
Abstract
Background
The aim of this study was to determine whether checkpoint kinase 1 inihibitor (CHK1), LY2603618, and gemcitabine prolong overall survival (OS) compared to gemcitabine alone in patients with unresectable pancreatic cancer.
Methods
Patients with Stage II-IV locally advanced or metastatic pancreatic cancer were randomized (2:1) to either 230 mg of LY2603618/1000 mg/m2 gemcitabine combined or 1000 mg/m2 gemcitabine alone. OS was assessed using both a Bayesian augment control model and traditional frequentist analysis for inference. Progression-free survival (PFS), overall response rate (ORR), duration of response, pharmacokinetics (PK), and safety (Common Terminology Criteria for Adverse Events [AEs] v 3.0) were also evaluated.
Results
Ninety-nine patients (n = 65, LY2603618/gemcitabine; n = 34, gemcitabine) were randomized (intent-to-treat population). The median OS (months) was 7.8 (range, 0.3–18.9) with LY2603618/gemcitabine and 8.3 (range, 0.8-19.1+) with gemcitabine. Similarly, in a Bayesian analysis, the study was not positive since the posterior probability that LY2603618/gemcitabine was superior to gemcitabine in improving OS was 0.3, which did not exceed the prespecified threshold of 0.8. No significant improvements in PFS, ORR, or duration of response were observed. Drug-related treatment-emergent AEs in both arms included nausea, thrombocytopenia, fatigue, and neutropenia. The severity of AEs with LY2603618/gemcitabine was comparable to gemcitabine. The LY2603618 exposure targets (AUC(0-∞) ≥21,000 ng∙hr/mL and Cmax ≥2000 ng/mL) predicted for maximum pharmacodynamic response were achieved after 230 mg of LY2603618.
Conclusions
LY2603618/gemcitabine was not superior to gemcitabine for the treatment of patients with pancreatic cancer.
Trial Registration
NCT00839332. Clinicaltrials.gov. Date of registration: 6 February 2009
Similar content being viewed by others

Background
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States [1]. Current therapeutic strategies for pancreatic cancer have a modest impact on disease course and prognosis [2]. The 5-year survial rate remains low (<5%) [3]. Until recently, gemcitabine was the standard of care for patients with advanced/metastatic pancreatic cancer. FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and 5-FU) and gemcitabine plus nab-paclitaxel (Abraxane®) are novel therapeutic regimens demonstrating survival advantages in patients with advanced pancreatic cancer [4–8]. Although these recent advances are promising, there is still a need for novel therapeutic targets to further improve and sustain clinical response in pancreatic cancer patients.
Checkpoint kinase 1 (CHK1) is a protein kinase involved in the DNA damage response. Activation of CHK1 initiates cell cycle arrest allowing for DNA repair and replication. Inhibition of CHK1 allows cells to enter mitosis without DNA repair, eventually leading to apoptosis [9]. Furthermore, inhibition of CHK1 sensitizes tumor cells to DNA-damaging agents making CHK1 a unique target for cancer therapy. Azorsa and colleagues recently identified CHK1 as a therapeutic target for sensitizing pancreatic cancer cells to gemcitabine therapy using a synthetic lethal RNAi screening approach [10].
LY2603618, a selective CHK1 inhibitor, enhances the activity of cytotoxic chemotherapy agents, including gemcitabine, in in vitro and in vivo nonclinical efficacy studies [11, 12]. Phase I of this Phase I/II study determined the recommended Phase II dose to be 230 mg [13]. Phase II, as presented here, determined if the overall survival (OS) in patients with Stage II-IV unresectable pancreatic cancer who were administered LY2603618 and gemcitabine exceeded the OS of patients treated with gemcitabine alone.
Methods
Study objectives
The primary objective of this Phase II study was to compare OS with LY2603618/gemcitabine to gemcitabine alone in patients with Stage II-IV unresectable pancreatic cancer. Secondary objectives included characterizing the safety and toxicity profile of LY2603618/gemcitabine and gemcitabine; estimating progression-free survival (PFS), duration of response, and change in tumor size; assessing response rates; evaluating the pharmacokinetics (PK) of LY2603618; investigating biomarker responses; and performing an exploratory assessment of Fridericia’s heart rate-corrected QT interval (QTcF).
