
Abstract
Background
Although over 60 non-syndromic deafness genes have been identified to date, the etiologic contribution of most deafness genes remained elusive. In this study, we addressed this issue by targeted next-generation sequencing of a large cohort of non-syndromic deaf probands.
Methods
Probands with mutations in commonly screened deafness genes GJB2, SLC26A4 and MT-RNR1 were pre-excluded by Sanger sequencing. The remaining 125 deaf probands proceeded through targeted exon capturing of 79 known deafness genes and Illumina HiSeq2000 sequencing.
Results
Bi-allelic mutations in 15 less commonly screened deafness genes were identified in 28 deaf probands, with mutations in MYO15A, GPR98, TMC1, USH2A and PCDH15 being relatively more frequent (≥3 probands each). Dominant mutations in MYO6, TECTA, POU4F3 and COCH were identified in 4 deaf families. A mitochondrial MTTS1 mutation was identified in one maternally inherited deaf family. No pathogenic mutations were identified in three dominant deaf families and two consanguineous families.
Conclusions
Mutations in the less commonly screened deafness genes were heterogeneous and contributed to a significant percentage (17.4%) of causes for non-syndromic deafness. Targeted next-generation sequencing provided a comprehensive and efficient diagnosis for known deafness genes. Complementary to linkage analysis or whole-exome sequencing of deaf families, pre-exclusion of known deafness genes by this strategy may facilitate the discovery of novel deafness genes.
Similar content being viewed by others

Background
Hearing impairment, or deafness in its most severe form, is the most common sensory disorder in humans. One in a thousand children develops congenital or prelingual deafness. Another one in a thousand children becomes deaf or severely hearing impaired before adulthood [1]. It was estimated that at least 50% to 60% of childhood hearing impairment was caused by genetic factors [2]. Among them, approximately 70% of cases are non-syndromic, meaning the hearing impairment is the only distinctive clinical feature, and the remaining 30% are syndromic with other abnormalities [3].
Identifying the genetic basis of deafness provides crucial information for diagnosis, intervention and treatment of the disease. The causes, however, are extremely heterogeneous. To date, mutations in 64 genes have been found to be associated with non-syndromic deafness (Hereditary Hearing Loss Homepage, http://webh01.ua.ac.be/hhh/). Most of those genes were originally identified through the linkage analysis of single or multiple deaf families. Expanded screening in large cohorts revealed that mutations in three genes, GJB2, SLC26A4 and MT-RNR1, were commonly found in deaf patients. In Chinese Hans, for example, bi-allelic GJB2 mutations were reported in 19.1% of patients with non-syndromic deafness, followed by bi-allelic SLC26A4 mutations in 12.1% and the mitochondrial A1555G mutation of MT-RNR in 1.6% [4–6]. These genes have been routinely screened during genetic testing and counseling of deafness.
On the other side, the causes remained unknown for majority of deaf individuals. The etiologic contribution of the less commonly screened deafness genes, in particular, has yet to be investigated systematically. Due to the large number and presumably low mutation frequencies of those genes, it would be highly expensive and time-consuming to address this issue by the conventional gene-by-gene Sanger sequencing. This limitation, however, may be overcome by the targeted next-generation sequencing (NGS) technologies. With genomic capturing of a wide array of known deafness genes, it is possible to screen those genes in high throughput by NGS. The feasibility of this strategy has been confirmed in principle in several pilot studies [7–9]. In this study, we performed a comprehensive mutations screening of 79 deafness genes in a large cohort of non-syndromic deaf probands using the targeted NGS. Our results provided a preliminary overview of the genetic etiology of non-syndromic deafness in Chinese Hans.
Methods
Recruitment of the subjects
Non-syndromic deaf probands were recruited from deaf patients seeking genetic testing and counseling in Xinhua Hospital, Shanghai, China. Family history and clinical questionnaire was obtained from all subjects or their parents. Physical and neurological examination was performed with special attention to renal, electrocardiac and ophthalmologic differences to exclude those with syndromic deafness. A comprehensive auditory evaluation was performed including otoscope examination, tympanometry, pure-tone audiometry (PTA), auditory brainstem response (ABR) in subjects with very young age, and in some cases computed tomography (CT) of the inner ear. The onset of hearing impairment was categorized as prelingual/early (≤ 6 years) and late (> 6 years). PTA was calculated as the average of the hearing level at 0.5, 1.0, 2.0 and 4.0 KHz for the better ear. The severity of hearing impairment was defined as mild (20~40 dB), moderate (41–70 dB), severe (71~95 dB) and profound (>95 dB). To enroll in this study, all probands should have bilateral, permanent sensorineural hearing impairment. The hearing level of the prelingual or early-onset deaf probands should exceed 70 dB. For the late-onset, progressive probands, their hearing level should exceed 40 dB at the age of 35 years. All subjects or their parents gave written, informed consent to participate in this study. This study was approved by the Ethics Committee of the Shanghai Jiaotong University School of Medicine, Xinhua Hospital.
