
Abstract
Background
Clinical options for lung squamous carcinoma (LUSC) are still quite limited. Carcinogenesis is an exceedingly complicated process involving multi-level dysregulations. Therefore, only looking into one layer of genomic dysregulation is far from sufficient.
Methods
We identified differentially expressed genes with consistent upstream genetic or epigenetic dysregulations in LUSC. Random walk was adopted to identify genes significantly affected by upstream abnormalities. Expression differentiation and survival analysis were conducted for these significant genes, respectively. Prognostic power of selected gene was also tested in 102 male LUSC samples through immunohistochemistry assay.
Results
Twelve genes were successfully retrieved from biological network, including ERα (ESRS1), EGFR, AR, ATXN1, MAPK3, PRKACA, PRKCA, SMAD4, TP53, TRAF2, UBQLN4 and YWHAG, which were closely related to sex hormone signaling pathway. Survival analysis in public datasets indicated ERα was significantly associated with a poor overall survival (OS) in male LUSC. The result of our immunohistochemistry assay also demonstrated this correlation using R0 resected tumors (n = 102, HR: 2.152, 95% CI: 1.089–4.255, p = 0.024). Although disease-free survival (DFS) difference was non-significant (n = 102, p = 0.12), the tendency of distinction was straight-forward. Cox analysis indicated ERα was the only independent prognostic factor for male patients’ OS after R0 resection (HR = 2.152, p = 0.037).
Conclusion
ERα was significantly related to a poor prognosis in LUSC, especially for male patients after radical surgery, confirmed by our immunohistochemistry data.
Similar content being viewed by others

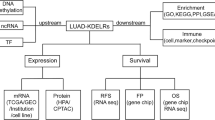

Background
Lung squamous carcinoma (LUSC) accounts for approximately 20 to 30% of all lung cancers and is associated with poor prognosis compared with non-squamous non-small-cell lung cancer (NSCLC) [1,2,3]. Up to date, unlike lung adenocarcinoma, targeted therapeutic choices for LUSC are still relatively scarce. Before the era of immunotherapy, platinum-based chemotherapy seemed to be the most proper option of first-line treatment for advanced stage LUSC patients [4, 5]. It has been widely reported that immune check point inhibitors, like pembrolizumab, atezolizumab, and nivolumab, can substantially improve LUSC patients’ prognosis [6,7,8]. However, the subsequent strategies after failure with immunotherapy were enormously limited. Therefore, prognostic biomarkers and candidate therapeutic targets are greatly needed for this deadly disease.
The functions of estrogen and estrogen receptors (ERs) have been intensively studied in reproductive organs, under most circumstances, in females. The importance of ER signaling pathway has also been extensively investigated in various pathologic functions during carcinogenesis, especially in breast cancer. There are two types of classic ERs. i.e. ER alpha (ERα, also known as ESR1), located on Chromosome 6, first cloned in 1986; ER beta (ERβ, also known as ESR2), located on Chromosome 14, first cloned in 1996 [9]. Estrogen is also believed to play an important role in lung carcinogenesis [10, 11]. Some researchers have already investigated the correlation between ER and lung cancer based on immunohistochemistry stain, but the results of these researches seem quite confusing. Some studies showed that ERα in lung cancer was mainly observed in the cytoplasm and associated with a poor prognosis. Some found that the nuclear ERβ predicted a better prognosis, and the expression level of cytoplasm was associated with a poor prognosis [12,13,14,15]. Nonetheless, opposing results have also been widely reported in various studies [16,17,18,19]. Moreover, the majority of these researches focused on female patients [20,21,22], probably subjected to the stereotypical thinking that estrogen plays a more important role in women. Additionally, according to literature review, ER-related investigations in lung cancer were majorly concentrating upon adenocarcinoma, seldom paying enough attention to LUSC, except for some subgroup analyses among NSCLC, let alone male LUSC patients. Therefore, the relationship between ER and the prognosis of male LUSC is greatly needed in order to provide a potential therapeutic target for these patients, since the majority of the LUSC patients are male.
It is putatively accepted that the process of carcinogenesis is due to multi-level abnormalities, including genetic alterations [majorly composed of somatic mutation and copy number variation (CNV)] [23, 24], aberrant methylomics [25, 26], and dysregulated transcriptomics [27, 28]. CNV, promoter methylation and somatic mutation could all influence gene activation or suppression, respectively, thereby leading to cancer transformation. CNVs may alter transcriptional dosage by influencing the number of copies of a gene located within a particular genomic region [29,30,31,32], explaining under most circumstances, CNV and corresponding gene expression are positively correlated in lung cancer [33,34,35]. Promoter hypomethylation might lead to gene activation, and promoter hypermethylation might cause gene suppression in most cases [36]. For instance, MGMT silencing, caused by its promotor methylation, can make glioma patients greatly benefit from temozolomide [37]. Somatic mutation could also result in activation or suppression of downstream signaling pathways [38]. In thyroid cancer, BRAF V600E mutation could activate MAPK pathway, thus influencing the massive dysregulation of molecular interactions [39]. Therefore, the multi-dimensional genomic abnormalities should be taken in to consideration aggregately, rather than only looking into the gene expression differentiation, in order to shed light upon the intricate bio-mechanisms of cancer transformation and progression. The integrative analysis of cancer genomics, methylomics and transcriptomics is a very wise choice to comprehensively dissect cancer etiology and provide potential clinical guidance of LUSC.
