
Abstract
Pathogenic variants in MYO15A are known to cause autosomal recessive nonsyndromic hearing loss (ARNSHL), DFNB3. We have previously reported on one ARNSHL family including two affected siblings and identified MYO15A c.5964+3G > A and c.8375 T > C (p.Val2792Ala) as the possible deafness-causing variants. Eight year follow up identified one new affected individual in this family, who also showed congenital, severe to profound sensorineural hearing loss. By whole exome sequencing, we identified a new splice-site variant c.5531+1G > C (maternal allele), in a compound heterozygote with previously identified missense variant c.8375 T > C (p.Val2792Ala) (paternal allele) in MYO15A as the disease-causing variants. The new affected individual underwent unilateral cochlear implantation at the age of 1 year, and 5 year follow-up showed satisfactory speech and language outcomes. Our results further indicate that MYO15A-associated hearing loss is good candidates for cochlear implantation, which is in accordance with previous report. In light of our findings and review of the literatures, 58 splice-site variants in MYO15A are correlated with a severe deafness phenotype, composed of 46 canonical splice-site variants and 12 non-canonical splice-site variants.
Similar content being viewed by others

Introduction
Hearing loss is one of the most common genetic sensory disorders, affecting one out of every 500–650 infants in the world [1]. Genetic factor accounts for approximately 50–60% of congenital sensorineural hearing loss cases [2]. It’s estimated that 70% of hereditary cases are nonsyndromic, meaning hearing loss is the only clinical manifestation. Hereditary hearing loss is extremely heterogeneous. To date, 124 deafness genes have been identified (http://hereditaryhearingloss.org/, updated 8/30/2021). The most prevalent type of hereditary hearing loss is autosomal recessive nonsyndromic hearing loss (ARNSHL), which accounts for about 80% of cases.
MYO15A (OMIM #602,666) variants have been shown to cause ARNSHL, DFNB3 (OMIM #600,316) in individuals from different populations worldwide [3]. In cochlea, myosin XVa, the protein encoded by MYO15A, is expressed at the tips of stereocilia in hair cells and plays as a motor protein that moves along actin filaments using energy from ATP hydrolysis. Transport of whirlin to the tips of the stereocilia by myosin XVa has been proved to be essential for the development and elongation of the stereocilia, which are essential for normal auditory function [4, 5]. In myosin XVa-deficient mice, no links between stereocilia were observed, implies that the mechano-transduction mechanism had been completely disrupted [6].
Previously, we reported on a family with two affected siblings who suffered severe to profound sensorineural hearing loss, DFNB3 [7]. Whole-exome sequencing (WES) of two affected siblings and unaffected parents was performed, and two compound heterozygous variants in MYO15A (NM_016239.4) were identified in the two affected siblings: c.8375 T > C (p.Val2792Ala) and c.5964+3G > A.
In the 8 year follow-up study, we identified one new affected individual in this family (III-1), who also showed congenital, profound sensorineural hearing loss, consistent with the DFNB3 phenotype. Bi-allelic variants in MYO15A were identified, including one novel splice-site variant c.5531+1G > C (maternal allele) and one previous identified missense variant c.8375 T > C (p.Val2792Ala) (paternal allele). In addition, an extensive genotype–phenotype correlation was conducted for MYO15A splice-site variants, which were filtered using the Professional edition of the Human Gene Mutation Database (HGMD) and summarized by a literature review.
Materials and methods
Subjects and clinical evaluations
The Chinese family of Han ethnicity with hearing loss reported here was followed up for 8 years after our initial report [7]. Medical history, temporal bone computed tomography (CT), otoscopy, pure tone audiometry (PTA) (for children under the age of six), auditory steady state response (for children under the age of six), acoustic immittance, auditory brainstem responses, and distortion product otoacoustic emission are all part of the clinical evaluation for hearing loss.