Pre-exclusion of mutations in common deafness genes
All deaf probands (n=190) were pre-screened against common deafness genes GJB2, SLC26A4 and MT-RNR1 by PCR amplification and bidirectional sequencing as previously described [4–6]. A total of 125 deaf probands were void of mutations in the three common deafness genes and proceeded through the targeted exon capturing and NGS.
Targeted genomic capturing and next-generation sequencing
A customized capture array (NimbleGen, Roche) was designed to capture all exons, splicing sites and immediate flanking intron sequences of 79 deafness genes (Additional file 1: Table S1) as previously described [10]. Genomic DNA were extracted from the blood samples using the Blood DNA kit (TIANGEN BIOTECH, Beijing, China) and fragmented to 200–300 base pairs using an ultrasonoscope (Covaris S2, Massachusetts, USA). End-repair, adenylation and adapter ligation were performed for library preparation following standard Illumina protocols. Targeted DNA fragments were captured by hybridization to the capture array and sequenced on Illumina HiSeq2000 Analyzers for 90 cycles per read. Image analysis, error estimation and base calling were performed using the Illumina Pipeline (version 1.3.4).
Data analysis and bioinformatics processing were performed as previously described [10]. Reads were aligned to NCBI37/hg19 assembly using the BWA Multi-Vision software package. SNPs and indels were identified using the SOAPsnp software and the GATK Indel Genotyper, respectively. Previously identified SNPs and their allele frequencies were determined using the NCBI dbSNP, 1000 Genomes and the in-house sequencing data of 200 Chinese Han normal hearing controls.
Confirmation of the potential mutations
Potential mutations detected by targeted NGS were verified by PCR amplification and Sanger sequencing in corresponding probands. For multiplex probands, segregation of the variants with the deaf phenotype was also verified in extended family members when available. The strength of the ectopic splicing sites created by two intronic variants was evaluated by the Human Splicing Finder program (http://www.umd.be/HSF/). Possible pathogenic effects of the missense mutations were evaluated by the Mutation Taster (http://www.mutationtaster.org), PROVEAN and SIFT (with cut-off scores set at −1.3 and 0.05, respectively, http://sift.jcvi.org) programs.
Results
Characterization of the deaf probands
A total of 190 probands with non-syndromic deafness was recruited in our study, including 137 simplex and 53 multiplex probands. Seven multiplex probands were from dominant deaf families; four from maternally inherited deaf families; forty-two had at least one first- or second-degree deaf relative and were consistent with a possible recessive inheritance.
Clinical information of the deaf probands was summarized in Table 1. The ages of the probands varied between a few months to over 50 years (mean 14.0 years, 95% CI 11.8-16.3 years). The 7 dominant probands had late-onset, progressive, moderate-to-severe sensorineural hearing impairment. All other probands had prelingual or early-onset, severe-to-profound sensorineural hearing impairment.
Pre-screening of the common deafness genes
Pre-screening of mutations in three common deafness genes, GJB2, SLC26A4 and MT-RNR1, was performed in all 190 deaf probands by Sanger sequencing. Bi-allelic mutations in GJB2 and SLC26A4 and the homoplasmic A1555G mutation of MT-RNR1 were detected in 36, 22 and 7 probands, respectively (Additional file 2: Table S2). Those probands were excluded from further screening by targeted NGS.
Targeted NGS sequencing of known deafness genes
The remaining 125 deaf probands, including 93 simplex probands, 7 dominant probands, 1 maternally inherited proband and 24 multiplex probands with presumably recessive inheritance went through targeted NGS screening of 79 deafness genes (Additional file 1: Table S1). Included in the list were 50 non-syndromic deafness genes (24 autosomal recessive, 17 autosomal dominant, 6 autosomal recessive/dominant, 2 X-linked and 1 mitochondrial), 7 non-syndromic/syndromic deafness genes (4 autosomal recessive, 2 autosomal recessive/dominant and 1 mitochondrial) and 22 syndromic deafness genes. A total of 1496 exons, 340995 bases (233790 bases in coding regions and 107205 bases in flanking intronic and untranslated regions) were captured and sequenced in our study. After alignment to the reference human genome (NCBI37/hg19), 47.28% of the clean reads could be uniquely matched to the targeted regions. The average depth for the targeted regions was 293.21-fold. 99.35% of the targeted regions were covered by 20 or more reads, demonstrating the high quality of the sequencing.