Patients
Adult patients who had given informed consent had adequate hematological, liver, and renal functions; histological or cytological evidence of a diagnosis of Stage II or III adenocarcinoma of the pancreas not amenable to resection with curative intent or Stage IV disease; and an Eastern Cooperative Oncology Group performance status (ECOG PS) 0–2. Patients with previous radical surgery for pancreatic cancer were eligible after progression was documented. Exclusion criteria included known hypersensitivity to gemcitabine; females who were pregnant or lactating; prior radiotherapy involving >25% of marrow-producing area; and treatment with any non-approved drug within 30 days of enrollment. Patients may have received previous adjuvant treatment with gemcitabine.
Study design and treatment plan
Prior to enrollment, the study protocol, patient informed consent, and any other written study documentation were approved by an ethics committee. This trial was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization. Phase II of this open-label, multicenter, randomized, 2-arm, Phase I/II trial was conducted in patients with locally advanced or metastatic pancreatic cancer. Patients were randomized (2:1) to either LY2603618/gemcitabine or gemcitabine. Gemcitabine (1000 mg/m2) was given as a 30-min infusion on days 1, 8, and 15 of a 28-day cycle. LY2603618 (230 mg) was administered as a 1-h infusion ~24 h after administration of gemcitabine. Patients continued on treatment until disease progression, unacceptable toxicity, or patient unwillingness to participate.
Statistical analysis
The primary objective was a comparison of OS on the intent-to-treat (ITT) population using a Bayesian posterior probability for the superiority of the combination over gemcitabine. Ninety-nine patients were planned, resulting in a frequentist design with ~60% power (1-sided, 0.2 type I error, no interim analysis) to detect a 2-month improvement in survival (7 months gemcitabine vs. 9 months LY2603618/gemcitabine). The Bayesian model [14, 15] incorporated historical gemcitabine data [16, 17] with prospective gemcitabine data to compare survival between the treatment arms and increase the power compared to the frequentist design. LY2603618 and gemcitabine would be considered superior to gemcitabine if the posterior probability of superiority exceeded 0.8. Simulation resulted in estimated power of 0.76 and type I error rate of 0.15. In addition to the Bayesian model, frequentist analysis of OS was also performed as a sensitivity analysis. The definition of secondary efficacy variables was consistent with standard conventions per RECIST (v 1.1) [18].
Exploratory analyses included: change from baseline in tumor size and carbohydrate antigen 19–9 (CA19-9) levels, and changes in QTcF from electrocardiograms (ECG) obtained at baseline and after LY2603618 administration on days 2 and 16 during cycle one.
Safety
All patients who received at least one dose of study drug were evaluated for safety and toxicity. AE severity was graded using the Common Terminology Criteria for AEs (CTCAE) v 3.0.
Pharmacokinetic/pharmacodynamic analysis
LY2603618 concentrations were quantified using a validated high-pressure liquid chromatography/mass spectrometry/mass spectrometry method. Whole blood samples were collected following the LY2603618 infusion on days 2 and 16 of cycle 1 before the start (<10 min) of infusion; immediately prior to the end of infusion (<5 min); and at 1, 3, and 24 h after the end of infusion. LY2603618 PK parameters were computed from the plasma concentration versus time data by standard noncompartmental analyses (Phoenix WinNonlin version 6.3, Pharsight, A Certara Company®; Princeton, NJ, USA). The PK parameters of maximum plasma concentration (Cmax) and area under the plasma concentration time-curve from time 0 to the time of the last measurable plasma concentration (AUC[0-tlast]) or infinity (AUC[0-∞]) on days 2 and 16 of cycle one were calculated, as well as the terminal elimination half-life (t1/2), volume of distribution at steady-state (Vss), systemic clearance (CL), percentage of AUC[0-∞] extrapolated (%AUC[tlast-∞]), and the intra- and intercycle accumulation ratios (RA).