Methods
Data collection and organization
The multi-level datasets of LUSC were directly downloaded from the Bioconductor package “RTCGA.rnaseq” and “TCGAbiolinks” at January 10th, 2020. Three levels of paired data (cancer and normal adjacent tissues) were retrieved from all patients, including 51 paired RNA sequencing data [raw counts and RNASeq by Expectation Maximization (RSEM) normalized read counts], 237 paired DNA CNV data (generated from Affymetrix SNP 6.0 platform, and segmented through circular binary segmentation method [40]), 40 paired DNA methylation data [generated from Illumina HumanMethylation450 platform, and the methylation level of each CpG site was calculated as the ratio (β value) of signal of methylated probes relative to the sum of all probes, ranging from 0 (absolutely unmethylated) to 1 (fully methylated)], and somatic mutation data of 548 patients (with mutational information of 17,225 genes). The clinical information of these The Cancer Genome Atlas (TCGA) LUSC patients was also directly downloaded from this Bioconductor package.
The mRNA expression profiles of LUSC, including GSE73403, GSE37745, GSE41271 and GSE50081, were downloaded from Gene Expression Omnibus (GEO) database, and the final normalized datasets were used in following analysis. All clinical information was extracted from the original publications.
Identification of significant dysregulated genes
Differentially expressed genes (DEGs) were identified using paired t test with RSEM normalized read counts (FDR < 0.01). Bioconductor package “CNTools” was used to process segmentation CNV data, and transform the segmentation data into a gene-level matrix, according to each genomic location of 27,270 genes. Dysregulated genes with significant amplification and deletion were also identified using paired t statistic test (FDR < 1*10− 5). Promoter region was defined as the genomic length between 1000 bps upstream transcription start site (TSS) and 300 bps downstream. In methylation analysis, the β value of the probe mapped to the certain CpG site within promoter region of a given gene was used to determine the methylation level of this probe. If multiple probes were projected onto the promoter region of a given gene, the mean value was used to define the methylation value of this gene. The methylation level of 20,110 genes were obtained in this manner, and significant hypo- or hyper-methylated genes were identified with paired t statistic test (FDR < 0.01).
Therefore, three groups of candidate genes of interest were identified for further analysis by virtue of different dysregulation patterns: (i) genes with both differential expression and consistent CNV (i.e. over-expressed genes with amplification, and under-expressed ones with deletion); (ii) genes with both differential expression and altered promoter methylation (i.e. over-expressed genes with promoter hypomethylation, and under-expressed ones with significant promoter hypermethylation); and (iii) genes with both differential expression and somatic mutation (mutation rate ≥ 5%).
Identification of significant genes using random walk algorithm in biological network
We downloaded protein–protein interaction network was from Human Protein Reference Database (HPRD) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Gene regulatory network was therefore constructed by merging HPRD and KEGG network together, including 10,479 nodes and 60,689 edges after eliminating self-loops and duplicated edges.
In this study, random walk algorithm was utilized to identify genes significantly affected by genomic and epigenetic alterations, taking advantage of aforementioned merged biological network [41]. The similar method has been successfully conducted in our previous study [42]. Genes of interest were referred to as information source (i.e., source nodes) and the rest as the information target (i.e., target nodes). The information flow originates from source nodes iteratively and randomly transmits to their neighbors with a probability proportional to their topological features. At each iteration, information flow can go back to source nodes and target nodes as well with the same probability. The final steady-state probability assigned to each gene stands for the integrated influence coming from source nodes while considering the network topology. Formally, the random walk with restart can be illustrated as followings:
Where W stands for the column-normalized adjacency matrix of connected network, and pt is a vector of probability the genes in the network holds at step t in the iteration process. Source nodes were weighted with initial probability vector p0 (the sum of its elements was normalized as 1), and r represents restart probability (r = 0.7 in the present study). All the genes in the network were eventually scored according to the values in the steady-state probability vector p∞. This was obtained at query time by repeating the iteration process until the difference between pt and pt + 1 (measured by the L1 norm) was lower than 10− 10. To obtain genes with significantly high steady-state probability, 10,000 permutations of node labels (with network topology remained the same) were performed to simulate the null distribution of final probability. The p value was termed as the probability of random values that were less than the calculated final probability. Genes with p < 0.02 were designated as the genes significantly afflicted by these genetic or epigenetic abnormalities.
Immunohistochemistry to identify the expression level of ER and androgen receptor (AR) in male LUSC tumors
One hundred and two formalin-fixed paraffin-embedded (FFPE) primary LUSC samples with OS and DFS information were collected after radical surgery in Department of Thoracic Surgery from January 2014 to December 2016. All the patients were male, ranging from IA to IIIA based on AJCC 7th staging system. The clinicopathological factors were carefully documented, including age, pathological tumor size, regional lymph node metastasis, AJCC stage, grade, whether adjuvant chemotherapy or radiotherapy were conducted. None of these patients received any kinds of neoadjuvant therapy. Anatomic pulmonary and mediastinal lymph node resection were all adopted in these 102 patients, and both squamous phenotype and R0 resection were confirmed by experienced pathologists.
