
Abstract
Background
It is a basic task in high-throughput gene expression profiling studies to identify differentially expressed genes (DEGs) between two phenotypes. RankComp, an algorithm, could analyze the highly stable within-sample relative expression orderings (REOs) of gene pairs in a particular type of human normal tissue that are widely reversed in the cancer condition, thereby detecting DEGs for individual disease samples measured by a particular platform.
Methods
In the present study, Gene Expression Omnibus (GEO) Series (GSE) GSE75540, GSE138206 were downloaded from GEO, by analyzing DEGs in oral squamous cell carcinoma based on online datasets using the RankComp algorithm, using the Kaplan-Meier survival analysis and Cox regression analysis to survival analysis, Gene Set Enrichment Analysis (GSEA) to explore the potential molecular mechanisms underlying.
Results
We identified 6 reverse gene pairs with stable REOs. All the 12 genes in these 6 reverse gene pairs have been reported to be associated with cancers. Notably, lower Interferon Induced Protein 44 Like (IFI44L) expression was associated with poorer overall survival (OS) and Disease-free survival (DFS) in oral squamous cell carcinoma patients, and IFI44L expression showed satisfactory predictive efficiency by receiver operating characteristic (ROC) curve. Moreover, low IFI44L expression was identified as risk factors for oral squamous cell carcinoma patients’ OS. IFI44L downregulation would lead to the activation of the FRS-mediated FGFR1, FGFR3, and downstream signaling pathways, and might play a role in the PI3K-FGFR cascades.
Conclusions
Collectively, we identified 6 reverse gene pairs with stable REOs in oral squamous cell carcinoma, which might serve as gene signatures playing a role in the diagnosis in oral squamous cell carcinoma. Moreover, high expression of IFI44L, one of the DEGs in the 6 reverse gene pairs, might be associated with favorable prognosis in oral squamous cell carcinoma patients and serve as a tumor suppressor by acting on the FRS-mediated FGFR signaling.
Similar content being viewed by others


Introduction
Head and neck cancer is the sixth most common malignant tumor in the world [1], and oral squamous cell carcinoma (OSCC) is the most common head and neck cancer [2]. There are more than 300,000 new cases of OSCC worldwide every year, and more than 140,000 patients die from OSCC every year [2, 3]. More importantly, the incidence of oral squamous cell carcinoma has been increasing in recent years [4,5,6]; however, for those who receive treatment with surgery and chemotherapy or radiation therapy, the five-year survival rate is still not ideal [7, 8]. Biomarkers could guide the selection of appropriate therapy by predicting disease activity and progression, by predicting which individuals will respond to a particular therapy, and by providing pharmacodynamic information to facilitate assessment of response to therapy [9, 10].
It is a basic task in high-throughput gene expression profiling studies to identify differentially expressed genes (DEGs) between two phenotypes. Nevertheless, it has proven difficult to identify DEGs that show slight differential expression between two phenotypes. In particular, it is hard to detect sufficient DEGs for future researches when the sample size is not large enough. However, it is often possible to find a variety of datasets related to the same biological questions from public repositories, including Gene Expression Omnibus (GEO) [11] and ArrayExpress [12]. By combining datasets generated by multiple laboratories, weak biological signals can be efficiently detected, thus improving statistical capacity. Nevertheless, direct combining of multiple datasets could be hindered by various random factors including measurement batch effects [13]. These problems also pose key obstacles for the analysis of transcriptional data in The Cancer Genome Atlas (TCGA) where there are many small-scale batches of data. Even if data is preprocessed, the measurement of highly sensitive samples cannot be applied to independent samples [13,14,15,16]. Considering these limitations, the clinical use of quantitative transcriptional characteristics is limited.
