH. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–31.
Sandler NG, Mentink-Kane MM, Cheever AW, Wynn TA. Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J Immunol. 2003;171:3655–67.
Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341–53.
Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204:19–28.
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–51.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7.
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210:535–49.
Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O'Kane CM, Krasnodembskaya AD. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med. 2017;196:1275–86.
Liu G, Ma H, Qiu L, Li L, Cao Y, Ma J, Zhao Y. Phenotypic and functional switch of macrophages induced by regulatory CD4+CD25+ T cells in mice. Immunol Cell Biol. 2011;89:130–42.
Liu X, Jiang S, Zhang Q, Xu S, Bao X, Cao W, Bai J, Tang L. Tim-3 regulates Tregs’ ability to resolve the inflammation and proliferation of acute lung injury by modulating macrophages polarization. Shock. 2017.
Van Kaer L, Parekh VV, Wu L. Invariant natural killer T cells: bridging innate and adaptive immunity. Cell Tissue Res. 2011;343:43–55.
Grabarz F, Aguiar CF, Correa-Costa M, Braga TT, Hyane MI, Andrade-Oliveira V, Landgraf MA, Camara NOS. Protective role of NKT cells and macrophage M2-driven phenotype in bleomycin-induced pulmonary fibrosis. Inflammopharmacology. 2018;26:491–504.
Wills-Karp M, Finkelman FD. Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci Signal. 2008;1:pe55.
Guo X, Li T, Xu Y, Xu X, Zhu Z, Zhang Y, Xu J, Xu K, Cheng H, Zhang X, Ke Y. Increased levels of Gab1 and Gab2 adaptor proteins skew interleukin-4 (IL-4) signaling toward M2 macrophage-driven pulmonary fibrosis in mice. J Biol Chem. 2017;292:14003–15.
Kral JB, Kuttke M, Schrottmaier WC, Birnecker B, Warszawska J, Wernig C, Paar H, Salzmann M, Sahin E, Brunner JS, et al. Sustained PI3K activation exacerbates BLM-induced lung fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci Rep. 2016;6:23034.
Tao B, Jin W, Xu J, Liang Z, Yao J, Zhang Y, Wang K, Cheng H, Zhang X, Ke Y. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol. 2014;193:2801–11.
Karmouty-Quintana H, Philip K, Acero LF, Chen NY, Weng T, Molina JG, Luo F, Davies J, Le NB, Bunge I, et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J. 2015;29:50–60.
Philip K, Mills TW, Davies J, Chen NY, Karmouty-Quintana H, Luo F, Molina JG, Amione-Guerra J, Sinha N, Guha A, et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J. 2017;31:4745–58.
Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase-mediated Mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016;44:582–96.
Osborn-Heaford HL, Murthy S, Gu L, Larson-Casey JL, Ryan AJ, Shi L, Glogauer M, Neighbors JD, Hohl R, Carter AB. Targeting the isoprenoid pathway to abrogate progression of pulmonary fibrosis. Free Radic Biol Med. 2015;86:47–56.
Osborn-Heaford HL, Ryan AJ, Murthy S, Racila AM, He C, Sieren JC, Spitz DR, Carter AB. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. 2012;287:3301–12.
Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4.
Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R. The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem. 2013;288:10750–8.
Gu L, Larson-Casey JL, Carter AB. Macrophages utilize the mitochondrial calcium uniporter for profibrotic polarization. FASEB J. 2017;31:3072–83.
He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem. 2013;288:20745–57.
He C, Larson-Casey JL, Gu L, Ryan AJ, Murthy S, Carter AB. Cu,Zn-Superoxide Dismutase-Mediated Redox Regulation of Jumonji Domain Containing 3 Modulates Macrophage Polarization and Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2016;55:58–71.
Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C. Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation. J Biol Chem. 2012;287:11629–41.
Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 2018;68-69:355–65.
Wang Y, Zhu J, Zhang L, Zhang Z, He L, Mou Y, Deng Y, Cao Y, Yang P, Su Y, et al. Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/transcription factor EC/IL-4 receptor alpha positive feedback loop in M2 macrophages. J Allergy Clin Immunol. 2017;140:1550–61. e1558
Inomata M, Kamio K, Azuma A, Matsuda K, Kokuho N, Miura Y, Hayashi H, Nei T, Fujita K, Saito Y, Gemma A. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respir Res. 2014;15:16.
Xiang J, Cheng S, Feng T, Wu Y, Xie W, Zhang M, Xu X, Zhang C. Neotuberostemonine attenuates bleomycin-induced pulmonary fibrosis by suppressing the recruitment and activation of macrophages. Int Immunopharmacol. 2016;36:158–64.
Ji WJ, Ma YQ, Zhou X, Zhang YD, Lu RY, Guo ZZ, Sun HY, Hu DC, Yang GH, Li YM, Wei LQ. Spironolactone attenuates bleomycin-induced pulmonary injury partially via modulating mononuclear phagocyte phenotype switching in circulating and alveolar compartments. PLoS One. 2013;8:e81090.
Ji WJ, Ma YQ, Zhang X, Zhang L, Zhang YD, Su CC, Xiang GA, Zhang MP, Lin ZC, Wei LQ, et al. Inflammatory monocyte/macrophage modulation by liposome-entrapped spironolactone ameliorates acute lung injury in mice. Nanomedicine (Lond). 2016;11:1393–406.
Acknowledgments
Our research was supported by the Natural Science Foundation of China (81530024, 9174920038 and 81770823), the Ministry of Science and Technology (2017ZX09304022 and 2016YFC1305002), the Department of Science and Technology of Hubei State (2017ACA096), and the Integrated Innovative Team for Major Human Disease Programs of Tongji Medical College, Huazhong University of Science and Technology.
Author information
Authors and Affiliations
Contributions
CYW contributed to the conception and edition of the manuscript. LZ, YW, GW, WX and WG drafted the manuscript. All authors reviewed the manuscript critically and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, L., Wang, Y., Wu, G. et al. Macrophages: friend or foe in idiopathic pulmonary fibrosis?. Respir Res 19, 170 (2018). https://doi.org/10.1186/s12931-018-0864-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-018-0864-2