Slides of the various male LUSC tumors were stained with anti-ERα (working reagent, Zhongshan Bio-chemistry, China), anti-AR (working reagent, Zhongshan Bio-chemistry, China). After incubation with anti-mouse secondary antibody (working reagent, Zhongshan Bio-chemistry, China), staining was developed using PV-6000 detecting system, and slides were counterstained with hematoxylin. Digital images of these slides were acquired using the Olympus BX37 microscope. Individual slides were scored by two independent, blinded and experienced pathologists.
Statistical analysis
All the data analyses were conducted using R programming (Version 3.3.1) and Bioconductor packages. Gene Ontology (GO) analysis was conducted via the DAVID bioinformatics tool (http://david.abcc.ncifcrf.gov/). The information of gene-to-gene interactions was retrieved from Bioconductor package “KEGGgraph” (Version 1.50.0). Network visualization was conducted using Cytoscape software (Version 3.4.0). In survival analysis of mRNA expression data, patients were divided into two groups based on median of expression values, and Kaplan–Meier analysis and log-rank test were performed to evaluate the prognostic difference between the two assigned groups.
Results
A schematic for the study is depicted in Fig. 1.

Schematic of methodology applied in this study
Collection of genes with differential expression, CNV, promoter methylation and somatic mutation with TCGA LUSC data
Paired samples were used to eliminate individual difference. DEGs were identified with paired t test, composed of 6416 up-regulated genes and 6338 down-regulated genes (Fig. 2A). Additionally, 5683 genes were identified as significantly amplified and 7093 genes were found deleted based on paired CNV data (Fig. 2A). Furthermore, 4674 genes with promoter hypermethylation and 3363 genes with hypomethylation were also identified as significant according to paired methylation profiles (Fig. 2A). Moreover, 1481 genes with mutation rate ≥ 5% were deemed as mutated genes with relatively high frequency.

Identification of three gene groups with multi-omics data. A Heat maps of DEGs, genes with CNV, and genes with aberrant promoter methylation, respectively. B Venn diagram illustrating three groups of candidate genes with differential expression and corresponding upstream altered molecular levels. C GO enrichment analysis of upregulated and downregulated DEGs with at least one types of consistent genetic or epigenetic dysregulations
Identification of candidate gene groups with different dysregulation patterns
Three different groups of DEGs with aberrant upstream dysregulations (Fig. 2B) were categorized as follows: (i) 1613 genes with over-expression and copy number gain, and 1939 genes with under-expression and copy number loss (altogether 3552 genes, termed as Group A); (ii) 1373 genes with over-expression and promoter hypomethylation, and 1147 genes with under-expression and promoter hypermethylation (altogether 2520 genes, termed as Group B); (iii) 577 DEGs (termed as Group C) with somatic mutation in more than 5% patients (362 over-expression and 215 under-expression). Genetic and epigenetic dysregulation of DEGs were shown in Fig. 2B. To comply with classic knowledge of gene regulation, promoter methylation was assumed to exert trans-regulation, while DNA copy number did cis-regulation effect upon transcriptomics.
Up-regulated and down-regulated DEGs, within Group A, B or C (with at least one types of consistent upstream dysregulations), were both used to conduct GO enrichment analysis, respectively. The results indicated that up-regulated DEGs were significantly associated with cell adhesion, positive regulation of GTPase activity, inflammatory response, and positive regulation of angiogenesis (Fig. 2C). Down-regulated DEGs were significantly related to epidermis development, mRNA exportation, cell division, and DNA repair (Fig. 2C).
Genetic alterations in LUSC
Segment Gain or Loss (SGOL) scores were calculated based on the segmentations mapping onto 22 autosomes. Since the TCGA samples we used were a mixture of male and female patients, we chose to drop the data on X and Y chromosomes to obliterate sexual difference. CNVs were observed quite frequently in Chromosome 3 and 5, which was highly consistent with previous reports [43,44,45,46,47] (Fig. 3A). Tumor mutational burden (TMB) analysis indicated that LUSC patients harbored the second mutation load among all the cancer types in TCGA datasets, with the log10 value of median mutations per cohort 2.30 (Fig. 3B). There were 335 genes with mutational rate > 10%, and 54 genes with mutational rate > 20% among all patients. For instance, the most frequently mutated gene was TP53, which mutated in 87.91% LUSC patients; while, the second one was TTN, with mutational rate 82.49% (Fig. 3C). The large-scale genetic alteration can be deemed as catastrophic in promoting LUSC formation and progression.