In order to make the best use of the information provided by different datasets, meta-analysis uses statistical methods to combine p-values [17], effect sizes [18, 19], ranks [20, 21] and other results from independent researches. However, due to small sample sizes and large heterogeneity, high false negative rates may occur [22]. The more complex hierarchical Bayesian method “borrows” the information of all genes to strengthen the inference of which genes are expressed differently [23,24,25,26]. Nevertheless, the crucial assumption of hierarchical models usually induces a bias to the estimation of gene differences [27]. Although batch effect adjustment methods have been used to normalize data across studies, the normalization process itself might lead to distortions of the true biological signals [28, 29] and even false inter-group differences, particularly when phenotypic groups are distributed unevenly across batches [14, 30]. To solve these problems, recently, Wang and colleagues have proposed RankComp, an algorithm, to analyze the highly stable within-sample relative expression orderings (REOs) of gene pairs in a particular type of human normal tissue that are widely reversed in the cancer condition, thereby detecting DEGs for individual disease samples measured by a particular platform [14, 31]. Since first reported, RankComp has been used to detect differentially expressed genes between different groups in lung, colorectal [31], and breast cancers [32] and osteosarcoma [33]. REOs of gene pairs are not sensitive to measurement batch effect [34] and quite consistent across distinct platforms [31], which facilitates RankComp to be used for cross-study comparison of gene expression.
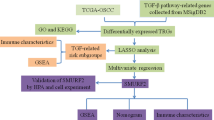
In the present study, by using RankComp algorithm based on training datasets GEO series GSE75540 and GSE138206, we attempted to identify differentially expressed gene pairs with highly stable REOs in oral squamous cell carcinoma and obtained 6 pairs of overlapping and stable reverse gene pairs. Through literature review, IFI44L, among the 6 gene pairs, was selected for further prognostic analysis and signaling pathway enrichment annotation (Fig. 1). Collectively, we confirmed the RankComp algorithm could identify reverse gene pairs with stable REOs in oral squamous cell carcinoma, providing potential prognostic markers and therapeutics targets for oral squamous cell carcinoma.

A schematic diagram showing the workflow of the present study. The workflow includes three major steps: the development of the REOs-based signature in the training datasets, the validation of the signature in validation datasets, and the signaling pathway enrichment annotation of the identified gene pairs
Materials and methods
Datasets and pre-processing
GSE75540, GSE138206 were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/). GSE75540 contained the expression profile of oral tongue squamous cell carcinoma and adjacent normal tissues. GSE138206 contained expression profile of oral squamous cell carcinoma and adjacent normal tissues. GSE75540 was based on the Illumina HumanHT-12 V4.0 expression beadchip (gene symbol), Illumina HumanMethylation450 BeadChip [UBC enhanced annotation v1.0], and Illumina HumanHT-12 WG-DASL V4.0 R2 expression beadchip [gene symbol version] platforms. GSE138206 was based on the [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array platform.
The software we used in this study include Python (v3.7.6; https://www.python.org/downloads/release/python-376/) and R studio (v4.0.2; https://www.rstudio.com/).
Stable REOs, the RankComp algorithm, and the concordance score
In each sample, the REO of a gene pair (A and B) is denoted as either GA > GB or GA < GB exclusively, where GA and GB represent the expression values of gene A and B, respectively. Stable gene pairs obtained from training sets can be used as markers of classification in any given sample. Based on these stable gene pairs, a classification model was constructed. For unclassified samples, the relative expression order relationships of all stable gene pairs were calculated. In a sample, the probability that GA < GB occurs less than or equal to k times in n pairs of stable gene pairs can be calculated by the following formula:
In the formula, n represents the number of samples and k represents the number of occurrences of GA < GB in n samples. If the P value is less than 0.05, it means that most of the samples can maintain the relationship of GA < GB, and we call it a significantly stable pair. For a sample with an unknown category, let k be the number of times that the genetic rank relationship of GA > GB appears in stable reversal pairs, and n is the logarithm of significantly stable pairs. Here, we define k as stable pair count. If P(k,n) < 0.05, then the sample is significantly in line with the characteristics of Type B and is classified as type B, and if P(nk,n) < 0.05, the sample is significantly in line with the characteristics of Type A and is classified as type A. In the present study, we use the reversal model of stable gene pairs for the classification of two types of samples. If GA < GB and GA > GB are significantly stable pairs in type A and type B, then we call (GA, GB) a stable reversal pair.
Survival analysis
The correlation between IFI44L expression and the disease-free survival (DFS) in patients with oral squamous cell carcinoma was analyzed using the Kaplan-Meier survival analysis by grouping the cases in GSE4676 or GSE75540 taking the median expression values of IFI44L as cut-off. We used univariate and multivariate Cox regression analysis to identify clinical risk parameters associated with survival using GSE34115 (contained the gene expression profile of archival tongue squamous cell carcinoma), GSE42023 (contained the gene expression profile of archival tongue squamous cell carcinoma), GSE84846 (contained the gene expression profile of oral squamous cell carcinoma).