Biomarker response
A nucleoside analog deoxyribonucleic acid (DNA) incorporation assay method measured the amount of gemcitabine incorporated into genomic DNA [19]. A sample for CA–19–9 analysis was collected at the start of each cycle.
Results
Patient disposition
Of the 107 enrolled patients, a total of 99 patients (n = 65, LY2603618/gemcitabine; n = 34, gemcitabine alone) were randomized and included in the ITT population. The first patient was enrolled on 26 February 2009. Patient demographics and disease characteristics at baseline are summarized in Table 1. The majority of the patient population (mean age, 64 years) presented with Stage IV disease (76.8%) and more than 90% had an ECOG PS 0–1 at study entry (Table 1). The primary reasons for study discontinuation in the LY2603618/gemcitabine arm and gemcitabine alone arm, respectively, included: progressive disease (70.8%; 61.8%), AE (12.3%; 17.6%), subject decision (4.6%; 14.7%), investigator decision (6.2%; 2.9%), death (4.6%; 2.9%), and protocol violation (1.5%; 0).
Clinical efficacy
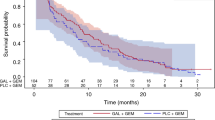
The Bayesian model was applied to compare OS between treatments. The posterior probability of superiority of LY2603618/gemcitabine over gemcitabine alone was 0.33, which did not exceed the pre-specified threshold of 0.8. These findings were confirmed by the frequentist analysis. The median OS was 7.8 months (range, 0.3–18.9 months) for LY2603618/gemcitabine and 8.3 months (range, 0.8–19.1+ months) for gemcitabine alone (Fig. 1a).

a Overall survival. Kaplan Meier survival curves of patients treated with LY2603618/gemcitabine combination therapy compared with historical gemcitabine studies. [1Jänne PA, Paz-Ares L, Oh Y, Eschbach C, Hirsh V, Enas N, Brail L, von Pawel J. Randomized, double-blind, phase II trial comparing gemcitabine-cisplatin plus the LTB4 antagonist LY293111 versus gemcitabine-cisplatin plus placebo in first-line non-small-cell lung cancer. J Thorac Oncol. 2014;9:126–31. 2Oettle H, Richards D, Ramanathan RK, van Laethem JL, Peeters M, Fuchs M, Zimmerman A, John W, Von Hoff D, Arning M, Kindler HL. A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann Oncol. 2005;16:1639–45.] b Progression-free survival. Kaplan Meier survival curves of patients treated with LY2603618/gemcitabine compared with gemcitabine monotherapy
Overall, LY2603618/gemcitabine was not statistically superior to gemcitabine alone when PFS, duration of response, ORR, and clinical benefit rate were assessed (Table 2). The median PFS was 3.5 months (range, 0–15.9 months) for LY2603618/gemcitabine and 5.6 months (range, 0–17.4 months) for gemcitabine (Table 2; Fig. 1b). No complete response (CR) was observed with either treatment. Although not statistically significant, a numerically higher percentage of patients had a partial response (PR) in the LY2603618/gemcitabine arm (21.5%; 95% confidence intervals [CI], 12.3–33.5) than in the gemcitabine arm (8.8%; 95% CI, 1.9–23.7). No distinguishing baseline characteristics were noted among patients with a response. Due to the overlapping confidence intervals, the clinical significance of the difference in response rates is unknown. The clinical benefit rate was 55.4% (95% CI, 42.5–67.7) and 64.7% (95% CI, 46.5–80.3) in the LY2603618/gemcitabine and gemcitabine arms, respectively. No significant difference in the change in tumor size (the sum of target lesions, per RECIST) from baseline to 8 weeks was observed between treatments (P = .6726).