Mutation identification and verification
Hundreds of variants of high sequencing quality were detected in each proband. To identify the most likely pathogenic mutations, we filtered out: 1) all previously identified SNPs with allele frequencies of 0.005 or higher, 2) synonymous variants in the coding region and 3) variants in the intronic or untranslated regions (with the exception of the splicing site mutations or variants that may create an ectopic splicing site).
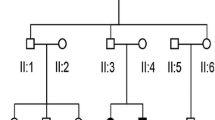
In 7 dominant probands, we identified 7 heterozygous, non-synonymous variants. None of them was found in 200 ethnically-matched normal hearing controls. Four of them were found co-segregating with the deaf phenotype in extended family members. In addition, a homoplasmic mitochondrial variant was identified in a maternally inherited deaf family with reduced penetrance (Table 2 and Figure 1). Among the five candidate mutations, T7511C in MT-TS1 and p.C1509G in TECTA have been reported to be associated with non-syndromic deafness in previous studies [11–14]. For the other three novel mutations, p.Q982X in MYO6 and c.603_604delGG in POU4F3 were predicted to lead to a prematurely stopped protein, while p.G87V in COCH changed an evolutionarily conserved amino acid (PhyloP score: 4.75) and was unanimously predicted to be disease-causing by the Mutation Taster, PROVEAN and SIFT programs. Our results suggested that these five mutations in MYO6, TECTA, POU4F3, COCH and MTTS1 were pathogenic.

Pedigrees and the identified mutations of the deaf families (Dominant: D372, D385, D882, P59, NT01, K11 and F9; mitochondrial inherited: D1037; consanguineous: D804 and C12). Probands of each family were pointed by the arrows.
A total of 254 non-synonymous or splicing site variants were identified in 117 simplex or recessive multiplex probands with allele frequencies of 0.0025 or lower (Additional file 3: Table S3). Consistent with the recessive inheritance, 48 candidate mutations in 15 deafness genes were present in homozygotes (6 probands) or compound heterozygotes (22 probands, Table 3). Three of them, p.R2728H in MYO15A, p.R445H and c.150delT in TMC1, have been previously reported to be associated with non-syndromic deafness [8, 15, 16]. Among the other 45 novel candidate mutations, sixteen were nonsense mutations, frame-shifting indels or splicing site mutations. In addition, two intronic variants, c.6956+9C>G in MYO15A and c.4961-3C>G in OTOF, were predicted to create a strong ectopic splicing site (HSF score of 80.6 and 75.0, respectively) by the Human Splicing Finder program. These 18 mutations were predicted to lead to a significantly truncated protein product. For the remaining 25 missense variants and 2 in-frame deletions, all of them changed evolutionarily conserved amino acids with positive PhyloP scores and were predicted to be disease-causing by at least one of the Mutation Taster, PROVEAN and SIFT programs. Nineteen of 25 missense mutations were unanimously predicted to be disease-causing by all three programs. None of the 48 candidate mutations were found in the 200 Chinese Han normal hearing controls. For 9 multiplex probands (noted with asterisks in Table 3), the bi-allelic mutations were also found in their deaf relatives. Based on these results, the 48 recessive candidate mutations were highly likely to be pathogenic.
Overall, our targeted NGS screening identified one mitochondrial mutation in MTTS1 and four dominant mutations in MYO6, TECTA, POU4F3 and COCH each. For recessive deafness genes, bi-allelic mutations were identified in 19 simplex and 9 multiplex probands, respectively. Mutations in MYO15A were most frequently detected (5/125), followed by mutations in USH2A, PCDH15, GPR98, TMC1 (3/125 each), CDH23 (2/125), OTOF, MYO7A, ESRRB, MARVELD2, TECTA, WHRN, MYO6, PTPRQ and POU3F4 (1/125 each).
Discussion
We investigated the genetic epidemiology of non-syndromic deafness in the present study. Like most deaf patients seeking genetic testing and counseling, the majority of our subjects (179/190) were simplex or recessive multiplex probands with non-syndromic, prelingual or early-onset, severe-to-profound hearing impairment. Unlike the syndromic deafness or the progressive, late-onset hearing impairment with representative audioprofiles, this type of hearing impairment is extremely heterogeneous and was often difficult to determine the genetic causes.