Gene set enrichment analyses (GSEA)
To explore the potential molecular mechanisms underlying our constructed prognostic gene signature, GSEA (Gene Set Enrichment Analyses) [35, 36] was performed to find differential characteristics of oral squamous cell carcinoma patients with high or low IFI44L expression. P < 0.01 and FDR (false discovery rate) q < 0.05 were considered statistically significant.
Results
RankComp algorithm was used to construct reverse gene pairs in oral squamous cell carcinoma with stable REOs
On the basis of REO robustness, two datasets, GSE75540 and GSE138206, were used to analyze DEGs between healthy and cancerous samples and identify reverse gene pairs with stable REOs. As shown in Table 1, GSE75540 contained a total of 22,153 DEGs between 51 normal samples and 100 cancerous samples; top 500 up- or down-regulated DEGs could form 80 reverse gene pairs with stable REOs. GSE138206 contained a total of 7133 DEGs between 10 normal samples and 5 cancerous samples; top 500 up- or down-regulated DEGs could form 114 reverse gene pairs with stable REOs. These gene pairs intersected in 6 reverse gene pairs shown in Table 2.
Functional annotations of overlapped reverse gene pairs
All the 12 DEGs in the 6 overlapped reverse gene pairs identified here have been reported to be associated with cancers. For example, Musculin (MSC) has been regarded as a component of a robust gene signature identified using a risk score model, and has been considered to be potential immunotherapy targets for hepatocellular carcinoma [37]. Multimerin-1 (MMRN1) has been recognized as a novel biomarker that may refine acute myelogenous leukemia risk stratification [38]. MMP-9 is known to be involved in carcinogenesis, inluding but not limited to invasive and metastatic abilities, and formation of blood vessels [39]. Tropomyosin 3 (TPM3) fusion with NTRK1 has been reported as one of the most well validated oncogenic events to date [40]. Laminin subunit beta-3 (LAMB3) relates to the invasion and metastasis of certain cancers, such as colon, pancreatic, pulmonary, cervical, gastric, and prostate cancer [41,42,43]. Aldehyde dehydrogenase 1 family member A1 (ALDH1A1) is a stemness marker and promotes the malignant behaviors in breast cancer [44, 45]. The low expression of secretogranin V (SCG5) could predict a poorer prognosis of pancreatic cancer [46]. Alcohol dehydrogenase 1B (ADH1B) polymorphisms have been reported to be associated with bladder cancer, gastric cancer, and breast cancer risk [47,48,49]. NR4A2 is a member of the Nur77 orphan receptor subfamily, which plays a critical role in human tumor cell survival [50,51,52,53]. HOXB2 serves as a tumor promotor in bladder cancer [54], colorectal cancer [55], and pancreatic cancer [56]. The oncogenic role of ID4 has also been reported in lung cancer [57], hepatocellular carcinoma [58], and breast cancer [59]. IFI44L serves as a tumor suppressor in hepatocellular carcinoma; in hepatocellular carcinoma patients, low IFI44L expression is associated with tumor size, recurrence, advanced stage and poor clinical survival [60].
Prognostic potential of IFI44L
To further investigate the clinical potential of the 12 DEGs, we analyzed the association of the 12 DEGs expression and the prognosis in oral squamous cell carcinoma patients. Kaplan-Meier survival analysis found that only IFI44L of the 12 DEGs was significantly associated with overall survival in patients with oral squamous cell carcinoma (P < 0.05; Table 3). GSE75540 included three types of samples: 75 cancerous tissues, 51 para-cancerous tissues, and 25 peripheral blood samples, for a total of 151 cases. In survival analysis, 51 para-cancerous tissues, 2 cancerous tissues with no survival information and 1 case with an overall survival of less than 30 days were first excluded. Thus, 72 cases of tissue samples and 25 cases of peripheral blood samples were assigned into high- or low-IFI44L expression group; however, although cases with higher IFI44L expression seemed to obtain better overall survival, the p value was > 0.05 (Fig. 2A). Considering that peripheral blood samples may differ from tissues and affect the analysis, peripheral blood samples were also excluded for the survival analysis. Finally, a total of 72 cases were included in survival analysis and assigned into low-IFI44L expression group and high-IFI44L expression group based on the median IFI44L expression; the Kaplan-Meier survival analysis showed that lower IFI44L expression was associated with poorer OS in oral squamous cell carcinoma patients (Fig. 2B). Then, we employed the receiver operating characteristic (ROC) curve [61] to test the prediction efficiency of the IFI44L. As shown in Fig. 2C-D, the area under the curve (AUC) for 3-,4-,5 years of OS were 0.69, 0.73 and 0.72, and for DFS were 0.70, 0.72, and 0.70. As revealed by the ROC curve, the IFI44L expression-based curve showed satisfactory predictive efficiency. In a larger cohort based on TCGA-HNSC data, lower IFI44L expression was associated with poorer OS (Fig. S1).