Safety
The median number of cycles completed was 2.0 (range, 0–16 cycles) for LY2603618/gemcitabine and 2.5 (range, 0–18 cycles) for gemcitabine. As shown in Table 3, study drug-related treatment-emergent AEs (TEAEs) were comparable between LY2603618/gemcitabine (89.2%) and gemcitabine (91.2%). The most frequently observed TEAEs in both arms were nausea, thrombocytopenia, fatigue, and neutropenia. Fewer patients experienced anemia with LY2603618/gemcitabine (13.8%) than with gemcitabine (26.5%). In contrast, a higher incidence of vomiting, decreased appetite, and stomatitis was observed for LY2603618/gemcitabine than for gemcitabine. For each arm, neutropenia and thrombocytopenia were the most common grade 3/4 TEAEs possibly related to treatment, in addition to anemia, which was also common to gemcitabine (Table 4). Serious adverse events (SAE) related to study treatment were reported among 13.8% and 23.5% of patients in the LY2603618/gemcitabine and gemcitabine arms, respectively.
Fourteen patients (n = 8, LY2603618/gemcitabine; n = 6, gemcitabine) discontinued the study due to AEs. Of the eight patients who discontinued in the LY2603618/gemcitabine arm, four events (grade 4 cerebrovascular accident, grade 1 left bundle branch block, grade 3 acute pulmonary oedema, and grade 3 atrial fibrillation) were possibly related to treatment. Of the six patients who discontinued in the gemcitabine arm, four possibly related events occurred (grade 3 thrombotic microangiopathy, grade 4 acute renal failure, grade 2 thrombocytopenia, and grade 3 hemolytic uraemic syndrome). Four deaths were reported during the study; three due to disease progression and one due to a non-related peripheral arterial ischemia event.
Pharmacokinetic/pharmacodynamic analyses
LY2603618 demonstrated consistent PK parameters after single (day 2) and repeat administration (day 16) during cycle 1 (Table 5). The LY2603618 plasma systemic exposure targets (i.e., AUC(0-∞) ≥21,000 ng hr/mL and Cmax ≥2000 ng/mL) that correlate with maximal pharmacodynamic (PD) effect observed in nonclinical HT-29 xenograft models (data on file) were achieved on a mean cohort basis after 230 mg of LY2603618 (Table 5). More specifically, 87% and 73% of the individual PK profiles on days 2 and 16 of cycle 1 were above the targets for Cmax and AUC(0-∞), respectively.
For the PD analyses, dFdC was incorporated into DNA following gemcitabine administration, with the levels declining to almost baseline by the end of each treatment cycle. The highest levels of dFdC incorporation were observed on days 8 and 15 across all doses. The increases in the amount of dFdC incorporation did not correspond to increasing doses of LY2603618.
Of the patients who had baseline CA19-9 levels > upper limit of normal, a similar percentage of patients (65.4% LY2603618/gemcitabine; 64% gemcitabine) experienced a >50% reduction from baseline in CA19-9 levels.
QTcF assessment
In a time-point exploratory QTcF assessment, no clinically significant trends in ECG parameter changes were reported. Five patients had a change in QTcF from baseline between 30 and 60 milliseconds (msec); no patients had a change in QTcF >60 msec.
Discussion
The current study was part of a Phase I/II study designed to compare the OS of LY2603618/gemcitabine to gemcitabine alone. This study used a Bayesian augmented control design to incorporate historical gemcitabine data, which minimized the number of patients needed for the treatment to be evaluated. Stage II or III patients not amenable to resection with curative intent or Stage IV disease were included in the current study to match the populations used in the historical studies used as reference data. The OS of LY2603618/gemcitabine was not superior to gemcitabine alone in patients with locally advanced or metastatic pancreatic cancer by either the Bayesian or frequentist approach. In addition, no significant differences between arms in any of the secondary endpoints were observed.
The safety profiles were comparable between arms, indicating that the addition of LY2603618 did not significantly change the safety profile of gemcitabine. This is consistent with the CHK1 inhibitor MK8776 [20], but in contrast to the data reported with the CHK1 selective inhibitors GDC-0425 and AZD7762 [21, 22]. In a Phase 1 study with AZD7762, unpredictable cardiac toxicity was observed [21]. Although it was demonstrated safe and feasible to administer GDC-0425 with gemcitabine, the CHK1 inhibitor appeared to increase some of the toxicities associated with gemcitabine [22].
