
Abstract
Background
Hexokinase 2 (HK2) is an enzyme that catalyses the conversion of glucose to glucose-6-phosphate, which has been found to be associated with malignant tumour growth. However, the potential immunological and clinical significance of HK2, especially in terms of prognostic prediction for patients with glioma, has not been fully elucidated.
Methods
To investigate the expression, immunological and clinical significance of HK2 in patients with glioma, several databases, including ONCOMINE, TIMER2.0, GEPIA, CGGA, UCSC, LinkedOmics, Metascape, STRING, GSCA, and TISIDB, as well as biochemical, cellular, and pathological analyses, were used in this study. In addition, we performed univariate, multivariate Cox regression and nomogram analyses of the hub genes positively and negatively correlated with HK2 to explore the potential regulatory mechanism in the initiation and development of glioma.
Results
Our results demonstrated that HK2 was highly expressed in most malignant cancers. HK2 expression was significantly higher in lower grade glioma (LGG) and glioblastoma (GBM) than in adjacent normal tissue. In addition, HK2 expression was significantly correlated with clinical parameters, histological manifestations, and prognosis in glioma patients. Specifically, the data from The Cancer Genome Atlas downloaded from UCSC Xena database analysis showed that high expression of HK2 was strongly associated with poor prognosis in glioma patients. The LinkedOmics database indicated that HK2-related genes were mainly enriched in immune-related cells. In LGG and GBM tissues, HK2 expression is usually correlated with recognized immune checkpoints and the abundance of multiple immune infiltrates. Similarly, the Metascape database revealed that HK2-related genes were mainly enriched and annotated in immune-related pathways and immune cells. Further investigations also confirmed that the inhibition of HK2 expression remarkably suppressed metastasis and vasculogenic mimicry (VM) formation in glioma cells through regulating the gene expression of inflammatory and immune modulators.
Conclusion
HK2 expression was closely associated with the malignant properties of glioma through activating multiple immune-related signalling pathways to regulate immune responses and the infiltration of immune cells. Thus, HK2 and its hub genes may be a potential target for the treatment of glioma.
Similar content being viewed by others


Background
Gliomas, which are intrinsic brain tumours that originate from neuroglial progenitor cells [1], are the most malignant and aggressive form of brain tumours and account for the majority of brain cancer-related deaths [2]. According to the World Health Organization (WHO) criteria, gliomas can be mainly classified as low-grade gliomas (LGG; grades I - III) and high-grade diffuse gliomas (grade IV), which are also known as glioblastoma (GBM). Conventional therapies, including surgery, chemotherapy and radiotherapy, have achieved limited improvements in the prognosis of glioma patients [3, 4]. Previous studies indicated that immunotherapy can be a promising strategy for the treatment of malignant tumours, especially tumours derived from the central neural system, because of their penetrability to the blood-brain barrier [1, 5]. However, clinical trials have indicated that the effect of these strategies is still limited for the treatment of gliomas. Several recent reports revealed that immune checkpoints might be safely targeted with high antitumour efficacy in glioma and achieve better anticancer efficacy through dual targeting with a variety of inhibitory molecules [6, 7].
Hexokinases (HKs) catalyse the first committed step in glucose metabolism [8]. A recent study indicated that HKs are involved in metabolic flux through glycolysis in hyperglycaemia [9], while the abnormally regulated glucose metabolism frequently results in increased glycolysis intermediates or glycolysis overload and ultimately induces tumorigenesis [10]. Several reports have demonstrated that highly expressed HKs (mostly of the HK2 isoform) are positively correlated with tumorigenesis in many malignant tumours, including glioma, bladder cancer, oral squamous cell carcinoma, colorectal cancer, and breast cancer [11,12,13,14,15]. In contrast, deletion of the HK2 gene can significantly inhibit the proliferation of cancer cells in animal models [16]. HK2 might be crucial for the Warburg effect, cell metabolism, cell survival, gluconic metabolic reprogramming, immune response, and inflammation [17, 18]. However, the correlation between HK2 and immune checkpoints has not been fully elucidated [18, 19].
In this study, we investigated the potential roles of HK2 expression in predicting prognosis on the basis of various databases and explored the correlation with clinicopathological features and the possible regulatory mechanism in glioma. Specifically, GSEA, the functional annotations and the signalling pathways of HK2 and HK2 related genes, and the correlation among HK2-related genes were analysed by the LinkedOmics database, Metascape, and protein-protein interaction (PPI) network, respectively. In addition, the screening of hub genes of HK2-related genes and the correlation between hub genes and the prognosis in glioma patients were analysed by cytoHubba plug-in and GEPIA databases, respectively. The top five hub genes of HK2-related genes were further confirmed by univariate and multivariate Cox regression and nomogram analysis. In addition, further biochemical, cellular, and pathological assays also indicated that HK2 expression was closely associated with tumour metastasis and vasculogenic mimicry (VM) formation in glioma cells through regulating the gene expression of inflammatory and immune modulators, as well as poor prognosis in glioma patients. Therefore, our findings suggested that HK2 might be a potential biomarker for investigating glioma development and predicting prognosis in glioma patients.
Methods
Analyses of differential expression of HK2 in various malignant tumours and glioma
To study the potential functions of HK2 in tumorigenesis, the ONCOMINE database (www.oncomine.org), which is an integrated online cancer microarray database for DNA/RNA sequence analyses [20], was first used to investigate the transcriptional expression of HK2 between different cancer tissues and their adjacent normal tissues. Differences in transcriptional expression were compared by Student’s t-test. The cut-off P-value and fold change were as follows: P-value is 0.01; fold change is 2; gene rank is 10%, and data type is mRNA.
To investigate the expression of HK2 in different glioma histologies, the choices were as follows: gene is HK2; analysis type is cancer vs. adjacent normal tissue; cancer type is glioblastoma; data type is mRNA; P-value < 0.05; gene rank is top 10%. The correlation between immune infiltrates and the HK2 expression level in glioma of the Cancer Genome Atlas (TCGA) cohort was analysed by tumour immune Estimation Resource 2.0 (TIMER2.0; http://timer.cistrome.org/) [21]. The correlation of HK2 expression between glioma and adjacent normal tissue was analysed by using the GEPIA database (http://gepia.cancer-pku.cn/) [22].
Chinese glioma genome atlas (CGGA) and UCSC Xena databases
The correlation between clinicopathological parameters and the mRNA level of HK2 in glioma patients was analysed by using the CGGA database (http://www.cgga.org.cn/), which is a user-friendly platform for data storage and analysis to explore brain tumour datasets from over 2000 specimens from Chinese cohorts. This database includes whole-exome sequencing (n = 286), DNA methylation (n = 159), mRNA sequencing (n = 1018), mRNA microarray (n = 301), and matched clinicopathological information, including gender, age, histologic grade, histology, IHD status, 1p/19q co-deletion and progression status [23].
To study the potential roles of HK2 expression in glioma, we investigated the correlation between HK2 expression and clinicopathologic characteristics, as well as the various prognostic parameters in patients with glioma (n = 669), by using a genome-related UCSC Xena (http://xena.ucsc.edu/) database [24].