Genetic alterations in LUSC patients. A SGOL scores of LUSC in 22 autosomes. B TMB analysis of all the cancer types in TCGA datasets. C Oncoprint plot of top 30 genes mutated most frequently in LUSC. D Display of related information regarding somatic mutation
Random walk in HPRD-KEGG merged biological network
We first projected all the upregulated and downregulated DEGs onto HRPD-KEGG merged biological network, and then the biggest connected component (BCC), containing 6140 nodes and 36,350 edges, was established as walking graph for random walk (Fig. 4A). Genes present in the BCC belonging to Group A, B or C (2,722 genes) were used as source nodes. Genes within only one gene group (afflicted with one consistent dysregulation, i.e. dysregulated promoter methylation, CNV or somatic mutation) were scored as 1 (n = 2236); genes simultaneously belonging to two gene groups (afflicted with two consistent dysregulations) were scored as 2 (n = 459); and genes within all three gene groups were scored as 3 (n = 27) (Fig. 4B). The initial probability vector was obtained by normalizing the score vector so that the sum of the vector was equal to 1. Therefore, 12 significant genes were successfully retrieved in respect to steady-stage probability through 10,000 permutations (Fig. 4C). These 12 significant genes were closely related to sex hormone signaling pathway, containing both ERα (ESRS1) and AR molecules.

Random walk of DEGs with at least one types of upstream dysregulations. A The biggest connected component containing 6140 DEGs and 36,350 edges. DEGs with genetic or epigenetic dysregulations were regarded as source nodes, and the rest were target nodes. Turquoise nodes represent source genes with score 1; blue ones represent source genes with score 2; red ones represent source genes with score 3; and grey ones represent source genes with score 0. B Pie chart of DEGs with different score. The number of DEGs with score 1 was 2236, the ones with score 2 was 4589, and DEGs with score 3 in the network was 27. The rest 3418 DEGs (scored as 0) were target nodes. C The subgraph composed of 12 significant genes retrieved via random walk with restart
Expression differentiation and survival analysis of ER and AR
Paired t test indicated that all these 12 significant genes were significantly differentiated between caner and normal adjacent tissues (Fig. 5A), and the same tendency were accordantly observed in both male and female LUSC patients, respectively (Supplementary Figures S1-S2). We collected 490 patients with OS information, and survival analysis showed that the expression level of ERα was significantly associated with LUSC patients’ OS (p = 0.0025, Supplementary Figure S3). Although the survival analysis didn’t reach significance threshold, AR’s expression showed a strong relation with patients’ OS (p = 0.13, Supplementary Figure S3). The survival distinction seemed blurred in 128 female LUSC patients (Supplementary Figure S4), while higher expressions of ER and AR were both significantly associated with poor OS (p = 0.0066 for ER, p = 0.026 for AR, respectively) in male patients (Fig. 5B). We thus collected the disease-free survival (DFS) information of 174 male LUSC patients, who were confirmed suffering with local disease and undergoing R0 radical resection according to TCGA clinical data. Survival analysis of AR and ER showed a tendency in predicting a poor DFS in male patients, despite not reaching significance level (p = 0.27 for AR, p = 0.24 for ER, respectively, Supplementary Figure S5).

Expression differentiation and OS association of 12 significant genes. A Boxplot of these 12 significant genes in paired samples, and corresponding p values of paired t tests were also displayed. B OS analysis of these 12 significant genes in male patients
Four LUSC expression profiles in GEO database were downloaded, including GSE73403, GSE33745, GSE41271, and GSE50081, in order to show the OS association of AR and ER expression. The result indicated that ER expressions were all strongly associated with male LUSC patients’ OS (GSE73403, n = 65, p = 0.033; GSE33745, n = 46, p = 0.1; GSE41271, n = 48, p = 0.063; GSE50081, n = 24, p = 0.061; Supplementary Figure S6). However, the associations between AR and male patients’ OS, or between these two sex hormone receptors and female patients’ OS, were uncertain according to these four GEO datasets, probably because of the small sample size (Supplementary Figure S6).
Immunohistochemistry results of ER expression in male patients
Out of 102 male LUSC FFPE tumors collected in our center, only 23 samples (22.55%) were regarded as ERα positive (expressed in ≥1% tumor cells). ERα expression was mainly observed the nucleus of LUSC tumor cells, which was consistent with previous researches [48, 49] (Fig. 6A). Table 1 showed the clinical characteristics of these patients [including age, pathologic tumor size (pT), regional lymph node invasion (pN), AJCC stage, pathologic grade, whether the patients underwent adjuvant chemotherapy (AdjCTX) or adjuvant radiotherapy (AdjRAD)], and the patient numbers in each factor were all balanced in ER (+) and ER (−) groups according to Chi-squared tests. Median follow-up time was 46.8 months, ranging from 3.9 to 82.7 months. Survival analysis indicated that male patients with ER positive were significantly associated with a poor OS (p = 0.024, Fig. 6B), and the overall HR was 2.152 (95% CI: 1.089–4.255), while the DFS difference between the two groups was not significant (p = 0.12, Fig. 6C). Cox analysis indicated that the ERα expression was an independent prognostic factor for male LUSC patients’ OS after radical R0 surgery (Table 2).