According to pure-tone audiometry (PTA) of the better ear, the average hearing threshold level at four air conduction frequencies (500, 1000, 2000, and 4000 Hz) was used to define the severity of hearing loss. According to the 2021 WHO classification of hearing loss, 20- < 35 dBHL was defined as mild, 35- < 50 dBHL was defined as moderate, 50- < 65dBHL was defined as moderate to severe, 65- < 80dBHL was defined as severe, and > 80 dBHL was defined as profound.
Molecular analysis
WES genetic analysis was performed in two new affected individuals, including II-3 and III-1. A blood DNA extraction kit was used to extract genomic DNA from peripheral blood according to the manufacturer's instructions (TianGen, Beijing, China). DNA was sheared, ligated to adaptors, extracted, and ligation-mediated PCR was used to amplify it. For enrichment, a 1 μg DNA library was combined with Buffer BL and GenCap probe (MyGenostics, Beijing, China). The Illumina NovaSeq 6000 platform was used to load each collected library. The fraction of mapped reads was 97–99% and average depth was 100 bp. After filtering out low-quality and duplicate reads, clean data were aligned to the human reference genome hg19 using the Burrows-Wheeler Aligner. Variants were called using four types of software (SOAPsnp, GATK, Samtools, and Platypus) and annotated by ANNOVAR. Then, variants were associated with multiple databases, including gnomAD, Inhouse database (MyGenostics), with minor allele frequencies (MAF) < 0.05. To check the possible pathogenicity of candidate variants, SIFT, PolyPhen-2, MutationTaster, and GERP++ software were used. Trio-based bioinformatic analysis of WES data were used for recessive, dominant, and X-linked conditions. Manually classification of those variants was conducted based on American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines for genetic hearing loss. Sanger sequencing was used to confirm potential pathogenic variants identified by these analyses. Primer sequences are provided in Additional file 1: Table S1. The sizes of PCR products are 654 bp (c.5531+1G > C), 652 bp (c.5964+3G > A) and 458 bp (c.8375 T > C).
Literature review of genotype–phenotype correlation of MYO15A splice-site variants
An extensive genotype–phenotype correlation was conducted for MYO15A splice-site variants. The Human Gene Mutation Database (HGMD) Professional edition was used to screen the variants, which were then evaluated through a literature review.
In silico validation of splice-site variants
To evaluate the splice site strength of different sequences, four prediction tools were used, including varSEAK (https://varseak.bio/), SpliceAI (https://github.com/Illumina/SpliceAI), CADD PHREAD (https://www.bio.tools/CADD_Phred#!), MaxEntScan (http://hollywood.mit.edu/burgelab/maxent/).
Results
Clinical findings
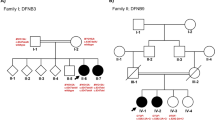
A three-generation Chinese pedigree (Family 4794), depicted in Fig. 1A, expanded 2 samples (II-3 and III-1) from previous reported pedigree [7]. This family included 4 patients with hearing loss (II-1, II-2, II-3 and III-1). Among those four patients, the molecular basis of two affected sibling (II-1 and II-2) were identified as c.8375 T > C (p.Val2792Ala) and c.5964+3G > A in MYO15A in our previous study [7] and their hearing loss was congenital, bilateral, severe to profound, and sensorineural. For II-3, wife of II-2, her hearing loss was postlingual, late onset (8 years old), sensorineural and progressive. For III-1, his hearing loss was congenital, bilateral, severe to profound, and sensorineural. The Audiograms of the two new affected individuals (II-3 and III-1) were depicted in Fig. 1B. The onset age of II-3 is different from other three patients and is inconsistent with reported DFNB3 phenotype. In any of the affected people, gross motor development was not noticeably slowed. All of the participants' physical evaluations indicated no symptoms of systemic disease or dysmorphic characteristics. In II-3 and III-1, high-resolution CT of the temporal bone revealed no abnormalities, ruling out middle and inner ear anomalies.