We attempted to solve this problem by targeted NGS screening of 79 deafness genes, including all known 57 non-syndromic deafness genes at the time when this study was designed. An additional 22 syndromic deafness genes were also included in the screening in case that any additional symptoms of syndromic deafness were either overlooked or absent during the clinical evaluation (see discussion below on USH2A mutations and Usher Syndrome type 2A). We focused on the presumably rare deafness genes by pre-exclusion of mutations in three commonly screened deafness genes GJB2, SLC26A4 and MT-RNR1. A strict criterion was applied to identify the possible pathogenic mutations including limiting the candidate mutations only to those with allele frequencies of 0.0025 or lower (based on our in-house sequencing data of 200 ethnically-matched normal hearing controls), recognizing only the bi-allelic variants as candidate recessive mutations, confirming the genotype-phenotype co-segregation with extended family members of the multiplex probands, and using a variety of analysis programs to predict the effects of the candidate mutations.
Consistent with our presumptions of the heterogeneity and the low mutation frequencies of the less commonly screened deafness genes, our targeted NGS screening identified a wide range of dominant, recessive and mitochondrial mutations in 20 deafness genes, whereas no individual gene had mutation detection rate over 3%. Overall, however, those less commonly screened deafness genes contributed to 17.4% (33/190) of causes in our deaf probands, illustrating the importance of genetic testing in those genes.
Several limitations need to be noted when interpreting our results. First, the majority of the probands were recruited through genetic testing and counseling of patients with hereditary hearing loss, which may lead to over-representation of the multiplex probands (27.9% in our subjects, estimated 22.3% in US in comparison[2]). The percentages of the identified genetic causes, therefore, may be slightly over-represented accordingly. Second, our mutation identification criteria selected only the bi-allelic variants as the candidate recessive mutations. It was possible that a small number of probands with mono-allelic mutations may have another undisclosed mutation in the untargeted regions of the genes such as the introns or the regulatory regions, resulting in slight under-representation of the identified genetic causes. Third, mutations in USH2A were associated with Usher Syndrome type 2A, a syndromic form of deafness characterized by congenital deafness and later development of progressive retinitis pigmentosa (RP). Because of the late onset of RP (usually postpubertal) in Usher Syndrome type 2A patients, it was not surprising that we identified bi-allelic USH2A mutations in three “non-syndromic” deaf probands, all under 6 years old.
Nevertheless, our study presented a preliminary overview of the genetic etiology of non-syndromic deafness in Chinese Hans (Figure 2). In addition to GJB2, SLC26A4 and MT-RNR1, a variety of less commonly screened deafness genes accounted for a significant portion of genetic causes of non-syndromic deafness. A total of 53 different types of mutations in 20 deafness genes were identified, with 48 of them being novel. Our results confirmed the effectiveness of the targeted NGS screening of the known deafness genes.

Estimates of genetic causes of non-syndromic deafness in Chinese Hans based on our study.
Interestingly, our comprehensive screening of 79 deafness genes did not identify any pathogenic mutations in three dominant families and two consanguineous families (Figure 1). We postulated that hearing impairment in these families was likely to be caused by mutations in novel deafness genes and warranted further studies by linkage analysis and/or whole-exome sequencing. Used as a complementary step, this pre-exclusion strategy against known deafness genes may facilitate the discovery of novel deafness genes.
Abbreviations
- NGS:
-
Next-generation sequencing
- PTA:
-
Pure-tone audiometry
- ABR:
-
Auditory brainstem response (ABR)
- CT:
-
Computed tomography
- RP:
-
Retinitis pigmentosa.
References
Morton NE: Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991, 630: 16-31. 10.1111/j.1749-6632.1991.tb19572.x.
Nance WE, Lim BG, Dodson KM: Importance of congenital cytomegalovirus infections as a cause for pre-lingual hearing loss. J Clin Virol. 2006, 35: 221-225. 10.1016/j.jcv.2005.09.017.
Nance WE: The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003, 9: 109-119. 10.1002/mrdd.10067.
Dai P, Yu F, Han B, Liu X, Wang G, Li Q, Yuan Y, Huang D, Kang D, Zhang X, et al: GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009, 7: 26-10.1186/1479-5876-7-26.