Survival analysis
The correlation between the expression of HK2 (or hub genes) and four prognostic indices, including overall survival (OS), progression-free survival (PFS), disease-free survival (DSS), and disease-free survival (DFS), was specifically analysed in brain tumours. In this context, the time from randomization to death (for any reason), the period between the beginning of treatment and the observation of disease progression or death from any cause in patients with tumour diseases, the terminal event was disease recurrence or death after treatment, and the death caused by a specific disease is the end event, were respectively defined as OS, PFS, DFS, and DSS. If it is not caused by a specific disease, it will not be included in the outcome index. The Kaplan-Meier plotter survival curve of HK2 (or hub genes) in TCGA dataset was analysed by the “survival” and “survminer” packages in R language (or GEPIA database). The receiver operating characteristic (ROC) curve was analysed by the “survival ROC” package. In addition, an immune-related prognostic model was established by using univariate and multivariate Cox regression analyses. The results were displayed using the forestplot package. All analytical methods above and R packages were performed using R software version v4.0.3.
LinkedOmics database analysis
The LinkedOmics database (http://www.linkedomics.org) [25], which contains multi-omics data and clinical data for 32 cancer types and a total of 11,158 patients from TCGA, was used to analyse the HK2-related genes and the signalling pathways in glioma. Briefly, the parameters were as follows: cancer cohort was glioma (LGG and GBM; n = 669); dataset was RNAseq; dataset attribute was HK2; RNAseq and statistical method was Pearson correlation method; selected tool was gene set enrichment analysis (GSEA); enrichment analysis was Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway; rank criteria were from LinkFinder module. KEGG (Kyoto Encyclopedia of Genes and Genomes) (https://www.genome.jp/kegg/) is a knowledge base for systematic analysis of gene functions, and it is the most well-known and universal signal pathway database [26].
Enrichment analysis and PPI network
Metascape (http://metascape.org), an integrated set of more than 40 gene function annotation databases [27], was used to investigate the enrichment analysis of HK2-related genes obtained from LinkedOmics (|Pearson’s rho| ≥ 0.5, P < 0.05). In addition, the analysis included gene ontology (GO) and KEGG enrichment analysis. We set min overlap as 3, min enrichment as 1.5, and P < 0.05 as significant.
To analyse the protein–protein interactions, the STRING online database (http://string-db.org; version: 11.0) was used to build a PPI network [28]. The molecular composite detection plug-in cytoHubba within Cytoscape was used to cluster the potential PPI network [29].
Evaluation of the correlation between immune infiltration and HK2 expression in glioma
The correlation between HK2 expression and immune infiltration in glioma was analysed using the TIMER2.0 database, which is a comprehensive resource for systematically analysing immune infiltrates in many malignant cancers [21]. In addition, Pearson analysis was performed to assess the correlations between HK2 expression and immune checkpoints, as well as mismatch repair (MMR) proteins. In addition, TISIDB (http://cis.hku.hk.TISIDB/) was performed to analyse the potential roles of HK2 in the regulation of immunomodulatory factors in glioma [30]. GSCALite database (http://bioinfo.life.hust.edu.cn/GSCA/#/) is a cross type comprehensive cancer analysis database. It explores the gene set cancer analysis relate to mRNA expression, mutation, immune infiltration and drug resistance, including 33 types of cancer data of TCGA and GDSC. The database contains the analysis of genome (gene expression, Single Nucleotide Variation (SNV), Copy Number Variation (CNV) and DNA methylation) and immune genome (including 24 immune cells). The TCGA mRNA expression and DNA methylation data were obtained from UCSC Xena. DNA methylation profile was measured experimentally using the Illumina Infinium HumanMethylation450 platform (about 1% of all human methylation sites, and it covers most CpG islands and promoter regions). DNA methylation values, described as beta values, are recorded for each array probe in each sample via BeadStudio software. DNA methylation beta values are continuous variables between 0 and 1, representing the ratio of the intensity of the methylated bead type to the combined locus intensity. Thus, the higher beta values represent higher level of DNA methylation, i.e. hypermethylation and lower beta values represent lower level of DNA methylation, i.e. hypomethylation. Finally, the correlations between the DNA methylation of HK2 or hub genes and immune infiltrates, as well as the potential roles of hub genes in the immune cell subset, were analysed by using the GSCALite database [31].
Cells and cell culture
The human glioma cell line T98 cells and the normal human brain astrocyte cell line HEB cells, were purchased from China Center for Type Culture Collection (Shanghai, China). All the cells were cultured in DMEM medium (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS (Gibco) and streptomycin/penicillin antibiotic mixture (Invitrogen, Thermo Fisher Scientific) at 37 °C with 5% CO2. The primary cell culture was slightly modified from the previously reported protocol [32,33,34]. The cell lines T98 and HEB cells were authenticated by short tandem repeat (STR) profiling of 15 loci and the amelogenin sex determination (X or XY) method (Promega, Madison, WI) according to our previous described [35]. The authenticated primary cell line (GBM1) that derived from GBM surgical specimens, was obtained from Procell Company (Wuhan, China). The primary cell line was maintained in primary serum-free cultures grown on laminin [36]. Before each experiment, the GBM1 cells were cultured in DMEM medium, supplemented with 10% FBS, streptomycin/penicillin antibiotic mixture, and 2 mM L-glutamine (Invitrogen) at 37 °C with 5% CO2. For transfection, the siRNAs targeting HK2 (si-HK2) were synthesized by RiboBio Biotech (Guangzhou, China). All cells were transfected with si-HK2 by using Lipofectamine 3000 reagent (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instruction.
Quantitative real-time PCR (qRT-PCR)
Total RNA of different groups of cells was extracted by using a Trizol kit (Invitrogen). The RNA was reverse transcribed using the StarScript II First-strand cDNA Synthesis Mix (GenStar, Shanghai, China). The subsequent real-time quantitative polymerase chain reaction was configured according to 2 × RealStar Green Fast Mixture instructions (GenStar). The RT-qPCR was detected using the ABI QS5 Real Time PCR software (ABI, Thermo Fisher Scientific) and normalized to actin by using 2-ΔΔCt. All primers were listed in Supplementary Table S1.
Western blot
The protein samples were extracted from different groups of cells by using cracking buffer solution RIPA (Solarbio, Beijing, China) at 4 °C for 15 min. The supernatants were centrifuged at 15,000 rpm, and the total protein was quantified by the BCA Kit (Solarbio). Thereafter, the protein samples were separated on 10% SDS-polyacrylamide gels and transferred to 0.45 μm PVDF membranes (Millipore, Billerica, MA). The blots were blocked with 5% Bovine Serum Albumin (BSA) in PBS buffer for 1 h, and then incubated with the appropriate primary antibodies (1:2000) at 4 °C overnight. After washing, the blots were incubated with the corresponding secondary antibodies (1:5000) at room temperature for 2 h. The signals were visualized by using a SuperSignal West Pico Substrate Kit (Pierce, Thermo Fisher Scientific), and analysed by using ImageJ software (National Institutes of Health, Bethesda, MD). The anti-Hexokinase 2 (22029-I-AP) and anti-STAT3 (10253–2-AP) antibodies were obtained from ProteinTech Group Inc. (Rosemont, IL), the anti-p-AKT (#4060) and anti-AKT (#4691) antibodies were purchased from Cell Signaling Technology (Danvers, MA), anti-p-STAT3 (BM4835) and anti-GAPDH (BM1623) antibodies, and all secondary antibodies were from Boster Biological Technology Ltd. (Wuhan, China).