Extended family pedigree, hearing phenotype and variant analysis. A Affected individuals are denoted in black. The arrow indicates the proband. The red dashed line indicates the two new affected individuals; B Audiogram of the two new affected individuals showing profound sensorineural hearing loss (red, right ear; blue, left ear); C Chromatogram of MYO15A (NM_016239.4): c.5531+1G > C and c.8375C > T in three affected individuals (II-2, II-3, III-1)
Genotyping
The remaining variants were manually filtered based on their frequency/presence in known SNP databases, previous association with disease, predicted functional impact, nucleotide/amino acid conservation, and the potential detrimental biochemistry. The analysis identified compound heterozygous MYO15A variants c.5531+1G > C and c.8375 T > C (p.Val2792Ala) in III-1. There were no other potential variants in known deafness genes. Sanger sequencing was used to confirm the two discovered variants, and parental testing was used to validate them (Fig. 1C). The boy inherited the heterozygous c.5531+1G > C variant in MYO15A from his mother (II-3) and c.8375 T > C (p.Val2792Ala) from his father (II-2). No de novo or compound heterozygous variants in other deafness genes were identified in II-3 according to the autosomal dominant or recessive pattern of inheritance, as there is no maternal family history of hearing loss.
c.5531+1G > C variant is located in the intron region of the 5’ splice donor sequence and results from a G to A substitution (Table 1, Fig. 2). Several software programs including CADD PHREAD, varSEAK, and SpliceAI were used to evaluate the effect of the c.5531+1G > C variant on the splice site. Each analysis predicted that the substitution results in the loss of the donor site, causing altered splicing. According to ACMG/AMP guidelines, this variant is classified as pathogenic (PVS1+PM2+PM3+PP1+PP3).

Locations of HGMD-reported splice-site variants in MYO15A (NM_016239.4)
Cochlear implantation
Individual III-1 had been treated with unilateral cochlear implantation (Cochlear, Nucleus® CI512) at the age of 1, and a 5 year follow-up demonstrated that his listening and language abilities had significantly improved, with a high degree of accuracy in speech perception and the development of near-normal language skills. Several studies have described the results of cochlear implantation in patients with DNFB3. Almost all reports suggested that cochlear implantation was satisfactory, similar with our case [49,50,51,52].
Genotype–phenotype analysis of MYO15A reported splice-site variants
According to this study and HGMD Professional database (prior to Oct 1st, 2021), there were 360 DFNB3-associated pathogenic variants in MYO15A, including 58 splice-site variants that comprise a significant 16.11% (58/360) of pathogenic variants (Table 1, Fig. 2). We performed genotype–phenotype correlation analysis by literature review. The majority of pathogenic splice-site variants disrupt exons inside the Motor domain, which are believed to decrease Myosin VA protein function by affecting the capacity of whirlin transport to the tips of hair cell stereocilia. Pathogenic splice-site variants at Myosin Xavi’s N-terminal extension are less identified. The variants mainly distributed from Motor domain to FERM (protein 4.1-ezrin-radixin-moesin) functional domains (Fig. 2). Only one pathogenic splice-site variant in N-terminal was reported, c.3609+985 A > G, lies in intron 2. Although, variants in MYO15A lead to variable hearing impairment phenotype, from mild to severe, splice-site variants have been linked to a severe hearing loss phenotype in all identified cases, except those hearing loss degrees were not described in the literatures (Table 1).
Assessment of pathogenicity to non-canonical splice-site variants
Among the 58 considered disease-causing splice-site variants, 46 were canonical that in general change the + 1, + 2, − 2 and − 1 residue of an intron, and the remaining 12 were presumably non-canonical splice-site variants, accounting for 20.69% (Table 2). As for 46 canonical splice-site variants in MYO15A, the number of donor and acceptor splice site variants was 30 (65.22%) and 16 (34.78%), respectively (Fig. 2). The remaining 16 were splice site variants and account for 34.78% (Fig. 2). Eight out of 12 non-canonical splice-site variants were absent in GnomAD (Table 1). Although c.5134-10C > G, c.5965-8C > T, c.7787+4A > G, c.8224+3A > G, c.8788+5G > T, were registered in HGMD as pathogenic variants, their interpretations of pathogenicity are conflicting. Only 1 sporadic patient was reported to be associated with these variants. These variants were classified as variants of unknown significance, according to ACMG/AMP guidelines, and their association with disease necessitated further investigation.