Ji YB, Han DY, Lan L, Wang DY, Zong L, Zhao FF, Liu Q, Benedict-Alderfer C, Zheng QY, Wang QJ: Molecular epidemiological analysis of mitochondrial DNA12SrRNA A1555G, GJB2, and SLC26A4 mutations in sporadic outpatients with nonsyndromic sensorineural hearing loss in China. Acta Otolaryngol. 2011, 131: 124-129. 10.3109/00016489.2010.483479.
Yuan Y, Guo W, Tang J, Zhang G, Wang G, Han M, Zhang X, Yang S, He DZ, Dai P: Molecular epidemiology and functional assessment of novel allelic variants of SLC26A4 in non-syndromic hearing loss patients with enlarged vestibular aqueduct in China. PLoS One. 2012, 7: e49984-10.1371/journal.pone.0049984.
Baek JI, Oh SK, Kim DB, Choi SY, Kim UK, Lee KY, Lee SH: Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J Rare Dis. 2012, 7: 60-10.1186/1750-1172-7-60.
Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, Parzefall T, Lev D, Shalev S, Frydman M, et al: Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011, 12: R89-10.1186/gb-2011-12-9-r89.
Shearer AE, DeLuca AP, Hildebrand MS, Taylor KR, Gurrola J, Scherer S, Scheetz TE, Smith RJ: Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc Natl Acad Sci U S A. 2010, 107: 21104-21109. 10.1073/pnas.1012989107.
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T, Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One. 2012, 6: e29500-
Chapiro E, Feldmann D, Denoyelle F, Sternberg D, Jardel C, Eliot MM, Bouccara D, Weil D, Garabedian EN, Couderc R, et al: Two large French pedigrees with non syndromic sensorineural deafness and the mitochondrial DNA T7511C mutation: evidence for a modulatory factor. Eur J Hum Genet. 2002, 10: 851-856. 10.1038/sj.ejhg.5200894.
Pfister M, Thiele H, Van Camp G, Fransen E, Apaydin F, Aydin O, Leistenschneider P, Devoto M, Zenner HP, Blin N, et al: A genotype-phenotype correlation with gender-effect for hearing impairment caused by TECTA mutations. Cell Physiol Biochem. 2004, 14: 369-376. 10.1159/000080347.
Sue CM, Tanji K, Hadjigeorgiou G, Andreu AL, Nishino I, Krishna S, Bruno C, Hirano M, Shanske S, Bonilla E, et al: Maternally inherited hearing loss in a large kindred with a novel T7511C mutation in the mitochondrial DNA tRNA(Ser(UCN)) gene. Neurology. 1999, 52: 1905-1908. 10.1212/WNL.52.9.1905.
Friedman RA, Bykhovskaya Y, Sue CM, DiMauro S, Bradley R, Fallis-Cunningham R, Paradies N, Pensak ML, Smith RJ, Groden J, et al: Maternally inherited nonsyndromic hearing loss. Am J Med Genet. 1999, 84: 369-372. 10.1002/(SICI)1096-8628(19990604)84:4<369::AID-AJMG12>3.0.CO;2-V.
Santos RL, Wajid M, Khan MN, McArthur N, Pham TL, Bhatti A, Lee K, Irshad S, Mir A, Yan K, et al: Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Hum Mutat. 2005, 26: 396-10.1002/humu.9374.
Yang T, Kahrizi K, Bazazzadeghan N, Meyer N, Najmabadi H, Smith RJ: A novel mutation adjacent to the Bth mouse mutation in the TMC1 gene makes this mouse an excellent model of human deafness at the DFNA36 locus. Clin Genet. 2010, 77: 395-398. 10.1111/j.1399-0004.2009.01338.x.
Acknowledgements
This research was supported by grants from National Basic Research Program of China (2011CB504501 to HW), National Science Foundation of China (81222010 to TY and 81170924 to HW), Shanghai Municipal Science and Technology Commission (11PJ1407000 to TY) and the Eastern Scholar program from Shanghai Institutions of Higher Learning (to TY).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TY and HW conceived and designed the experiments. TY, HW, YC and LL ascertained the probands and collected the DNA samples. XW performed the targeted NGS screening and bioinformatics analyses. YC and LL performed the auditory evaluation and other laboratory experiments. TY wrote the paper. All authors read and approved the final manuscript.
Tao Yang, Xiaoming Wei contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yang, T., Wei, X., Chai, Y. et al. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis 8, 85 (2013). https://doi.org/10.1186/1750-1172-8-85
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-8-85