Wound healing assays
All the cells were seeded and scratched with a 10 μL pipette tip in 24-well plate. The sound width was photographed at 0 and 24 h under a phase-contrast microscope, respectively.
Transwell assay (cell invasion assay)
An 8-μm pore size of Falcon™ cell culture inserts (BD Biosciences, Franklin Lakes, NJ), coated with Matrigel (1:20, BD Biosciences) in the upper chambers, was placed in a 24-well Corning transwell microplates (Corning Incorporated, NY). A total 5 × 104 cells were seeded into with serum-free DMEM medium. The DMEM medium with 10% FBS were filled in the bottom chambers. After 16 h of incubation, the cells on the upper surface were carefully removed with a cotton swab, and the membranes were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 20 min, respectively. The invasive cells were examined under a phase-contrast microscope.
Tube formation assay
The cold Matrigel (BD Biosciences) was added into a μ-Slide angiogenesis (ibidi GmbH, Gräfelfing, Germany) with a pre-chilled micropipette, and incubated 5% CO2-humidified incubator at 37 °C for 1 h for solidification. The cells (5 × 104) were seeded in solidified Matrigel and incubated with 2% FBS for 3 h. The length of tubes in each field was photographed and the average of tubules from 3 random fields of view.
Tissue microarray (TMA) and immunohistochemistry (IHC)
The glioma TMA, which was obtained from Outdo Biotech Ltd. (Shanghai, China), was stained with anti-Hexokinase 2 polyclonal antibody (1:50; 22,029-I-AP, Proteintech Group Inc.) at 4 °C overnight and analysed with Image-Pro Plus software (Media Cybernetics, Rockville, MD).
Statistical analysis
The overview of our workflow was shown in Supplementary Fig. S1. The clinical significance of HK2 expression in glioma patients was further validated in the ONCOMINE, GEPIA, and TIMER2.0 databases. Briefly, the correlation between HK2 and hub gene expression was analysed by the GEPIA database. The HK2-related functional annotation and signalling pathways were identified using LinkedOmics and Metascape, respectively. In addition, the survival rates were displayed with HRs, 95% Cis, and log-rank P-value. Univariate and multivariate Cox regression was used to analyse the correlation between the mRNA level of hug genes and the prognosis of glioma patients. Statistical analyses of group differences were performed by using Student’s t test and ANOVA. All experiments were conducted with three independent replicates and the results were presented as means ± SEM. The differences in patient’s prognosis between different groups were analysed with Kaplan-Meier analyses with log-rank tests. All data were analysed by Rv4.0.3. P < 0.05 was regarded as statistically significant. All experiments were repeated three times with similar results.
Results
The expression of HK2 positively correlates with the malignancy of glioma
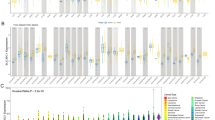
To get insight the potential roles of HK2 expression in tumorigenesis and tumour development, the ONCOMINE database was used to validate the expression level of HK2 in various malignant cancers. Compared with adjacent normal tissues, HK2 was found to be highly expressed in bladder cancer, oesophageal cancer, gastric cancer, head and nest cancer, kidney cancer, lymphoma, ovarian cancer, pancreatic cancer, sarcoma, and brain and central nervous system (CNS)-related cancer (Fig. 1A), suggesting that HK2 may play an important role in tumorigenesis. Moreover, the alignment between adjacent normal tissues from GTEx and tumour specimens from TCGA database reflected that high expression of HK2 was determined in most malignant cancers (Fig. 1B). Specifically, HK2 expression was significantly increased in ACC (adrenocortical carcinoma), BLCA (bladder urothelial carcinoma), BRCA (breast invasive carcinoma), CESC (cervical squamous cell carcinoma and endocervical adenocarcinoma), CHOL (cholangiocarcinoma), COAD (colon adenocarcinoma), ESCA (oesophageal carcinoma), GBM, HNSC (head and neck squamous cell carcinoma), KIRC (kidney renal clear cell carcinoma), KIRP (kidney renal papillary cell carcinoma), LGG, LIHC (liver hepatocellular carcinoma), LUAD (lung adenocarcinoma), LUSC (lung squamous cell carcinoma), OV (ovarian serous cystadenocarcinoma), PAAD (pancreatic adenocarcinoma), PRAD (prostate adenocarcinoma), SKCM (skin cutaneous melanoma), STAD (stomach adenocarcinoma), TGCT (testicular germ cell tumours), THCA (thyroid carcinoma), UCEC (uterine corpus endometrial carcinoma) and UCS (uterine carcinosarcoma). Notably, compared with the adjacent normal tissue, further GEPIA analysis then confirmed that HK2 expression was significantly increased in LGG and GBM tissues and positively correlated with the malignancy of gliomas (Fig. 1C). ROC showed that the expression of HK2 mRNA in glioma and normal tissue was 0.92 (95% CI: 0.907–0.933) (Fig. 1D) and the best cut-off value of HK2 was 1.483 (TPM; Transcript per million). Similarly, our results also indicated significant upregulation of HK2 expression in T98 and GBM1 cells, compared with that in HEB normal control (Fig. 1E). In addition, the expression levels of HK2 were significantly upregulated in different glioma subgroups, including glioblastoma, oligodendroglioma, anaplastic astrocytoma, anaplastic oligoastrocytoma and anaplastic oligodendroglioma, in TCGA, Bredel, Mural, Sun, and French datasets (Fig. 1F-K, Supplementary Table S2), suggesting that HK2 may play an important role in glioma tumorigenesis and development.

HK2 expression is positively correlated with the malignancy of human glioma. A The expression of HK2 in different cancers by using ONCOMINE database analysis. B The expression levels of HK2 in different cancer types from TCGA and GTEx datasets. C Compared with adjacent normal tissues, the expression of HK2 was much higher in LGG and GBM from GEPIA database. Box plots of differential expression of HK2 in different histologies of glioma. D The ROC curve analysis between patients and healthy controls to HK2. E The expression levels of HK2 expression in HEB, T98, and GBM1 cell lines. F-K The expression levels of HK2 were upregulated in different glioma subgroups (e.g., GBM, oligodendroglioma, anaplastic astrocytoma, and anaplastic oligoastrocytoma) from different datasets (e.g., TCGA, Bredel, Mural, Sun, and French datasets). Data shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
High expression of HK2 is associated with poor prognosis in glioma patients
We then evaluated the expression of HK2 with the CGGA database and several clinical parameters in glioma patients [37, 38]. Our results indicated that HK2 expression was positively correlated with histological manifestations (P = 5.9e-23), malignancy (P = 7.6e-19), IDH mutation (P = 2.6e-14), 1p/19q co-deletion (P = 2e-32), IDH mutation & 1p/19q co-deletion (P = 1.7e-26), and 1p/19q co-deletion in different grades (P = 0.012, P = 1.5e-18, and P = 6.6e-07, respectively) in glioma patients (Supplementary Fig. S2A-F). In addition, there was no significant difference between HK2 expression and gender (P = 0.28), age (P = 0.88), or progression (P = 0.084) in glioma (Supplementary Fig. S2G-I). Similarly, 669 LGG and GBM specimens in TCGA dataset from the UCSC Xena database (Supplementary Table S3) revealed that HK2 expression was closely correlated with the clinicopathological features of glioma patients, including malignancy (P < 0.001), IDH mutation (P < 0.001), O-6-methylguanine DNA methyltransferase (MGMT) promoter status (P < 0.001), transcriptome subtype (P < 0.001), histology (P < 0.001), and 1p/19q co-deletion (P < 0.001) (Supplementary Table S4).