Correlation of IFI44L expression with the prognosis in patients with oral squamous cell carcinoma according to GSE75540 A Overall survival analysis on cancer tissues (n = 72) and peripheral blood samples (n = 25); 2 cancerous tissues with no survival information and 1 case with an overall survival of less than 30 days were excluded. B overall survival analysis on cancer tissues (n = 72). C ROC curves
Moreover, based on the aforementioned 72 cases in GSE75540, we performed univariate and multivariate Cox regression analysis to analyze the association of age, gender, stage, and IFI44L expression with the OS in oral squamous cell carcinoma patients. As shown in Fig. 3 and Table 4, among these four factors, low IFI44L expression (HR = 2.63; 95% CI = 0.90-7.70) might predict higher risk for oral squamous cell carcinoma patients’ OS, although the p value was 0.0785. Based on TCGA-HNSC data, IFI44L is differentially expressed in subjects with different clinical parameters, including downregulated in male subjects (Fig. S2A), downregulated in subjects with tumor (Fig. S2B), downregulated in subjects with progression after therapy (Fig. S2C), and downregulated in higher tumor stages (not significantly, Fig. S2D).

Univariate and multivariate Cox regression of oral squamous cell carcinoma patients
Functional annotation of IFI44L
Since low IFI44L expression showed to be associated with poor OS and DFS in oral squamous cell carcinoma patients, next, we performed GSEA functional annotation analysis on different characteristics in high- and low-IFI44L cases, attempting to identify signaling pathways related to IFI44L function. As shown in Fig. 4A-C, IFI44L downregulation would lead to the activation of the FRS-mediated FGFR1, FGFR3, and downstream signaling pathways; low IFI44L expression also plays a role in the PI3K-FGFR cascades.

Gene Set Enrichment Analysis (GSEA) functional annotation analysis on IFI44L A The low expression of IFI44L activates FRS-mediated FGFR1 and FGFR3 signaling pathways. B The low expression of IFI44L activates the downstream signaling pathways of FGFR1 and FGFR3. C The low expression of IFI44L plays a role in the PI3K and FGFR cascades
Discussion
In the present study, by analyzing DEGs in oral squamous cell carcinoma based on online datasets using the RankComp algorithm, we identified 6 reverse gene pairs with stable REOs. All the 12 genes in these 6 reverse gene pairs have been reported to be associated with cancers. Notably, lower IFI44L expression was associated with poorer OS and DFS in oral squamous cell carcinoma patients, and IFI44L expression showed satisfactory predictive efficiency by ROC curve. Moreover, low IFI44L expression were identified as risk factors for oral squamous cell carcinoma patients’ OS. IFI44L downregulation would lead to the activation of the FRS-mediated FGFR1, FGFR3, and downstream signaling pathways, and might play a role in the PI3K-FGFR cascades.
Totally different from traditional meta-analysis methods and batch-correction methods, RankComp, an algorithm based on the cross-platform significantly stable REOs for a particular normal tissue, is an economic and efficient method which can readily and accurately identify DEGs in any disease sample measured by any of the platforms [62]. Regarding other algorithms, both batch effect correction and normalization method might result in a distortion of true biological signals between two phenotypes, leading to false differences between groups [14, 28,29,30]; as for the RankComp algorithm, which has a high accuracy and is insensitive to measurement batch effect and data normalization, could normalize microarray samples measured by different platforms [62]. Herein, by using RankComp algorithm based on GSE75540 and GSE138206, we successfully identified 6 reverse gene pairs with stable REOs. As we have mentioned, all the 12 genes involved in the 6 reverse gene pairs have been reported to be associated with multiple cancers, suggesting that these reverse gene pairs might possess prognostic potential in oral squamous cell carcinoma.