A trend towards a lower LY2603618 systemic exposure and more rapid CL associated with a larger interpatient variability in Phase II (Table 5) compared to Phase I was observed [13]. This is likely a result of the more limited PK sampling schedule (sampling to only 24 h post dose) used in the Phase II study (i.e., larger AUC(tlast-∞) (%) values; Table 5), thereby limiting the capability of the conventional PK analysis method to accurately quantify the terminal elimination phase of LY2603618 and resulting in an underestimate of AUC(0-∞) and overestimate of CL. In contrast, the LY2603618 PK profiles over the first 24 h from Phase II demonstrated a high degree of concordance with the PK profiles from Phase I. The average t1/2 following administration of 230 mg LY2603618 was consistent with a t1/2 suitable for achieving and maintaining the desired target human exposures while minimizing the intra- and intercycle accumulation (Table 5). Gemcitabine did not appear to affect the PK of LY2603618, as the PK parameters reported in this study were similar to the PK parameters calculated after LY2603618 monotherapy [23].
The study had inherent limitations that may have contributed to the negative clinical outcome observed. In addition, due to the lack of a clinically-validated PD marker to quantify direct CHK1 inhibition by LY2603618, the magnitude and duration of CHK1 target inhibition at 230 mg is neither known nor has it been correlated to clinical responses. Therefore, it is possible the PK surrogate targets (i.e., AUC(0-∞) ≥21,000 ng•hr/mL and Cmax ≥2000 ng/mL) derived from nonclinical xenograft models for maximal PD response were not appropriate thresholds to predict clinical responses in humans. In addition, inclusion of only patients with Stage IV disease may have yielded a more favorable clinical outcome.
One Phase III randomized trial comparing gemcitabine with FOLFIRINOX reported statistically significant improvements in OS (hazard ratio [HR] 0.57, P < .0001), PFS (HR 0.47, P < .0001), and ORR (P = .0001) in chemo-naïve patients with ECOG PS 0 and 1 [5]. Despite the clincial efficacy observed with FOLFIRINOX, this treatment was associated with more frequent and more severe toxicity [5]. As a result, only patients with an adequate PS are typically eligible for FOLFIRINOX treatment. Recent meta-analyses have reported that patients with poorer performance had less OS benefit from combined therapies for metastatic pancreatic cancer [24, 25]. Since FOLFIRINOX emerged as a treatment option during the conduct of this study, there was concern that a greater proportion of patients with low PS status who were not eligible for FOLFIRINOX would be included. However, only 9.2% and 8.8% of patients on the experimental arm and control arm, respectively, were PS 2 and so this consideration was unlikely to have affected the outcome.
The Metastatic Pancreatic Adenocarcinoma Clinical Trial (MPACT) demonstrated improved clinical efficacy with gemcitabine/nab-paclitaxel than gemcitabine (median OS, 8.5 months vs. 6.7 months, respectively); however, fatigue, neuropathy, and neutropenia were more common among patients receiving combination therapy than monotherapy [4, 8]. Interestingly, the overall incidence of grade III/IV study drug-related TEAEs in the current study was not increased in the LY2603618/gemcitabine arm compared with the gemcitabine arm, except for the incidence of grade 3 leukocytes and platelets, which was higher in the LY2603618/gemcitabine arm.
Conclusion
OS was not improved with the addition of LY2603618/gemcitabine compared with gemcitabine alone. The safety and PK profiles were comparable between treatment arms. As a result of this finding, LY2603618/gemcitabine will not be further developed for the treatment of patients with pancreatic cancer.