We further investigated the potential role of HK2 expression in predicting prognosis in glioma patients. Our results indicated that the survival rates decreased with increasing HK2 expression in OS, PFS, and DSS (Fig. 2A-C), whereas there was no correlation between HK2 expression and DFS in patients with gliomas (Fig. 2D). In addition, the mRNA level of HK2 was always accompanied by poorer OS (P < 0.0001), PFS (P < 0.0001), and DSS (P < 0.0001) (Fig. 2E-G) but not DFS (P = 0.19) in glioma patients (Fig. 2H). Analysis of TCGA dataset (n = 699) also illustrated the potential role of HK2 expression in predicting prognosis in glioma patients. As shown in Supplementary Table S5, compared with lower expression of HK2, high expression of HK2 was significantly associated with poor prognosis in glioma patients (P < 0.001). Multivariate Cox regression analysis indicated that histological grade (P < 0.001) and IDH mutation (P < 0.001) can be biomarkers for predicting poor survival when HK2 expression (P = 0.177), MGMT promoter status (P = 0.919), transcriptome subtype (P = 0.435), and 1p/19q co-deletion (P = 0.637) were considered. Furthermore, ROC curves of four prognostic models, including OS, PFS, DSS, and DFS, revealed that high expression of HK2 was positively correlated with poor prognosis in glioma (Fig. 2I-L).

High expression of HK2 is associated with poor prognosis in gliomas. A-D Heatmap of HK2 expression profiles, risk type distribution, and survival time in glioma patients. E-H The correlation between HK2 expression and patient prognosis (OS, PFS, DSS and DFS) of glioma in TCGA database. I-L ROC curves for the 1-, 3-, and 5-year survival according to the HK2 risk signature in glioma. ****P < 0.0001
Notably, according to the classification of central nervous system tumours divided adult gliomas based on IDH status and 1p19q codeletion status [39], similar results from TCGA and CGGA databases were also determined that the highest mRNA expression of HK2 was found in GBM (Fig. 3A-B). With the higher histological grade of glioma, the mRNA expression of HK2 tended to be higher. Further, we used the Kaplan-Meier plotter to analyse the prognostic values of the mRNA expression of HK2 in new histological manifestations of glioma. The results from TCGA database showed that HK2 mRNA expression showed no correlation with the prognosis of Oligodendroglioma (P = 0.97). However, higher mRNA expression of HK2 was associated with poorer prognosis in Astrocytoma (P = 0.018) and glioblastoma (P = 0.026) (Fig. 3C-E). CGGA database shows the same results. The mRNA expression of HK2 also showed no correlation with the prognosis of Oligodendroglioma (P = 0.65). In contrast, the higher mRNA expression of HK2 was associated with poorer prognosis in Astrocytoma (P = 0.00074) and glioblastoma (P < 0.0001) (Fig. 3F-H).

Evaluate HK2 gene expression with box plots according to clinical parameters in glioma patients using data from TCGA and CGGA databases. A-B The mRNA expression of HK2 was remarkably correlated with different histological manifestations of glioma in TCGA (A) and CGGA databases (B). C-H The higher mRNA expression of HK2 was associated with shorter OS in different histological manifestations of glioma. I-J Mutation rate of IDH1 in LGG (I) and GBM (J). K-L Mutation rate of IDH2 in LGG (K) and GBM (L). M-N IDH1 mutation in LGG (M) and GBM (N) was associated with OS. O-P HK2 expression was markedly decreased in the IDH mutation group compared to IDH wide type group in TCGA (O) and CGGA databases (P). Q Representative images of HK2 expression in glioma patient specimens at low (10 ×; upper panels) and high (100 ×; lower panels) magnification. R Quantification of the correlation between HK2 expression and malignancy of the glioma specimens. LGG, I and II grades; HGG, III and IV grades. S Overall and disease-free survival rates in low- (n = 40) or high-expression (n = 41) of HK2 in glioma patients. *P < 0.05, **P < 0.01, ***P < 0.001. ****P < 0.0001
Since previous studies have shown that IDH mutations are closely related to the prognosis of glioma [39], we next analysed the role of IDH1 and IDH2 mutations in glioma by using the GSCA database. The mutation rate of IDH1 (R132H) in LGG was 77.57%, and the mutation rate in glioblastoma was 6.45%. IDH2 (R172K) mutation rate in LGG was 3.99% and in glioblastoma was 0.25% (Fig. 3I-L). The results showed that IDH1 mutation was dominant in glioma compared with IDH2. Next, we analysed IDH1 mutation and its prognostic value in LGG and GBM patients. The results from Kaplan-Meier plot showed that IDH1 mutation was associated with better OS in LGG (Fig. 3M) and glioblastoma (Fig. 3N). Finally, we analysed the relationship between HK2 expression and IDH mutation. The results of TCGA and GSCA databases showed that compared with IDH mutation group, the level of HK2 gene expression was significantly higher in IDH wild type group (Fig. 3O-P).
Next, we subjected glioma TMA for immunostaining by applying anti-HK2 antibody (Fig. 3Q). The results revealed that the lower expression of HK2 was mainly determined in lower-grade gliomas (I and II; n = 40). In contrast, HK2 staining intensity was observed to be elevated in higher-grade gliomas (HGG; III and IV; n = 41), indicating that HK2 expression is positively correlated with the malignancy of glioma patients (Fig. 3R). Moreover, we correlated HK2 expression with clinicopathologic characteristics of glioma (Table 1). Correlation analysis revealed that HK2 expression was positively associated with high-grade gliomas (P < 0.001) and tumour recurrence (P < 0.001). There was no significant difference between HK2 expression and age (P = 0.735) and gender (P = 0.939). In addition, we then compared patients’ survival in relation to the HK2 expression in LGG and HGG cohorts. We found that the worse prognosis was positively related to the expression level of HK2 in glioma patients (Fig. 3S). Age, gender, grades and tumour recurrence were included in the Cox multivariate survival analysis (Table 2). The results showed that grades (P = 0.016) and tumour recurrence (P = 0.01) were risk factors for the prognosis of glioma patients. Together, our findings suggested that HK2 expression might be a potential biomarker for predicting prognosis of the patients with glioma.