Figure 3 summarizes the results of the 58 splice-site variants in MYO15A that predict to produce a great variety of splicing outcomes. Variants that destroy natural donor sequences seem to cause the skipping of their associated exon while variants in acceptor sequence are associated with intron retention. It should be noted that in a considerable number of cases, additional events can also take place. Pathogenic non-canonical mRNA alterations, which are normally associated with common events like intron retention or selective exon skipping, can also include cryptic events that occur outside of conventionally designated exons and unconventional splicing processes that regulate gene expression.

The types of MYO15A reported splice-site variants. Green boxes are exons and white boxes are introns. A yellow asterisk indicates the site of variant. A Canonical donor splice-site variant leads to intro retention; B Canonical acceptor splice-site variant leads to exon skipping; C Non-canonical splice-site variant. Deep intronic variants creating new splice sites
Discussion and conclusion
Since our initial report of MYO15A variants as the ARNSHL-associated gene among individuals with hearing loss in the Chinese population in 2013, several pathogenic variants of this gene have been identified in case–control studies with Chinese participants [53, 48, 54]. Our recent study of 511 Chinese individuals with hearing loss identified a genetic spectrum and showed that the disease-causing variants in MYO15A were the third most common cause (0.92%) of ARNSHL, behind GJB2 and SLC26A4 variants [32].
The variant c.5531+1G > C in MYO15A has never been reported in cases with hearing loss and was not presented in the public database. c.5531+1G > C occurs in trans with the reported pathogenic variant c.8375 T > C in MYO15A. It is well known that individuals with MYO15A-associated hearing loss (DFNB3) often present with nonsyndromic, congenital, severe to profound sensorineural hearing loss with normal middle and inner ear structure. Given the fact that II-3’s hearing loss was late-onset and progressive, which is atypical of DFNB3, it is possible that other genes or other factors are responsible for II-3’s hearing loss. Although the etiology of the II-3 hearing loss was not confirmed, there is a at least 50% chance the couple’s (II-2 and II-3) children will have MYO15A-associated hearing loss, as she is a heterozygous variant carrier of MYO15A. Pre-implantation genetic testing may be used to assess the risk for hearing loss.
MYO15A contains 67 exons and allows for a wide range of transcriptional variability, with the longest mRNA transcript being 3,530 amino acids. It encodes a N-terminal extension domain, ATPase motor domain, two myosin-tail homology 4 (MyTH4) domains, a Src-homology-3 (SH3) domain, and a band 4.1 superfamily (FERM) domain (Fig. 2).
According to this study and HGMD professional database, 360 pathogenic variants of MYO15A have been identified. According to a recent study, 27% of splicing variations linked to severe dominant developmental disorders are not found inside the canonical splice site [55], which is similar to 20.69% obtained in this study. The most common MYO15A mutation type is missense alteration in the exonic region. Nonsense, in-frame deletion, splice-site variations, intragenic deletions and duplications are less common forms [12]. Between introns 2–65, 58 identified splice-site variations have been reported, accounting for 16.11% (58/360) of pathogenic variants in MYO15A (Table 1).
Spliceosomes are responsible for pre-mRNA splicing in humans [56]. The donor splice-site variants were more common than the acceptor splice-site variants, according to the literature review (ratio 1.5:1). We have observed that, in MYO15A, splice-site variants affect the 5' splice donor site (70.59%) more frequently than the 3ʹ donor site (29.41%).