Abbreviations
- %AUC[tlast-∞] :
-
Percentage of AUC[0-∞] extrapolated
- 5-FU:
-
Fluorouracil
- AE:
-
Adverse event
- AUC[0-tlast] :
-
Area under the plasma concentration time-curve from time 0 to the time of the last measurable plasma concentration
- AUC[0-∞] :
-
Area under the plasma concentration time-curve from time 0 to infinity
- AUC(0–24) :
-
Area under the plasma concentration time-curve from time 0 to 24 h
- AUC(tlast - ∞) :
-
Fraction of AUC(0 - ∞) extrapolated from the time of the last measurable plasma concentration (tlast) to infinity
- CA19-9:
-
Carbohydrate antigen 19–9
- Cav,24 :
-
Average plasma concentration over 24 h calculated using AUC(0–24)
- CHK1:
-
Checkpoint kinase 1
- CI:
-
Confidence intervals
- CL:
-
Systemic clearance
- Cmax :
-
Maximum plasma concentration
- CR:
-
Complete response
- CTCAE:
-
Common terminology criteria for adverse events
- CV%:
-
Percent coefficient of variation
- dFdC 2’:
-
2’-difluorodeoxycytidine
- DNA:
-
Deoxyribonucleic acid
- ECG:
-
Electrocardiogram
- ECOG PS:
-
Eastern cooperative oncology group performance status
- FOLFIRINOX:
-
Oxaliplatin, irinotecan, leucovorin, and 5-FU
- ITT:
-
Intent-to-treat
- m2 :
-
Meters squared
- mg:
-
Milligram
- MPACT:
-
Metastatic pancreatic adenocarcinoma clinical trial
- msec:
-
Millisecond
- NC:
-
Not calculated
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PD:
-
Pharmacodynamic
- PFS:
-
Progression-free survival
- PK:
-
Pharmacokinetics
- PR:
-
Partial response
- PS:
-
Performance status
- QTcF:
-
Fridericia’s heart rate-corrected QT interval
- RA :
-
Intra- and intercycle accumulation ratios
- RNAi:
-
Ribonucleic acid interference
- SAE:
-
Serious adverse event
- t1/2 :
-
Terminal elimination half-life
- TEAE:
-
Treatment-emergent AEs
- tmax :
-
Time of maximum observed plasma concentration
- Vss :
-
Volume of distribution at steady-state
References
American Cancer Society. American Cancer Society Cancer Facts & Figures. 2014. http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf. accessed 18 Aug 2015.
Puleo F, Maréchal R, Demetter P, Bali M, Calomme A, Closset J, Bachet JB, Deviere J, Van Laethem JL. New challenges in perioperative management of pancreatic cancer. World J Gastroenterol. 2015;21:2281–93.
Kuhlmann KF, de Castro SM, Wesseling JG, ten Kate FJ, Offerhaus GJ, Busch OR, van Gulik TM, Obertop H, Gouma DJ. Surgical treatment of pancreatic adenocarcinoma; actual survival and prognostic factors in 343 patients. Eur J Cancer. 2004;40:549–58.
Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, Zhang H, Soon-Shiong P, Shi T, Rajeshkumar NV, Maitra A, Hidalgo M. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–54.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25.
Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, Tuveson DA. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2:260–9.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante JR, Moore MJ, Seay TE, Tjulandin S, Ma WW, Saleh MN, Harris M, Reni M, Ramanathan RK, Tabernero J, Hidalgo M, Van Cutsem E, Goldstein D, Wei X, Iglesias JL, Renschler MF. Randomized phase III study of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone in patients with metastatic adenocarcinoma of the pancreas (MPACT) [ASCO abstract LBA148]. J Clin Oncol. 2013;31 Suppl 4:LBA148.
McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142:1–10.
Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, Kiefer JA, Henderson MC, Trent JM, Von Hoff DD, Mousses S. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J Transl Med. 2009;7:43.
King C, Diaz H, Barnard D, Barda D, Clawson D, Blosser W, Cox K, Guo S, Marshall M. Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Invest New Drugs. 2014;32:213–26.
Calvo E, Chen VJ, Marshall M, Ohnmacht U, Hynes SM, Kumm E, Diaz HB, Barnard D, Merzoug FF, Huber L, Kays L, Iversen P, Calles A, Voss B, Lin AB, Dickgreber N, Wehler T, Sebastian M. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Invest New Drugs. 2014;32:955–68.