The inhibition of HK2 expression suppresses tumour metastasis and VM formation in glioma cells
Since the aberrant HK2 expression was critical for glioma development in vivo and in vitro, we next examined whether the inhibition of HK2 could suppress tumour development in glioma cells. Therefore, we attempted to establish HK2-knockdown glioma cell lines through respectively transfecting two siRNAs targeted to HK2 in T98 and GBM1 cells. Western blot assay indicated that the transfection of either si-HK2 miRNA (si-HK2–1 and si-HK2–2) could efficiently inhibit HK2 expression in these two cell lines (Fig. 4A). Further wound healing and transwell assays revealed that the metastatic capabilities were significantly inhibited in HK2-silenced T98 and GBM1 cells (Fig. 4B-C). In addition, we also found that the VM Formation was also inhibited after inhibiting HK2 expression in glioma cells (Fig. 4D), suggesting that the HK2 is potentially related to tumorigenesis and tumour development in the patients with glioma.

Knockdown of HK2 expression results in the suppression of tumour metastasis and VM Formation in glioma cells. A Representative images and quantification of the inhibition of HK2 expression in si-RNAs targeted HK2 (si-HK2–1 and si-HK2–2)-mediated HK2 knockdown in T98 and GBM1 cell lines. B-C The suppression of HK2 expression can inhibit tumour metastasis by using wound healing (B) and transwell (C) assays in T98 and GBM1 cell lines. D Tube formation assay shows HK2 knockdown could induce the inhibition of VM Formation in glioma cells. Bars, 200 μm. Data shown as mean ± SEM.*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
HK2 is involved in glioma development through regulating immune infiltration and immune checkpoints
The TIMER2.0 database-based analyses illustrated that HK2 expression may be associated with immune infiltration, indicating the potential prognostic role of HK2 in glioma patients. In LGG and GMB specimens, HK2 expression was related to infiltration of CD4+ T cells, M1 macrophages, regulatory T cells (Tregs), mast cells, macrophages/monocytes, neutrophils and myeloid dendritic cells (Fig. 5A). Specifically, in LGG specimens, the analysis of the GSCA database revealed that HK2 expression was positively correlated with the infiltration score, cytotoxicity, dendritic cells (DCs), macrophages, monocytes, and T helper 1 and 2 (Th1/2) cells but negatively correlated with B cells, naïve CD4 T cells (CD4_naïve), naïve CD8 T cells (CD8_naïve), CD8 T cells (CD8_T), central memory and neutrophils. Additionally, we found positive correlations between HK2 expression and the infiltration score, macrophage, monocyte, natural killer T cells (NKT), T-helper 17 (Th17) cells, and B cells and negative correlations between the nTreg, CD4_naïve, CD8_naïve, and CD8_T groups in GBM patients (Fig. 5B-C). In addition, CD8_T, CD8_naïve, central memory, and B cells were found to be associated with the DNA methylation of HK2 in both LGG and GBM groups (FDR < 0.05) (Fig. 5D-E).

The correlation between HK2 expression and immune infiltration level in LGG and GBM. A Evaluation of the correlation between HK2 expression and immune infiltration by using the TIMER 2.0 database. B-E The correlation between the expression (B-C) or DNA methylation (D-E) of HK2 expression and immune infiltration in LGG and GBM specimens from the GSCA database. F The correlation between the expression of HK2 and immune score in gliomas. G The correlation between HK2 and the confirmed immune checkpoints in gliomas. H The mRNA levels of TNF-α, IL-2, IL-3, IL-4, and IL-12 in HK2-knockdown and control groups in T98 and GBM1 cell lines. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001, ns, not significant
We then evaluated the correlation between HK2 expression and immune score by using the TCGA dataset. The ESTIMATE algorithm revealed that HK2 expression was positively related to immune invasion in glioma specimens (Fig. 5F). Interestingly, we found that immune checkpoints were also involved in this process (Fig. 5G). TISIDB database analysis showed a positive correlation between HK2 expression and DNA methylation in the LGG and GBM groups with immune stimulators, immune inhibitors or MHC molecules (Supplementary Fig. S3A-F). Therefore, to further confirm our hypothesis, we next examined the expression of several key inflammatories and immune modulators, including TNF-α, IL-2, IL-3, IL-4, and IL-12, in HK2-silenced T98 and GBM1 glioma cells. qRT-PCR assay demonstrated the upregulation of TNF-α, IL-2, IL-3, and IL-4 expression, whereas the downregulation of IL-12 expression in HK2-knockdown glioma cells (Fig. 5H), suggesting that HK2 is essential for glioma development through regulating immune infiltration.
Identification and analysis of genes related to HK2 expression in glioma
Due to the potential roles of HK2 in glioma development, we hypothesized that HK2 might represent a new target for the treatment of glioma. We identified the gene expression related to HK2 using the LinkedOmics database. The most significantly positively or negatively related genes (n = 50) and signalling pathways (n = 32), which were regulated by HK2, were identified by using heatmaps (Fig. 6A-B, Supplementary Table S6) and KEGG pathway enrichment analysis (Fig. 6C, Supplementary Table S7). Specifically, eukaryotic orthologous groups (KOG) identified the top six immune-related signalling pathways, including NF-κB, complement and coagulation cascades, Toll-like receptor, NOD-like receptor, Th17 cell differentiation, and JAK-STAT signalling pathways, which are regulated by HK2 (Fig. 6D-I).

GO term, KEGG pathway, and GSEA enrichment analyses of HK2-related genes. A-B Heatmap of the top 50 positively and negatively related genes with HK2. C KEGG pathway enrichment analysis using the GSEA database. D-I Gene set enrichment plots of the NF-κB (D), complement and coagulation cascades (E), Toll-like receptor (F), NOD-like receptor (G), Th17 cell differentiation (H), and JAK-STAT (I) signalling pathways. J-M GO and KEGG enrichment analyses of HK2 positively or negatively related genes (Pearson’s rho ≥0.5, P < 0.05). N Expression levels of p-STAT3, STAT3, p-Akt, Akt in HK2-knockdown and control groups in T98 and GBM1 cell lines. Data shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Next, GO and KEGG cluster analyses were performed to identify potential HK2-regulated downstream signalling (|Pearson’s rho| ≥ 0.5, P < 0.05). GO term enrichment indicated that positively correlated HK2-related genes enriched in positive regulation of interleukin− 8 production, T cell activation involved in immune response, CD4-positive, alpha-beta T cell differentiation, T cell proliferation, interleukin− 8 production, nterleukin-1 production, toll−like receptor signalling pathway, cell activation involved in immune response, positive regulation of inflammatory response and immune response−regulating signalling pathways, were positively regulated (Fig. 6J). In contrast, negatively correlated HK2-related genes were enriched in neural nucleus development, neurotransmitter transport, regulation of dendrite development, regulation of cell morphogenesis involved in differentiation, regulation of G protein−coupled receptor signalling pathway, negative regulation of nervous system development, regulation of neuron projection development, negative regulation of kinase activity, brain development and cell morphogenesis involved in differentiation (Fig. 6K). In addition, KEGG pathway analysis revealed that Staphylococcus aureus infection, leishmaniasis, osteoclast differentiation, tuberculosis, phagosome and systemic lupus erythematosus, synaptic vesicle cycle, epithelial cell signalling in Helicobacter pylori infection, GABAergic synapse and neuroactive ligand−receptor interaction signals were involved in HK2-associated pathways (Fig. 6L-M). Moreover, it is noted that similar results were also determined by using western blot analyses that the inactivation of PI3K/Akt signalling pathways after HK2 knockdown in T98 and GBM1 glioma cells (Fig. 6N).