Among these 12 genes, little is known about IFI44L, which was found to exert moderate impact upon Hepatitis C virus infection [63]. Notably, the expression level of IFI44L has also been implicated in cancers [60, 64]. IFI44L has been recognized as a novel tumor-suppressor gene in human hepatocellular carcinoma that regulates met/Src signaling to affect cancer stemness, metastasis, and drug resistance [60]. However, the role of IFI44L in oral squamous cell carcinoma has never been investigated. Moreover, according to TCGA data, in glioma patients, higher IFI44L expression predicted higher survival probability (Fig. 5). Similarly, in the present study, according to GSE75540, lower IFI44L expression was associated with poorer OS and DFS in oral squamous cell carcinoma patients. Moreover, by using univariate and multivariate Cox regression analysis based on GSE75540, we identified the low IFI44L expression as a risk factor for oral squamous cell carcinoma patients’ OS. These data indicate that high IFI44L expression might be a favorable biomarker for oral squamous cell carcinoma patients.

Sangerbox online analysis (http://sangerbox.com/) were performed to analyze the correlation of IFI44L expression with in glioma prognosis. A The correlation between IFI44L expression and survival probability of patients with oral squamous cell carcinoma. B The specificity and sensitivity of IFI44L expression being a prognostic marker
Regarding possible molecular mechanism, GSEA analysis indicated that IFI44L downregulation would lead to the activation of the FRS-mediated FGFR1, FGFR3, and downstream signaling pathways; low IFI44L expression also plays a role in the PI3K-FGFR cascades. Increasing evidence demonstrated that FGFR aberrations are tied to oncogenesis, driving mutations where the acquisition of somatic molecular alterations could directly stimulate the growth and proliferation of tumor cells, promoting neovascularization and resistance to anticancer therapies [65,66,67,68,69]. The field of FGFR targeting has advanced rapidly with the recent development of new drugs repressing FGFs/FGFRs, thereby exhibiting a manageable safety profile in early clinical trials [70]. FGFR inhibitors have been reported to be effective in tumors with abnormal FGFR signaling, providing new treatment strategies within the era of precision medicine [71, 72]. Considering these previous findings, IFI44L might be a promising agent serving as a tumor suppressor in oral squamous cell carcinoma, possibly through acting on the FRS-mediated FGFR1, FGFR3, and downstream signaling pathways.
Collectively, we identified 6 reverse gene pairs with stable REOs in oral squamous cell carcinoma, which might serve as gene signatures playing a role in the diagnosis in oral squamous cell carcinoma. Moreover, high expression of IFI44L, one of the DEGs in the 6 reverse gene pairs, might be associated with favorable prognosis in oral squamous cell carcinoma patients and serve as a tumor suppressor by acting on the FRS-mediated FGFR signaling. Regarding the limitations of the present study, the RankComp algorithm might not be sufficient enough to identify genes whose differential expression results in slight alterations in the ranking. Moreover, based on the sensitivity of gene expression ordering to the microarray platforms to a certain extent, herein, the present study only analyzed the microarray data from the same platform. Future research should be performed to exclude gene pairs without stable ordering in datasets from multiple platforms, and identified promising factor should be investigated for specific effects in vitro and in vivo.
Availability of data and materials
The datasets GSE75540, GSE138206 analysed during the current study are available in the GEO repository(https://www.ncbi.nlm.nih.gov/geo/).
References
Lala M, et al. Clinical outcomes with therapies for previously treated recurrent/metastatic head-and-neck squamous cell carcinoma (R/M HNSCC): a systematic literature review. Oral Oncol. 2018;84:108–20.
Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma--an update. CA Cancer J Clin. 2015;65(5):401–21.
Sasahira T, Kirita T. Hallmarks of cancer-related newly prognostic factors of oral squamous cell carcinoma. Int J Mol Sci. 2018:19(8).
Tota JE, et al. Rising incidence of oral tongue cancer among white men and women in the United States, 1973-2012. Oral Oncol. 2017;67:146–52.
Moore SR, et al. The epidemiology of tongue cancer: a review of global incidence. Oral Dis. 2000;6(2):75–84.
Hussein AA, et al. Global incidence of oral and oropharynx cancer in patients younger than 45 years versus older patients: a systematic review. Eur J Cancer. 2017;82:115–27.
Brenner H. Long-term survival rates of cancer patients achieved by the end of the 20th century: a period analysis. Lancet. 2002;360(9340):1131–5.