Immunohistochemistry results of ER expression in male patients. A ERα expression in male LUSC patients. ERα expression was mainly observed the nucleus of tumor cells, and photographs of different ER expression levels were displayed. B OS analysis of ERα expression in 102 male patients. C DFS analysis of ERα expression in 102 male patients
Discussion
High-throughput and multi-dimensional genomic data usher us into a new era with overwhelming amount of information, and thus the complicated molecular mechanisms of carcinogenesis can be perceived and analyzed in a more integrative perspective. In this study, multi-level genetic or epigenetic data of LUSC, including CNV, somatic mutation, DNA promoter methylation and gene expression, were systemically utilized to discover novel and pivotal molecules and signaling pathways in a more comprehensive manner. First of all, paired samples were collected to identify significant genes with expression differentiation, CNV, promoter methylation and somatic mutation, respectively, and thereby three groups of DEGs were identified with different upstream deregulatory patterns complying with putatively accepted regulatory principles. Additionally, we adopted a simple and effective computational strategy to randomly walk DEGs with significant genetic or epigenetic alterations in HPRD and KEGG merged biological network. Random walk with restart was used to identify significant genes with the most influence from upstream abnormalities in priori knowledge-based network, of which the performance was proven to be much more superior to other methods, such as neighborhood approaches. We adopted with this strategy because it can subtly combine the multi-omics information with biological regulatory network, tracing down the information flow which meant to accumulate in significant genes. As we know, the dysregulated molecular interactions were the joint effort made by multi-level genomic malfunctions. It should be much better to take all the multi-omics data available into consideration, rather than analyze unidimensional data separately each time.
GO enrichment analysis indicated that the up-regulated DEGs with consistent upstream dysregulations were significantly associated with cell adhesion, inflammatory response, and angiogenesis, while down-regulated DEGs were mainly related to cell division and DNA repair. In some occasion, cancer cells need to adhere avidly to basement membrane matrix through upregulation of related genes, leading to vigorous cell proliferation [50]. Cell adhesion molecules were also reported to be upregulated to facilitate cancer cells to migrate toward surrounding normal tissues and promote tumor invasion [51]. In some types of cancer, an oncogenic change can induce an inflammatory microenvironment that promotes the development of tumors, and “smoldering” inflammation in the tumor microenvironment has many tumor-promoting effects, such as aiding in the proliferation and survival of malignant cells, promoting angiogenesis and facilitating metastasis [52]. Pathological angiogenesis is a hallmark of cancer and concentrated efforts in this area of research are leading to the discovery of a growing number of anti-angiogenic molecules to treat cancer [53], for example, bevacizumab [54]. As for the GO terms associated with down-regulated DEGs, disarranged cell division, especially disfunction in mitotic nuclear division, might be held accountable for cellular aneuploidy, which is consistently observed in virtually all cancers [55, 56]. Additionally, dysregulation of DNA damage repair and signaling to cell cycle checkpoints, known as the DNA damage response, is associated with a predisposition to cancer and affects responses to DNA-damaging anticancer therapy [57, 58]. The result of GO analysis implied that upstream genomic abnormalities surely promoted the process of carcinogenesis, by influencing all the key biological and molecular signaling pathways through disarrangement of tumor transcriptomics. Furthermore, LUSC tumor harbored the second amount of somatic mutational burdens among all the cancer types according to TCGA mutation data, with the ranking between skin cutaneous melanoma (SKCM) and lung adenocarcinoma (LUAD, Fig. 3B). Four genes, including TTN, TP53, MUC16 and CSMD3, mutated in even more than half of the patients (Fig. 3C), and the majority of the mutational type was missense mutation (Fig. 3D). The mutational burden of LUSC indicated that the cascade of genomic catastrophe might cause large-scale transcriptional dysregulation, leading to uncontrolled tumor cell cycles.
The significant genes identified from random walk were strongly associated with sex-hormone signaling pathways (Fig. 4). All these genes were all significantly differentiated between cancer and adjacent normal tissues, independent of different genders. ERα was significantly related to all-gender and male patients’ OS, except for female. As we mentioned before, the correlation between ER and lung cancer’s prognosis varies tremendously. The profound controversy might be due to the differences in detecting methodology, i.e., which antibody is used, heterogeneous definitions of positivity, and patient population selected for research, i.e., pathology, stage, gender, and so on [12, 13, 59]. Currently, the relationship between ER and lung cancer has been widely reported in female patients and lung adenocarcinoma according to literature review. As we know, there has been no study specifically focusing upon the relationship between ER and the prognosis of male LUSC patients.
The relationship between ER and EGFR has also been widely studied in lung adenocarcinoma. EGFR has been reported to directly phosphorylate ER at specific serine residues in 87.5% of the ER-positive lung tumors [60, 61]. In addition to MEK/ERK signaling pathway, estrogen also activates the PI3K/AKT signaling pathway to promote lung cancer cell metastasis through epithelial mesenchymal transition in lung adenocarcinoma [62]. A combination of ER antagonist and EGFR tyrosine kinase inhibitor has been shown to be effective in decreasing cell proliferation and tumor growth in lung adenocarcinoma [63, 64]. These studies indicated that the intimate interaction between ER and EGFR pathways supported a rationale to use the combined therapy lung adenocarcinoma, especially in EGFR mutated patients. In our study, the interaction between ER and EGFR was also spotted to be greatly essential in LUSC, implying the combined therapy of EFGR and ER, for instance, the combination usage of cetuximab and fulvestrant, might be effective in treating LUSC patients. The correlation between ERα and OS seemed to be much stronger in male LUSC patients, and our data also confirmed this relationship through immunohistochemistry assay. More samples might be needed to verify the significant association between ERα and DFS.