The mRNA levels of hub genes of HK2-related genes might be independent prognostic biomarkers in glioma patients
The STRING database showed an interactive network of most genes with strong correlations to HK2 expression (|Pearson’s rho| ≥ 0.5, P < 0.05) (Supplementary Fig. S4A-B). Notably, the interactive network of the top 10 hub genes (Supplementary Tables S8 & S9) was analysed by the CytoHubba plug-in (Fig. 7A-B). The results showed that the top 5 positive HK2-related hub genes were ITGB2, CD53, C3AR1, CYBB and ITGAM, whereas the top 5 negative HK2-related hub genes were SYP, CPLX1, SLC6A1, GABRG2 and SCRT1. GEPIA analysis then confirmed that the expression of all these hub genes was identically regulated in LGG and GBM specimens, except for SLC6A1 expression, which was not significantly different in the LGG group (Fig. 7C). In addition, the GEPIA database also showed correlations between the individual expression of these hub genes and HK2 (Supplementary Fig. S5A-J), suggesting that HK2 is probably involved in glioma development by regulating its hub genes.

Graphic representation of hub genes of the PPI network and the correlation between the top 10 hub genes and the prognostic prediction in glioma patients. A-B The interactive network of the top 10 hub genes in HK2 positive correlation genes and top 10 hub genes in HK2 negative correlation genes by using the algorithm. C The expression of the top 10 hub genes positively and negatively correlated with HK2 in LGG and GBM specimens. D The correlation between the expression of these hub genes and the prognosis (OS and FPS) of glioma patients. *P < 0.05
We also found that the high expression of the top 5 positively HK2-related hub genes was associated with poorer OS and PFS, whereas the negative HK2-related hub genes showed better prognosis in an expression level-dependent manner in gliomas (Fig. 7D), suggesting that the expression of these hub genes was involved in the prognosis of glioma patients. In addition, univariate Cox regression analyses indicated significant correlations between the expression of these hub genes (ITGB2, CD53, C3AR1, CYBB, ITGAM, SYP, CPLX1, GABRG2, SLC6A1 and SCRT1) and the prognosis of glioma (Supplementary Fig. S6A-B). Moreover, further multivariate Cox regression analysis revealed that the expression of CYBB (P = 0.00087), CD53 (P = 0.04883), SCRT1 (P = 0.00071) and SYP (P = 0.02491) can be independent factors of the prognosis of gliomas (Supplementary Fig. S6C-D) and predict the survival of glioma patients (Supplementary Fig. S6E-F).
We subsequently evaluated the correlation between the expression of these hub genes and immune infiltration. Our results indicated that the expression or DNA methylation of all these HK2-related hub genes is closely correlated with immune cell subtypes (Fig. 8A-H). In addition, we also found a correlation between hub genes and immune score in glioma specimens (Fig. 8I), suggesting that the expression of hub genes plays a role in glioma development by regulating immune infiltration.

Correlations between hub genes expression or DNA methylation and immune cells. A-H The correlation between the expression (A-D) or DNA methylation (E-H) of all these hub genes and immune infiltration in LGG and GBM specimens. I The correlation between the expression of hub genes and immune score in glioma. *P < 0.05, **P < 0.01
Discussion
HK2 is usually expressed in skeletal, cardiac muscle and adipose tissues [40], as well as many malignant tumour tissues [41]. Recent study indicated that the regulation of cancer cell metabolism may be partially induced by the recruitment of the immune and inflammatory cells [42]. In addition, the metabolic reprogramming is not only closely associated with tumorigenesis, angiogenesis, tumour invasion and migration, but also involved in the process of proliferation, differentiation, and execution of effector functions, which is important to the immune response [43,44,45]. In the tumour microenvironment, the aberrant or intermediates metabolites of metabolism in cancer cells could be associated with the regulation of activation, differentiation, and function of immune cells [46, 47]. A very recent study indicated that the cancer cells are able to reduce the metabolic fitness of tumour-infiltrating immune cells, and subsequently escape anti-tumour immune response [48, 49]. Therefore, the overlapping immune response and metabolic reprogramming in cancer cells would be critical for anti-tumour immune response in cancer. In the present study, we found that HK2 is highly expressed in gliomas and that HK2 expression is positively correlated with OS, PFS, and DSS in glioma patients, suggesting that it may be a potential clinical target for the treatment of glioma. Our results also indicated that HK2 expression is associated with recognized immune checkpoints and the abundance of multiple immune infiltrates, including CD4+ T cells, M1 macrophages, regulatory T cells, mast cells, monocytes, neutrophils and myeloid dendritic cells, in LGG and GBM specimens. There was a slight difference in the TIMER 2.0 and GSCA databases, which might result from different algorithms of different databases. Multiple database analyses can provide more comprehensive and reliable evidence in this study. Our results also demonstrated that the high expression of HK2 is associated with poor prognosis of glioma. We expected that HK2 may regulate innate immune cells and abnormally activate the immune response, resulting in the poor prognosis of gliomas. However, the underlying mechanism needs to be further investigated. Moreover, although there are certain correlations between HK2 expression and immune stimulators, immune inhibitors and MHC molecules in LGG and GBM patients, the underlying mechanism by which HK2 activates immune cells still needs to be further investigated.
A previous study indicated that PD-L1 can enhance glycolysis by regulating HK2 expression in non-small cell lung cancer (NSCLC), indicating that HK2 may play a role in tumour immunity [50], which is consistent with our results also showing that the HK2-regulated genes were mainly enriched in immune-related signalling pathways and the activation of immune cells (Fig. 6C-I). Further investigations revealed that the positive HK2-regulated genes were mainly associated with immune cells, the immune response and the secretion of inflammatory factors (Fig. 6J), suggesting that certain HK2-mediated immune response processes were involved in glioma development. The identification of the downstream signals of HK2 demonstrated that the high expression of hub genes, including ITGB2, CD53, C3AR1, CYBB and ITGAM, maybe a potential target for the treatment of glioma. In contrast, the upregulation of the top 5 negative HK2-regulated hub genes, such as SYP, CPLX1, GABRG2, SLC6A1 and SCRT1, was accompanied by a better prognosis of gliomas. Moreover, multivariate Cox regression revealed that CYBB, CD53, SCRT1 and SYP might be independent factors for predicting the prognosis of gliomas. It is known that CYBB encodes the gp91-phox protein of phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [51]. Previous studies indicated that NADPH oxidase 2, also known as CYBB/NOX2, in conventional DCs (cDCs) regulates endocytosed MOG (myelin oligodendrocyte protein) antigen processing and supports MOG antigen presentation to CD4+ T cells through LC3-associated phagocytosis (LAP) [52, 53]. CD53 is a member of the tetraspanin superfamily expressed exclusively within the immune compartment. CD53 is highly expressed in a variety of immune cells, including B cells, CD4+ T cells, CD8+ T cells, dendritic cells, macrophages and natural killer cells [54]. SCRT1 participates in gene regulation, metabolic processes and immune responses as a tissue-specific transcription factor [55]. CTLA4-associated SYP could regulate the RAS pathway and T cell activation [56]. Notably, our results also showed that HK2 and its hub genes activate different immune cells by regulating the expression of different immune-related genes, thereby affecting the prognosis of gliomas, suggesting that HK2 and hub genes probably play important roles in different immune processes and may be potential targets for the treatment of glioma.