Shiboski CH, Schmidt BL, Jordan RC. Tongue and tonsil carcinoma: increasing trends in the U.S. population ages 20-44 years. Cancer. 2005;103(9):1843–9.
Almangush A, et al. Prognostic biomarkers for oral tongue squamous cell carcinoma: a systematic review and meta-analysis. Br J Cancer. 2017;117(6):856–66.
Hussein AA, et al. A review of the most promising biomarkers for early diagnosis and prognosis prediction of tongue squamous cell carcinoma. Br J Cancer. 2018;119(6):724–36.
Barrett T, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Parkinson H, et al. ArrayExpress--a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007;35(Database issue):D747–50.
Leek JT, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–9.
Nygaard V, Rodland EA, Hovig E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics. 2016;17(1):29–39.
Guan Q, et al. Quantitative or qualitative transcriptional diagnostic signatures? A case study for colorectal cancer. BMC Genomics. 2018;19(1):99.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–27.
Rhodes DR, et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62(15):4427–33.
Hu P, Greenwood CM, Beyene J. Integrative analysis of multiple gene expression profiles with quality-adjusted effect size models. BMC Bioinformatics. 2005;6:128.
Choi JK, et al. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl 1):i84–90.
Hong F, et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22(22):2825–7.
Zintzaras E, Ioannidis JP. Meta-analysis for ranked discovery datasets: theoretical framework and empirical demonstration for microarrays. Comput Biol Chem. 2008;32(1):38–46.
Cahan P, et al. Meta-analysis of microarray results: challenges, opportunities, and recommendations for standardization. Gene. 2007;401(1-2):12–8.
Conlon EM, Song JJ, Liu JS. Bayesian models for pooling microarray studies with multiple sources of replications. BMC Bioinformatics. 2006;7:247.
Scharpf RB, et al. A Bayesian model for cross-study differential gene expression. J Am Stat Assoc. 2009;104(488):1295–310.
Ruan L, Yuan M. An empirical Bayes' approach to joint analysis of multiple microarray gene expression studies. Biometrics. 2011;67(4):1617–26.
Conlon EM, et al. A Bayesian model for pooling gene expression studies that incorporates co-regulation information. PLoS One. 2012;7(12):e52137.
Li B, et al. Bayesian inference with historical data-based informative priors improves detection of differentially expressed genes. Bioinformatics. 2016;32(5):682–9.
Wang D, et al. Extensive up-regulation of gene expression in cancer: the normalised use of microarray data. Mol BioSyst. 2012;8(3):818–27.
Loven J, et al. Revisiting global gene expression analysis. Cell. 2012;151(3):476–82.
Lazar C, et al. Batch effect removal methods for microarray gene expression data integration: a survey. Brief Bioinform. 2013;14(4):469–90.
Guan Q, et al. Differential expression analysis for individual cancer samples based on robust within-sample relative gene expression orderings across multiple profiling platforms. Oncotarget. 2016;7(42):68909–20.
Cai H, et al. A qualitative transcriptional signature to reclassify estrogen receptor status of breast cancer patients. Breast Cancer Res Treat. 2018;170(2):271–7.
Hu G, et al. Identification of potential key genes associated with osteosarcoma based on integrated bioinformatics analyses. J Cell Biochem. 2019;120(8):13554–61.
Geman, D., et al. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol, 2004. 3: p. Article19.
Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Zhang FP, et al. Construction of a risk score prognosis model based on hepatocellular carcinoma microenvironment. World J Gastroenterol. 2020;26(2):134–53.
Laszlo GS, et al. Multimerin-1 (MMRN1) as novel adverse marker in pediatric acute myeloid leukemia: a report from the Children's oncology group. Clin Cancer Res. 2015;21(14):3187–95.
Huang, H., Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors (Basel), 2018. 18(10).
Khotskaya YB, et al. Targeting TRK family proteins in cancer. Pharmacol Ther. 2017;173:58–66.
Wang Y, et al. Upregulated LAMB3 increases proliferation and metastasis in thyroid cancer. Onco Targets Ther. 2018;11:37–46.
Laroussi N, et al. Identification of a novel mutation of LAMB3 gene in a Lybian patient with hereditary epidermolysis bullosa by whole exome sequencing. Ann Dermatol. 2017;29(2):243–6.