Conclusions
ERα was significantly related to a poor prognosis in LUSC, especially for male patients after radical surgery, confirmed by our immunohistochemistry data.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- LUSC:
-
Lung squamous carcinoma
- NSCLC:
-
Non-small-cell lung cancer
- ER:
-
Estrogen receptor
- CNV:
-
Copy number variation
- DEGs:
-
Differentially expressed genes
- TCGA:
-
The Cancer Genome Atlas
- TSS:
-
Transcription start site
- HPRD:
-
Human Protein Reference Database
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- FFPE:
-
Formalin-fixed paraffin-embedded
- GO:
-
Gene Ontology
- SGOL:
-
Segment Gain or Loss
- TMB:
-
Tumor mutational burden
- BCC:
-
Biggest connected component
- AR:
-
Androgen receptor
- AdjCTX:
-
Adjuvant chemotherapy
- AdjRAD:
-
Adjuvant radiotherapy
References
Lortet-Tieulent J, Soerjomataram I, Ferlay J, Rutherford M, Weiderpass E, Bray F. International trends in lung cancer incidence by histological subtype: adenocarcinoma stabilizing in men but still increasing in women. Lung Cancer. 2014;84(1):13–22. https://doi.org/10.1016/j.lungcan.2014.01.009.
Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C, Reid ME. The international epidemiology of lung Cancer: latest trends, disparities, and tumor characteristics. J Thorac Oncol. 2016;11(10):1653–71. https://doi.org/10.1016/j.jtho.2016.05.021.
Socinski MA, Obasaju C, Gandara D, Hirsch FR, Bonomi P, Bunn PA Jr, et al. Current and emergent therapy options for advanced squamous cell lung Cancer. J Thorac Oncol. 2018;13(2):165–83. https://doi.org/10.1016/j.jtho.2017.11.111.
Scagliotti GV, De Marinis F, Rinaldi M, Crino L, Gridelli C, Ricci S, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(21):4285–91. https://doi.org/10.1200/JCO.2002.02.068.
Le Chevalier T, Scagliotti G, Natale R, Danson S, Rosell R, Stahel R, et al. Efficacy of gemcitabine plus platinum chemotherapy compared with other platinum containing regimens in advanced non-small-cell lung cancer: a meta-analysis of survival outcomes. Lung Cancer. 2005;47(1):69–80. https://doi.org/10.1016/j.lungcan.2004.10.014.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung Cancer. N Engl J Med. 2018;379(21):2040–51. https://doi.org/10.1056/NEJMoa1810865.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301. https://doi.org/10.1056/NEJMoa1716948.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in advanced non-small-cell lung Cancer. N Engl J Med. 2019;381(21):2020–31. https://doi.org/10.1056/NEJMoa1910231.
Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392(1):49–53. https://doi.org/10.1016/0014-5793(96)00782-X.
Zang EA, Wynder EL. Differences in lung cancer risk between men and women: examination of the evidence. J Natl Cancer Inst. 1996;88(3–4):183–92. https://doi.org/10.1093/jnci/88.3-4.183.
Siegfried JM. Women and lung cancer: does oestrogen play a role? Lancet Oncol. 2001;2(8):506–13. https://doi.org/10.1016/S1470-2045(01)00457-0.
Baik CS, Eaton KD. Estrogen signaling in lung cancer: an opportunity for novel therapy. Cancers (Basel). 2012;4(4):969–88. https://doi.org/10.3390/cancers4040969.
Kawai H. Estrogen receptors as the novel therapeutic biomarker in non-small cell lung cancer. World J Clin Oncol. 2014;5(5):1020–7. https://doi.org/10.5306/wjco.v5.i5.1020.
Kawai H, Ishii A, Washiya K, Konno T, Kon H, Yamaya C, et al. Estrogen receptor alpha and beta are prognostic factors in non-small cell lung cancer. Clin Cancer Res. 2005;11(14):5084–9. https://doi.org/10.1158/1078-0432.CCR-05-0200.
Schwartz AG, Prysak GM, Murphy V, Lonardo F, Pass H, Schwartz J, et al. Nuclear estrogen receptor beta in lung cancer: expression and survival differences by sex. Clin Cancer Res. 2005;11(20):7280–7. https://doi.org/10.1158/1078-0432.CCR-05-0498.
Raso MG, Behrens C, Herynk MH, Liu S, Prudkin L, Ozburn NC, et al. Immunohistochemical expression of estrogen and progesterone receptors identifies a subset of NSCLCs and correlates with EGFR mutation. Clin Cancer Res. 2009;15(17):5359–68. https://doi.org/10.1158/1078-0432.CCR-09-0033.
Liu CM, Chiu KL, Chen TS, Chang SM, Yang SY, Chen LH, et al. Potential therapeutic benefit of combining gefitinib and tamoxifen for treating advanced lung adenocarcinoma. Biomed Res Int. 2015;2015:642041–11. https://doi.org/10.1155/2015/642041.