Conclusions
In summary, integrated bioinformatics approaches indicated that HK2 expression may mediate immune infiltration to affect the prognosis of glioma patients and may be a potential prognostic biomarker. In addition, HK2 and its hub genes mediate the activation of immune cells and the regulation of immune-related factors and are involved in the prognosis of glioma patients. Therefore, our findings demonstrated that HK2 and its hub genes are significantly correlated with prognosis and immune infiltration in glioma and may be potential targets for the treatment of patients with glioma.
Availability of data and materials
Publicly available datasets were analysed in this study and can be found in UCSC Xena (http://xena.ucsc.edu/), the Cancer Genome Atlas (https://portal.gdc.cancer.gov/), and the CGGA database (http://www.CGGA.org.cn/).
Abbreviations
- HK2:
-
Hexokinase 2
- LGG:
-
Lower Grade Glioma
- GBM:
-
Glioblastoma
- VM:
-
Vasculogenic Mimicry
- HKs:
-
Hexokinases
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- DSS:
-
Disease-free survival
- DFS:
-
Disease-free survival
- ROC:
-
receiver operating characteristic
- PPI:
-
Protein-protein interaction
- GSEA:
-
Geneset enrichment analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- MMR:
-
Mismatch repair
- si-HK2:
-
siRNAs targeting HK2
- BSA:
-
Bovine Serum Albumin
- TMA:
-
Tissue microarray
- IHC:
-
Immunohistochemistry
- DCs:
-
Dendritic cells
- FDR:
-
False discovery rate
References
Xu S, Tang L, Li X, Fan F, Liu Z. Immunotherapy for glioma: current management and future application. Cancer Lett. 2020;476:1–12.
Ludwig K, Kornblum HI. Molecular markers in glioma. J Neuro-Oncol. 2017;134(3):505–12.
Tom MC, Cahill DP, Buckner JC, Dietrich J, Parsons MW, Yu JS. Management for Different Glioma Subtypes: are all low-grade gliomas created equal? Am Soc Clin Oncol Educ Book. 2019;39:133–45.
Braunstein S, Raleigh D, Bindra R, Mueller S, Haas-Kogan D. Pediatric high-grade glioma: current molecular landscape and therapeutic approaches. J Neuro-Oncol. 2017;134(3):541–9.
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–42.
Wang X, Guo G, Guan H, Yu Y, Lu J, Yu J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J Exp Clin Cancer Res. 2019;38(1):87.
Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9.
Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. 2018;77(11):1636–43.
Rabbani N, Thornalley PJ. Hexokinase-2 glycolytic overload in diabetes and ischemia-reperfusion injury. Trends Endocrinol Metab. 2019;30(7):419–31.
Ashour A, Xue M, Al-Motawa M, Thornalley PJ, Rabbani N. Glycolytic overload-driven dysfunction of periodontal ligament fibroblasts in high glucose concentration, corrected by glyoxalase 1 inducer. BMJ Open Diabetes Res Care. 2020;8(2):e001458.
Zhang B, Chen J, Cui M, Jiang Y. LncRNA ZFAS1/miR-1271-5p/HK2 promotes glioma development through regulating proliferation, migration, Invasion and Apoptosis. Neurochem Res. 2020;45(12):2828–39.
Zou Y, Du Y, Cheng C, Deng X, Shi Z, Lu X, et al. FAT10 promotes the progression of bladder cancer by upregulating HK2 through the EGFR/AKT pathway. Exp Cell Res. 2021;398(1):112401.
Zhu X, Du J, Gu Z. Circ-PVT1/miR-106a-5p/HK2 axis regulates cell growth, metastasis and glycolytic metabolism of oral squamous cell carcinoma. Mol Cell Biochem. 2020;474(1–2):147–58.
Du W, Liu N, Zhang Y, Liu X, Yang Y, Chen W, et al. PLOD2 promotes aerobic glycolysis and cell progression in colorectal cancer by upregulating HK2. Biochem Cell Biol. 2020;98(3):386–95.
Zang H, Li Y, Zhang X, Huang G. Knockdown of circRAD18 mitigates breast Cancer progression through the regulation of miR-613/HK2 Axis. Cancer Manag Res. 2020;12:3661–72.
Garcia SN, Guedes RC, Marques MM. Unlocking the potential of HK2 in Cancer metabolism and therapeutics. Curr Med Chem. 2019;26(41):7285–322.
Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–26.
Zhong WJ, Yang HH, Guan XX, Xiong JB, Sun CC, Zhang CY, et al. Inhibition of glycolysis alleviates lipopolysaccharide-induced acute lung injury in a mouse model. J Cell Physiol. 2019;234(4):4641–54.
Tan VP, Miyamoto S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. 2015;11(6):963–4.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–14.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–60.
Zhao Z, Zhang KN, Wang Q, Li G, Zeng F, Zhang Y, et al. Chinese glioma genome atlas (CGGA): a comprehensive resource with functional genomic data from Chinese glioma patients. Genom Proteom Bioinform. 2021;19(1):1–12.
Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–8.
Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956–63.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Ru B, Wong CN, Tong Y, Zhong JY, Zhong SSW, Wu WC, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35(20):4200–2.
Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34(21):3771–2.
Li YY, Wang HZ, Sun T, Chen JM, Guo LC, Shen HT, et al. Biological characteristics of a new human glioma cell line transformed into A2B5(+) stem cells. Mol Cancer. 2015;14:75.
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–83.
Luo H, Chen ZX, Wang S, Zhang R, Qiu WJ, Zhao L, et al. c-Myc-miR-29c-REV3L signalling pathway drives the acquisition of temozolomide resistance in glioblastoma. Brain. 2015;138:3654–72.
Barash U, Spyrou A, Liu P, Vlodaysky E, Zhu CC, Luo JJ, et al. Heparanase promotes glioma progression via enhancing CD24 expression. Int J Cancer. 2019;145(6):1596–608.
Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4(6):568–80.
Wang Y, Qian T, You G, Peng X, Chen C, You Y, et al. Localizing seizure-susceptible brain regions associated with low-grade gliomas using voxel-based lesion-symptom mapping. Neuro-Oncology. 2015;17(2):282–8.