Hou J, Wang L, Wu D. The root of Actinidia chinensis inhibits hepatocellular carcinomas cells through LAMB3. Cell Biol Toxicol. 2018;34(4):321–32.
Wang Q, et al. Tamoxifen enhances stemness and promotes metastasis of ERalpha36(+) breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018;28(3):336–58.
Ciccone V, et al. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1alpha/VEGF signalling in MCF-7 breast cancer cells. J Exp Clin Cancer Res. 2018;37(1):311.
Xu JS, et al. Combining bioinformatics techniques to explore the molecular mechanisms involved in pancreatic cancer metastasis and prognosis. J Cell Mol Med. 2020.
Masaoka H, et al. Aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase 1B (ADH1B) polymorphisms exacerbate bladder cancer risk associated with alcohol drinking: gene-environment interaction. Carcinogenesis. 2016;37(6):583–8.
Ishioka K, et al. Association between ALDH2 and ADH1B polymorphisms, alcohol drinking and gastric cancer: a replication and mediation analysis. Gastric Cancer. 2018;21(6):936–45.
Lilla C, et al. Alcohol dehydrogenase 1B (ADH1B) genotype, alcohol consumption and breast cancer risk by age 50 years in a German case-control study. Br J Cancer. 2005;92(11):2039–41.
Guo J, et al. Clinicopathological significance of orphan nuclear receptor Nurr1 expression in gastric cancer. Clin Transl Oncol. 2015;17(10):788–94.
Han Y, et al. Nuclear orphan receptor NR4A2 confers chemoresistance and predicts unfavorable prognosis of colorectal carcinoma patients who received postoperative chemotherapy. Eur J Cancer. 2013;49(16):3420–30.
Wang J, et al. Orphan nuclear receptor nurr1 as a potential novel marker for progression in human prostate cancer. Asian Pac J Cancer Prev. 2013;14(3):2023–8.
Inamoto T, et al. 1,1-Bis(3′-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol Cancer Ther. 2008;7(12):3825–33.
Liu J, et al. HOXB2 is a putative tumour promotor in human bladder Cancer. Anticancer Res. 2019;39(12):6915–21.
Li, H., et al. miR-4324 functions as a tumor suppressor in colorectal cancer by targeting HOXB2. J Int Med Res, 2020. 48(3): p. 300060519883731.
Segara D, et al. Expression of HOXB2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clin Cancer Res. 2005;11(9):3587–96.
Qi K, et al. Id4 promotes cisplatin resistance in lung cancer through the p38 MAPK pathway. Anti-Cancer Drugs. 2016;27(10):970–8.
Zhang Y, et al. Id4 promotes cell proliferation in hepatocellular carcinoma. Chin J Cancer. 2017;36(1):19.
Zhang X, et al. ID4 promotes breast cancer chemotherapy resistance via CBF1-MRP1 pathway. J Cancer. 2020;11(13):3846–57.
Huang WC, et al. IFI44L is a novel tumor suppressor in human hepatocellular carcinoma affecting cancer stemness, metastasis, and drug resistance via regulating met/Src signaling pathway. BMC Cancer. 2018;18(1):609.
Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143(1):29–36.
Wang H, et al. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics. 2015;31(1):62–8.
Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–5.
Li H, et al. Integrated expression profiles analysis reveals novel predictive biomarker in pancreatic ductal adenocarcinoma. Oncotarget. 2017;8(32):52571–83.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–29.
Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18(7):1855–62.
Dienstmann R, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25(3):552–63.
Dieci MV, et al. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3(3):264–79.
Zhou Y, et al. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer. 2020;11(8):2000–7.
Wu YM, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–47.
Touat M, et al. Targeting FGFR signaling in Cancer. Clin Cancer Res. 2015;21(12):2684–94.
Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318–32.
Stafford F, et al. Organisation and provision of head and neck cancer surgical services in the United Kingdom: United Kingdom National Multidisciplinary Guidelines. J Laryngol Otol. 2016;130(S2):S5–8.
Funding
This study was supported by Guangzhou Scientific Research Program Project (201904010045).
Author information
Authors and Affiliations
Contributions
Deming Ou wrote the main manuscript text and Ying Wu coordinated the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Ou, D., Wu, Y. The prognostic and clinical significance of IFI44L aberrant downregulation in patients with oral squamous cell carcinoma. BMC Cancer 21, 1327 (2021). https://doi.org/10.1186/s12885-021-09058-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-09058-y