Skjefstad K, Grindstad T, Khanehkenari MR, Richardsen E, Donnem T, Kilvaer T, et al. Prognostic relevance of estrogen receptor alpha, beta and aromatase expression in non-small cell lung cancer. Steroids. 2016;113:5–13. https://doi.org/10.1016/j.steroids.2016.05.008.
Li W, Tse LA, Wang F. Prognostic value of estrogen receptors mRNA expression in non-small cell lung cancer: a systematic review and meta-analysis. Steroids. 2015;104:129–36. https://doi.org/10.1016/j.steroids.2015.09.005.
Honma N, Hosoi T, Arai T, Takubo K. Estrogen and cancers of the colorectum, breast, and lung in postmenopausal women. Pathol Int. 2015;65(9):451–9. https://doi.org/10.1111/pin.12326.
Ganti AK, Sahmoun AE, Panwalkar AW, Tendulkar KK, Potti A. Hormone replacement therapy is associated with decreased survival in women with lung cancer. J Clin Oncol. 2006;24(1):59–63. https://doi.org/10.1200/JCO.2005.02.9827.
Chlebowski RT, Schwartz AG, Wakelee H, Anderson GL, Stefanick ML, Manson JE, et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women's Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet. 2009;374(9697):1243–51. https://doi.org/10.1016/S0140-6736(09)61526-9.
Ferguson LR, Chen H, Collins AR, Connell M, Damia G, Dasgupta S, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35:S5–S24. https://doi.org/10.1016/j.semcancer.2015.03.005.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. https://doi.org/10.1016/j.ccr.2010.05.026.
Shenker N, Flanagan JM. Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer. 2012;106(2):248–53. https://doi.org/10.1038/bjc.2011.550.
Akhavan-Niaki H, Samadani AA. DNA methylation and cancer development: molecular mechanism. Cell Biochem Biophys. 2013;67(2):501–13. https://doi.org/10.1007/s12013-013-9555-2.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. https://doi.org/10.1038/nature10983.
Domany E. Using high-throughput transcriptomic data for prognosis: a critical overview and perspectives. Cancer Res. 2014;74(17):4612–21. https://doi.org/10.1158/0008-5472.CAN-13-3338.
Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39(10):1256–60. https://doi.org/10.1038/ng2123.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65. https://doi.org/10.1038/nature09708.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–12. https://doi.org/10.1038/nature08516.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54. https://doi.org/10.1038/nature05329.
Park YR, Bae SH, Ji W, Seo EJ, Lee JC, Kim HR, et al. GAB2 amplification in squamous cell lung Cancer of non-smokers. J Korean Med Sci. 2017;32(11):1784–91. https://doi.org/10.3346/jkms.2017.32.11.1784.
Liu B, Yang L, Huang B, Cheng M, Wang H, Li Y, et al. A functional copy-number variation in MAPKAPK2 predicts risk and prognosis of lung cancer. Am J Hum Genet. 2012;91(2):384–90. https://doi.org/10.1016/j.ajhg.2012.07.003.
Yang L, Liu B, Huang B, Deng J, Li H, Yu B, et al. A functional copy number variation in the WWOX gene is associated with lung cancer risk in Chinese. Hum Mol Genet. 2013;22(9):1886–94. https://doi.org/10.1093/hmg/ddt019.
Ouadid-Ahidouch H, Rodat-Despoix L, Matifat F, Morin G, Ahidouch A. DNA methylation of channel-related genes in cancers. Biochim Biophys Acta. 2015;1848(10):2621–8. https://doi.org/10.1016/j.bbamem.2015.02.015.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. https://doi.org/10.1056/NEJMoa043331.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13(3):184–99. https://doi.org/10.1038/nrc3431.
Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–72. https://doi.org/10.1093/biostatistics/kxh008.
Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82(4):949–58. https://doi.org/10.1016/j.ajhg.2008.02.013.
An N, Yang X, Cheng S, Wang G, Zhang K. Developmental genes significantly afflicted by aberrant promoter methylation and somatic mutation predict overall survival of late-stage colorectal cancer. Sci Rep. 2015;5(1):18616. https://doi.org/10.1038/srep18616.
Chen X, Chang CW, Spoerke JM, Yoh KE, Kapoor V, Baudo C, et al. Low-pass whole-genome sequencing of circulating cell-free DNA demonstrates dynamic changes in genomic copy number in a squamous lung Cancer clinical cohort. Clin Cancer Res. 2019;25(7):2254–63. https://doi.org/10.1158/1078-0432.CCR-18-1593.
Qiu ZW, Bi JH, Gazdar AF, Song K. Genome-wide copy number variation pattern analysis and a classification signature for non-small cell lung cancer. Gene Chromosome Canc. 2017;56(7):559–69. https://doi.org/10.1002/gcc.22460.
Garnis C, Davies JJ, Buys TP, Tsao MS, MacAulay C, Lam S, et al. Chromosome 5p aberrations are early events in lung cancer: implication of glial cell line-derived neurotrophic factor in disease progression. Oncogene. 2005;24(30):4806–12. https://doi.org/10.1038/sj.onc.1208643.
Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet Cytogenet. 2008;182(1):1–11. https://doi.org/10.1016/j.cancergencyto.2007.12.004.