Liu X, Li Y, Qian Z, Sun Z, Xu K, Wang K, et al. A radiomic signature as a non-invasive predictor of progression-free survival in patients with lower-grade gliomas. Neuroimage Clin. 2018;20:1070–7.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology. 2021;23(8):1231–51.
Gatica D, Klionsky DJ. TOS-sing aside the glycolytic role of HK2/hexokinase-II to activate autophagy. Autophagy. 2015;11(6):865–6.
DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9(1):446.
Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, et al. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther. 2019;195:162–71.
Terry S, Engelsen AST, Buart S, Elsayed WS, Venkatesh GH, Chouaib S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020;492:1–10.
Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2(2):132–41.
Chen B, Gao A, Tu B, Wang Y, Yu X, Wang Y, et al. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255:120187.
Karayama M, Masuda J, Mori K, Yasui H, Hozumi H, Suzuki Y, et al. Comprehensive assessment of multiple tryptophan metabolites as potential biomarkers for immune checkpoint inhibitors in patients with non-small cell lung cancer. Clin Transl Oncol. 2021;23(2):418–23.
Yan Y, Chang L, Tian H, Wang L, Zhang Y, Yang T, et al. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J Immunother Cancer. 2018;6(1):148.
Hurley HJ, Dewald H, Rothkopf ZS, Singh S, Jenkins F, Deb P, et al. Frontline science: AMPK regulates metabolic reprogramming necessary for interferon production in human plasmacytoid dendritic cells. J Leukoc Biol. 2021;109(2):299–308.
Guerra L, Bonetti L, Brenner D. Metabolic modulation of immunity: a new concept in cancer immunotherapy. Cell Rep. 2020;32(1):107848.
Kim S, Jang JY, Koh J, Kwon D, Kim YA, Paeng JC, et al. Programmed cell death ligand-1-mediated enhancement of hexokinase 2 expression is inversely related to T-cell effector gene expression in non-small-cell lung cancer. J Exp Clin Cancer Res. 2019;38(1):462.
Newburger PE, Malawista SE, Dinauer MC, Gelbart T, Woodman RC, Chada S, et al. Chronic granulomatous disease and glutathione peroxidase deficiency, revisited. Blood. 1994;84(11):3861–9.
Keller CW, Kotur MB, Mundt S, Dokalis N, Ligeon LA, Shah AM, et al. CYBB/NOX2 in conventional DCs controls T cell encephalitogenicity during neuroinflammation. Autophagy. 2021;17(5):1244–58.
Tarazona-Santos E, Bernig T, Burdett L, Magalhaes WC, Fabbri C, Liao J, et al. CYBB, an NADPH-oxidase gene: restricted diversity in humans and evidence for differential long-term purifying selection on transmembrane and cytosolic domains. Hum Mutat. 2008;29(5):623–32.
Dunlock VE. Tetraspanin CD53: an overlooked regulator of immune cell function. Med Microbiol Immunol. 2020;209(4):545–52.
Lim KS, Chang SS, Choi BH, Lee SH, Lee KT, Chai HH, et al. Genome-wide analysis of allele-specific expression patterns in seventeen tissues of Korean cattle (Hanwoo). Animals (Basel). 2019;9(10):727.
Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272(5265):1170–3.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (81872070), Science and Technique Foundation of Guangdong Province (210728156901639), Natural Science Foundation of Guangdong Province (2022A1515012424), and Guangxi Natural Science Foundation for Guangdong - Guangxi United Program (2021GXNSFDA075014).
Author information
Authors and Affiliations
Contributions
YH contributed to the collection and assembly of data, data analysis and interpretation, manuscript writing, methodology, and software. FO, FY and NZ contributed data analysis and interpretation. WZ reviewed and helped the manuscript writing. HX and XY supervised, obtained funding, and revised the manuscript. All authors read and approved the manuscript for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical approvals were from the Medical Ethics Committee of Shantou University Medical College (SUMC2020–73).
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
An overview of the proposed workflow.
Additional file 2: Fig. S2.
GCCA database was used to evaluate HK2 gene expression with box plots according to clinical parameters in glioma patients. A-I The respective correlation between HK2 expression and histology (A), grade (B), IDH mutation status (C), 1p/19q co-deletion status (D), IDH mutation status & 1p/19q co-deletion status (E), 1p/19q co-deletion status in different grades (F), gender (G), age status (H) and progression status (I). *P < 0.05, **P < 0.01, ***P < 0.001.
Additional file 3: Fig. S3.
The respective correlation between HK2 expression or methylation and immune stimulators, immune inhibitors, and MHC molecules in LGG and GBM specimens. A-F The respective correlation between HK2 expression and immunostimulatory (A-B), immunoinhibitory (C-D), and MHC molecules (E-F) is shown in LGG and GBM specimens. *P < 0.05, **P < 0.01, ***P < 0.001.
Additional file 4: Fig. S4.
Construction of the PPI network using HK2 positively A and negatively B related genes (Pearson’s rho ≥ 0.5, *P < 0.05).
Additional file 5: Fig. S5.
The correlation between the expression of HK2 and hub genes. A-J. The hub genes Including ITGB2, CD53, C3AR1, CYBB, ITGAM, SYP, CPLX1, SLC6A1, GABRG2, and SCRT1, in the LGG and GBM groups. *P < 0.05, **P <0.01, ***P < 0.001.
Additional file 6: Fig. S6.
The correlation between hub genes and the clinicopathological features of glioma. A-D Univariate (A-B) and multivariate (C-D) Cox regression showed that the top 5 positively and negatively HK2-related hub genes were associated with glioma prognosis. E-F A nomogram was constructed by CYBB, CD53, and grade (E), as well as SCRT1, SYP, and gender (F), for predicting the survival of glioma. HR, Hazard ratio; 95% CI, 95% confidence interval. *P < 0.05, **P < 0.01, ***P < 0.001.
Additional file 7:.
The uncropped immunoblotting images of full-length blots.
Additional file 8: Table S1.
List of primer sequences for qRT-PCR.
Additional file 9: Table S2.
Different datasets to analyze HK2 expression in pathological classification of gliomas (ONCOMINE).
Additional file 10: Table S3.
The information of clinicopathological characteristics of patient with gliomas.
Additional file 11: Table S4.
Correlation between HK2 expression and clinicopathologic characteristics of glioma patients.
Additional file 12: Table S5.
Univariate and multivariate analyses of various prognostics parameters in patients with glioma Cox-regression analysis.
Additional file 13: Table S6.
The identification of the top 50 positively and negatively related genes with HK2.
Additional file 14: Table S7.
KEGG pathway enrichment analysis of HK2 related genes.
Additional file 15: Table S8.
The functional roles of HK2 positively related hub genes.
Additional file 16: Table S9.
The functional roles of HK2 negatively related hub genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Y., Ouyang, F., Yang, F. et al. The expression of Hexokinase 2 and its hub genes are correlated with the prognosis in glioma. BMC Cancer 22, 900 (2022). https://doi.org/10.1186/s12885-022-10001-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-10001-y