Normal pre-mRNA splicing that define exon–intron boundaries at the + 1, + 2, − 2 and − 1 residue of an intron is usually disrupted by these canonical splice-site variants, and lead to the development of a slew of hereditary diseases [57]. However, because these intronic cis-elements are not always highly conserved and their modifications do not always impair the splicing processes, it is unclear whether non-canonical splice-site variants would result in RNA-splicing errors [58]. They may yield new cryptic exons as well as splice variants in retained intron. Despite the fact that c.3609+985A > G is positioned deep within intron 2 (more than 100 base pairs away from exon–intron boundaries), several lines of evidence suggest that it has a negative impact on the gene product. This mutation was projected to result in the loss of this putative exon’s donor site. The variant cosegregated with hearing loss in at least 8 Palestinian ARNSHL families and was not present in any public database. The reference base pair was conserved among multiple species. The 150-bp genomic sequence immediately proximal to the variant site was predicted to have exotics potential based on conservation analysis. It is predicted that c.3609+985A > G leads to the loss of this hypothetical exon’s donor site [8].
It is accessible to acquire MYO15A RNA from patients’ inner ear to assess the effect of variants on expression directly. Multiple in silico prediction computer algorithms have been developed to predict the results of non-canonical splice-site variants [59, 60]. Due to the high complexion of splicing regulation, in silico prediction methods lack sufficient specificity and sensitivity for reliable application. By combining the outputs of multiple predictive tools, a more accurate prediction can be achieved. However, such in silico tools, even for combination, can only be used as a single piece of integrated supporting evidence in the evaluation of pathogenicity [55, 61]. The in vitro minigene splicing assay provides a useful tool for analysis of splice events, including RT-PCR, cell-based minigene assays, and massive parallel reporter assays [61]. A transient minigene experiment for c.9083+6 T > A revealed the abnormal splicing pattern, which could be caused by disruption of U1 snRNP binding to the 5ʹ splice-site, which prevents splicing initiation and results in exon 52 skipping [45, 62]. The Human Splicing Finder program predicted that c.6956+9C > G would result in a strong ectopic splicing site (HSF score of 80.6) [27]. In order to provide a better understanding of alternative tissue-specific splicing mechanism, in vivo minigene assay have been applied in the zebrafish and C. elegans [63, 64]. It’s not completely understood how some splice-site variants disrupt normal translation and produce unusual transcriptional products in the inner ear. The precise medical care for DFNB3 patients will benefit from a better understanding of mRNA processing from mutant MYO15A.
Web resources
varSEAK, https://varseak.bio/. SpliceAI, https://github.com/Illumina/SpliceAI. CADD, https://www.bio.tools/CADD_Phred#!. MaxEntScan, http://hollywood.mit.edu/burgelab/maxent/.
Availability of data and materials
The patients’ phenotype and detected variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under the accession numbers SCV001332616.1, SCV001332617.1, and SCV001332618.1.
Abbreviations
- ARNSHL:
-
Autosomal recessive nonsyndromic hearing loss
- WES:
-
Whole exome sequencing
- ACMG:
-
American college of medical genetics and genomics
- AMP:
-
Association for molecular pathology
References
Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med. 2006;354(20):2151–64.
Smith RJ, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365(9462):879–90.
Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Morton CC, Morell RJ, Noben-Trauth K, Camper SA, et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280(5368):1447–51.
Belyantseva IA, Boger ET, Friedman TB. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A. 2003;100(24):13958–63.
Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol. 2005;7(2):148–56.
Beyer LA, Odeh H, Probst FJ, Lambert EH, Dolan DF, Camper SA, Kohrman DC, Raphael Y. Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J Neurocytol. 2000;29(4):227–40.
Gao X, Zhu QY, Song YS, Wang GJ, Yuan YY, Xin F, Huang SS, Kang DY, Han MY, Guan LP, et al. Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J Transl Med. 2013;11:284.
Abu Rayyan A, Kamal L, Casadei S, Brownstein Z, Zahdeh F, Shahin H, Canavati C, Dweik D, Jaraysa T, Rabie G, et al. Genomic analysis of inherited hearing loss in the Palestinian population. Proc Natl Acad Sci U S A. 2020;117(33):20070–6.