Wang J, Qian J, Hoeksema MD, Zou Y, Espinosa AV, Rahman SM, et al. Integrative genomics analysis identifies candidate drivers at 3q26-29 amplicon in squamous cell carcinoma of the lung. Clin Cancer Res. 2013;19(20):5580–90. https://doi.org/10.1158/1078-0432.CCR-13-0594.
Kadota K, Eguchi T, Villena-Vargas J, Woo KM, Sima CS, Jones DR, et al. Nuclear estrogen receptor-alpha expression is an independent predictor of recurrence in male patients with pT1aN0 lung adenocarcinomas, and correlates with regulatory T-cell infiltration. Oncotarget. 2015;6(29):27505–18. https://doi.org/10.18632/oncotarget.4752.
Rouquette I, Lauwers-Cances V, Allera C, Brouchet L, Milia J, Nicaise Y, et al. Characteristics of lung cancer in women: importance of hormonal and growth factors. Lung Cancer. 2012;76(3):280–5. https://doi.org/10.1016/j.lungcan.2011.11.023.
Moro L, Arbini AA, Marra E, Greco M. Up-regulation of Skp2 after prostate cancer cell adhesion to basement membranes results in BRCA2 degradation and cell proliferation. J Biol Chem. 2006;281(31):22100–7. https://doi.org/10.1074/jbc.M604636200.
Li R, Wheeler T, Dai H, Ayala G. Neural cell adhesion molecule is upregulated in nerves with prostate cancer invasion. Hum Pathol. 2003;34(5):457–61. https://doi.org/10.1016/S0046-8177(03)00084-4.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44. https://doi.org/10.1038/nature07205.
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57. https://doi.org/10.1038/35025220.
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–9. https://doi.org/10.1200/JCO.2007.14.9930.
Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432(7015):338–41. https://doi.org/10.1038/nature03099.
van Jaarsveld RH, Kops G. Difference makers: chromosomal instability versus aneuploidy in Cancer. Trends Cancer. 2016;2(10):561–71. https://doi.org/10.1016/j.trecan.2016.09.003.
Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–17. https://doi.org/10.1038/nrc3399.
Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16(1):35–42. https://doi.org/10.1038/nrc.2015.4.
Siegfried JM, Stabile LP. Estrongenic steroid hormones in lung cancer. Semin Oncol. 2014;41(1):5–16. https://doi.org/10.1053/j.seminoncol.2013.12.009.
Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491–4. https://doi.org/10.1126/science.270.5241.1491.
Marquez-Garban DC, Chen HW, Fishbein MC, Goodglick L, Pietras RJ. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72(2):135–43. https://doi.org/10.1016/j.steroids.2006.11.019.
Zhao XZ, Liu Y, Zhou LJ, Wang ZQ, Wu ZH, Yang XY. Role of estrogen in lung cancer based on the estrogen receptor-epithelial mesenchymal transduction signaling pathways. Onco Targets Ther. 2015;8:2849–63. https://doi.org/10.2147/OTT.S90085.
Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005;65(4):1459–70. https://doi.org/10.1158/0008-5472.CAN-04-1872.
Pietras RJ, Marquez DC, Chen HW, Tsai E, Weinberg O, Fishbein M. Estrogen and growth factor receptor interactions in human breast and non-small cell lung cancer cells. Steroids. 2005;70(5–7):372–81. https://doi.org/10.1016/j.steroids.2005.02.017.
Acknowledgements
We would like to express our sincere gratitude to Prof. Zhao Jie for the advice of analysis of tissue samples.
Funding
This work was supported by the National Natural Science Foundation of China (81802271 to N.A.), the National Natural Science Foundation of China (81801734 to X.Y.), and by the Natural Science Foundation of Shandong Province, China (ZR2019QH003 to X.Y.).
Author information
Authors and Affiliations
Contributions
(I) Conception and design: Ning An, Xue Yang. (II) Administrative support: Ning An, Xue Yang. (III) Provision of study materials or patients: Xiangfeng Jin, Rongjian Xu. (IV) Collection and assembly of data: Xiangfeng Jin, Rongjian Xu. (V) Data analysis and interpretation: Ning An, Xue Yang, Zhuang Yu. (VI) Manuscript writing: All authors. (VII) Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by ethics board of The Affiliated Hospital of Qingdao University (NO.QYFY WZLL 25595) and individual consent for this retrospective analysis was waived.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure S1.
Boxplot of these 12 significant genes in paired male samples. Supplementary Figure S2. Boxplot of these 12 significant genes in paired female samples. Supplementary Figure S3. OS analysis of these 12 significant genes in all-gender patients. Supplementary Figure S4. OS analysis of these 12 significant genes in female patients. Supplementary Figure S5. DFS analysis of ERα and AR in male LUSC patients. Supplementary Figure S6. OS analysis of ER and AR in both male and female LUSC patients, respectively using 4 independent cohorts.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, X., Jin, X., Xu, R. et al. ER expression associates with poor prognosis in male lung squamous carcinoma after radical resection. BMC Cancer 21, 1043 (2021). https://doi.org/10.1186/s12885-021-08777-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08777-6