Sakuma N, Moteki H, Takahashi M, Nishio SY, Arai Y, Yamashita Y, et al. An effective screening strategy for deafness in combination with a next-generation sequencing platform: a consecutive analysis. J Hum Genet. 2016;61(3):253–61.
Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet. 2001;109(5):535–41.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50.
Wu CC, Tsai CY, Lin YH, Chen PY, Lin PH, Cheng YF, Wu CM, Lee CY, Erdenechuluun J, Liu TC, et al. Genetic epidemiology and clinical features of hereditary hearing impairment in the Taiwanese population. Genes (Basel). 2019;10(10):772.
Figueroa-Ildefonso E, Bademci G, Rajabli F, Cornejo-Olivas M, Villanueva RDC, Badillo-Carrillo R, et al. Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population. Genes (Basel). 2019;10(8).
Nal N, Ahmed ZM, Erkal E, Alper OM, Luleci G, Dinc O, et al. Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat. 2007;28(10):1014–9.
Hou YC, Yu HC, Martin R, Cirulli ET, Schenker-Ahmed NM, Hicks M, et al. Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc Natl Acad Sci U S A. 2020;117(6):3053–62.
Bazazzadegan N, Vazehan R, Fadaee M, Fattahi Z, Abolhassani A, Parsimehr E, et al. Brief report of variants detected in hereditary hearing loss cases in Iran over a 3-year period. Iran J Public Health. 2019;48(10):1910–5.
Sloan-Heggen CM, Babanejad M, Beheshtian M, Simpson AC, Booth KT, Ardalani F, et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet. 2015;52(12):823–9.
Woo HM, Park HJ, Baek JI, Park MH, Kim UK, Sagong B, et al. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet. 2013;14:72.
Motavaf M, Soveizi M, Maleki M, Mahdieh N. MYO15A splicing mutations in hearing loss: a review literature and report of a novel mutation. Int J Pediatr Otorhinolaryngol. 2017;96:35–8.
Zhang J, Guan J, Wang H, Yin L, Wang D, Zhao L, Zhou H, Wang Q. Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med Genet. 2019;20(1):60.
Liang P, Chen F, Wang S, Li Q, Li W, Wang J, et al. Whole exome sequencing of six Chinese families with hereditary non-syndromic hearing loss. Int J Pediatr Otorhinolaryngol. 2021;148:110817.
Sommen M, Schrauwen I, Vandeweyer G, Boeckx N, Corneveaux JJ, van den Ende J, et al. DNA diagnostics of hereditary hearing loss: a targeted resequencing approach combined with a mutation classification system. Hum Mutat. 2016;37(8):812–9.
Bademci G, Foster J 2nd, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet Med. 2016;18(4):364–71.
Sun Y, Yuan J, Wu L, Li M, Cui X, Yan C, et al. Panel-based NGS reveals disease-causing mutations in hearing loss patients using BGISEQ-500 platform. Medicine (Baltimore). 2019;98(12):e14860.
Atik T, Onay H, Aykut A, Bademci G, Kirazli T, Tekin M, et al. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS One. 2015;10(11):e0142154.
Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomarkers. 2011;15(1–2):29–33.
Yang T, Wei X, Chai Y, Li L, Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;8:85.
Safka Brozkova D, Poisson Markova S, Meszarosova AU, Jencik J, Cejnova V, Cada Z, et al. Spectrum and frequencies of non GJB2 gene mutations in Czech patients with early non-syndromic hearing loss detected by gene panel NGS and whole-exome sequencing. Clin Genet. 2020;98(6):548–54.
Schrauwen I, Sommen M, Corneveaux JJ, Reiman RA, Hackett NJ, Claes C, et al. A sensitive and specific diagnostic test for hearing loss using a microdroplet PCR-based approach and next generation sequencing. Am J Med Genet A. 2013;161A(1):145–52.
Sun T, Xu K, Ren Y, Xie Y, Zhang X, Tian L, Li Y. Comprehensive molecular screening in Chinese usher syndrome patients. Invest Ophthalmol Vis Sci. 2018;59(3):1229–37.
Wang L, Zhao L, Peng H, Xu J, Lin Y, Yang T, et al. Targeted next-generation sequencing identified compound heterozygous mutations in MYO15A as the probable cause of nonsyndromic deafness in a Chinese Han family. Neural Plast. 2020;2020:6350479.
Yuan Y, Li Q, Su Y, Lin Q, Gao X, Liu H, Huang S, Kang D, Todd NW, Mattox D, et al. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls. Eur J Hum Genet. 2020;28(2):231–43.
Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, et al. Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum Mutat. 2016;37(10):991–1003.
Yan D, Tekin D, Bademci G, Foster J 2nd, Cengiz FB, Kannan-Sundhari A, et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum Genet. 2016;135(8):953–61.
Downie L, Halliday J, Burt R, Lunke S, Lynch E, Martyn M, et al. Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur J Hum Genet. 2020;28(5):587–96.
Zazo Seco C, Wesdorp M, Feenstra I, Pfundt R, Hehir-Kwa JY, Lelieveld SH, et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet. 2017;25(3):308–14.
Belguith H, Aifa-Hmani M, Dhouib H, Said MB, Mosrati MA, Lahmar I, et al. Screening of the DFNB3 locus: identification of three novel mutations of MYO15A associated with hearing loss and further suggestion for two distinctive genes on this locus. Genet Test Mol Biomarkers. 2009;13(1):147–51.
Riahi Z, Bonnet C, Zainine R, Louha M, Bouyacoub Y, Laroussi N, et al. Whole exome sequencing identifies new causative mutations in Tunisian families with non-syndromic deafness. PLoS One. 2014;9(6):e99797.
Han JJ, Nguyen PD, Oh DY, Han JH, Kim AR, Kim MY, et al. Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population. Sci Rep. 2019;9(1):1604.
Boudewyns A, van den Ende J, Sommen M, Wuyts W, Peeters N, Van de Heyning P, et al. Role of targeted next generation sequencing in the etiological work-up of congenitally deaf children. Otol Neurotol. 2018;39(6):732–8.
Budde BS, Aly MA, Mohamed MR, Breß A, Altmüller J, Motameny S, et al. Comprehensive molecular analysis of 61 Egyptian families with hereditary nonsyndromic hearing loss. Clin Genet. 2020;98(1):32–42.
Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O, Ulubil-Emiroglu M, et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A. 2007;143A(20):2382–9.
Cabanillas R, Dineiro M, Cifuentes GA, Castillo D, Pruneda PC, Alvarez R, et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics. 2018;11(1):58.
García-García G, Berzal-Serrano A, García-Díaz P, Villanova-Aparisi R, Juárez-Rodríguez S, de Paula-Vernetta C, et al. Improving the Management of Patients with Hearing Loss by the Implementation of an NGS Panel in Clinical Practice. Genes (Basel). 2020;11(12).
Danial-Farran N, Brownstein Z, Gulsuner S, Tammer L, Khayat M, Aleme O, Chervinsky E, Zoubi OA, Walsh T, Ast G, et al. Genetics of hearing loss in the Arab population of Northern Israel. Eur J Hum Genet. 2018;26(12):1840–7.
Khan A, Han S, Wang R, Ansar M, Ahmad W, Zhang X. Sequence variants in genes causing nonsyndromic hearing loss in a Pakistani cohort. Mol Genet Genomic Med. 2019;7(9):e917.
Chen Y, Wang Z, Chen D, Chai Y, Pang X, Sun L, et al. Targeted next-generation sequencing in Uyghur families with non-syndromic sensorineural hearing loss. PLoS One. 2015;10(5):e0127879.
Zhang F, Xu L, Xiao Y, Li J, Bai X, Wang H. Three MYO15A mutations identified in one Chinese family with autosomal recessive nonsyndromic hearing loss. Neural Plast. 2018;2018:5898025.
Chang MY, Kim AR, Kim NK, Lee C, Lee KY, Jeon WS, Koo JW, Oh SH, Park WY, Kim D, et al. Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing. Mol Cells. 2015;38(9):781–8.
Chang MY, Lee C, Han JH, Kim MY, Park HR, Kim N, Park WY, Oh DY, Choi BY. Expansion of phenotypic spectrum of MYO15A pathogenic variants to include postlingual onset of progressive partial deafness. BMC Med Genet. 2018;19(1):29.
Liu WH, Chang PY, Chang SC, Lu JJ, Wu CM. Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS ONE. 2019;14(1): e0211261.
Usami SI, Nishio SY, Moteki H, Miyagawa M, Yoshimura H. Cochlear Implantation from the perspective of genetic background. Anat Rec (Hoboken). 2020;303(3):563–93.
Xia H, Huang X, Guo Y, Hu P, He G, Deng X, Xu H, Yang Z, Deng H. Identification of a novel MYO15A mutation in a Chinese family with autosomal recessive nonsyndromic hearing loss. PLoS ONE. 2015;10(8): e0136306.
Ma D, Shen S, Gao H, Guo H, Lin Y, Hu Y, Zhang R, Wang S. A novel nonsense mutation in MYO15A is associated with non-syndromic hearing loss: a case report. BMC Med Genet. 2018;19(1):133.
Wai H, Douglas AGL, Baralle D. RNA splicing analysis in genomic medicine. Int J Biochem Cell Biol. 2019;108:61–71.
Hang J, Wan R, Yan C, Shi Y. Structural basis of pre-mRNA splicing. Science. 2015;349(6253):1191–8.
Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32.
Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28(21):4364–75.
Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–3.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods. 2005;37(4):331–40.
Fischer B, Luthy K, Paesmans J, De Koninck C, Maes I, Swerts J, Kuenen S, Uytterhoeven V, Verstreken P, Versees W. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol. 2016;23(11):965–73.
Wani S, Kuroyanagi H. An emerging model organism Caenorhabditis elegans for alternative pre-mRNA processing in vivo. Wiley Interdiscip Rev RNA. 2017;8(6):e1428.
Markmiller S, Cloonan N, Lardelli RM, Doggett K, Keightley MC, Boglev Y, Trotter AJ, Ng AY, Wilkins SJ, Verkade H, et al. Minor class splicing shapes the zebrafish transcriptome during development. Proc Natl Acad Sci U S A. 2014;111(8):3062–7.
Acknowledgements
We sincerely thank all the family members for their participation and cooperation in this study.
Funding
This work was supported by grants from Beijing Natural Science Foundation (7191011), National Natural Science Foundation of China (82171158, 81730029, 81873704, 81870731, 82171155, 81900953 and 61827805), and Natural Science Foundation of Hainan Province (819MS110). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Y-YY and PD conceived of the study and participated in its design. XG, J-YY, W-QW drafted the manuscript. KY, S-SH and D-YK participated in the next generation sequencing and literature review. M-YH, YS, G-JW, and J-CX participated in the data analysis and results discussion. XL, YF and XZ participated in the collection of clinical data and blood samples. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Chinese PLA General Hospital Research Ethics Committee. Parents of minor subject (5 year-old) signed informed consent forms for participation in clinical and genetic research. In this study, all methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
We obtained fully informed written consent from parents of minor subjects for publication of their clinical data.
Competing interests
The authors declare that there is no competing interest in this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers sequences.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, JY., Wang, WQ., Han, MY. et al. Addition of an affected family member to a previously ascertained autosomal recessive nonsyndromic hearing loss pedigree and systematic phenotype-genotype analysis of splice-site variants in MYO15A. BMC Med Genomics 15, 241 (2022). https://doi.org/10.1186/s12920-022-01368-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01368-9