Calvo E, Braiteh F, Von Hoff DD, McWilliams R, Becerra C, Galsky MD, Jameson G, Lin J, McKane S, Wickremsinhe ER, Hynes SM, Bence Lin AK, Hurt K, Richards D. Phase I study of CHK1 inhibitor LY2603618 in combination with gemcitabine in patients with solid tumors. Oncology. 2016;91:251–60.
Congdon P. Applied bayesian modeling. 2nd ed. Hoboken: Wiley; 2003.
Ibrahim J, Chen M-H, Sinha D. Bayesian survival analysis. New York: Springer; 2001.
Oettle H, Richards D, Ramanathan RK, van Laethem JL, Peeters M, Fuchs M, Zimmermann A, John W, Von Hoff D, Arning M, Kindler HL. A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann Oncol. 2005;16:1639–45.
Janne PA, Paz-Ares L, Oh Y, Eschbach C, Hirsh V, Enas N, Brail L, von Pawel J. Randomized, double-blind, phase II trial comparing gemcitabine-cisplatin plus the LTB4 antagonist LY293111 versus gemcitabine-cisplatin plus placebo in first-line non-small-cell lung cancer. J Thorac Oncol. 2014;9:126–31.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Wickremsinhe ER, Lutzke B, Jones B, Schultz GA, Freeman AB, Pratt SE, Bones AM, Ackermann BL. Quantification of gemcitabine incorporation into human DNA by LC/MS/MS as a surrogate measure for target engagement. Anal Chem. 2010;82:6576–83.
Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, Venook AP, Loechner S, Rosen LS, Shanahan F, Parry D, Shumway S, Grabowsky JA, Freshwater T, Sorge C, Kang SP, Isaacs R, Munster PN. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol. 2015;33:1060–6.
Sausville E, LoRusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, Zabludoff S, Agbo F, Oakes P, Senderowicz A. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014;73:539–49.
Infante J, Hollebecque A, Postel-Vinay S, Bauer T, Blackwood B, Evangelista M, Mahrus S, Peale F, Lu X, Sahasranaman S, Zhu R, Chen Y, Ding X, Murray E, Schutzman J, Lauchle J, Soria J-C, LoRusso P. Phase I study of GDC-0425, a checkpoint kinase 1 inhibitor, in combination with gemcitabine in patients with refractory solid tumors [AACR abstract CT139]. Cancer Res. 2015;75:CT139.
Weiss GJ, Donehower R, Iyengar T, Ramanathan RK, Lewandowski K, Westin E, Hurt K, Hynes SM, Anthony SP, McKane S. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest New Drugs. 2013;31:136–44.
Collins DC, Morris PG. Systemic therapy for advanced pancreatic cancer: individualising cytotoxic therapy. Expert Opin Pharmacother. 2015;16:851–61.
Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C. Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer. 2008;8:82.
Acknowledgements
The authors wish to acknowledge Chastity Bradley, PhD for her medical writing assistance, Elizabeth Kumm for statistical support, Ignacio Garcias-Ribas and Eric Westin for their contributions to the CHK1 clinical program, and Rodney L. Decker for his assistance with the PK analysis. We wish to also thank the patients who participated in this trial and the study coordinators, nurses, nurse practitioners, clinical research assistants, and doctors who assisted with the research.
Funding
The study was sponsored by Eli Lilly and Company. Eli Lilly and Company was responsible for all aspects of this study, including study concept/design, data analysis, and the interpretation and writing of the manuscript with the use medical writing resources.
Availability of data and materials
At this time, Eli Lilly and Company does not provide access to individual patient data for compounds whose development has terminated. Eli Lilly and Company provides access to the individual patient data from studies on approved medicines and indications as defined by the sponsor specific information on ClinicalStudyDataRequest.com.
Authors’ contributions
As per ICMJE guidelines, all authors were involved in the study design, research, analysis, and/or interpretation of the data; likewise, all authors shared the responsibility of writing/editing the submitted manuscript and are fully accountable for the integrity of the data. In addition, all authors have read and approved the manuscript. The individual contributions of each author have been outlined below: Conception of the work: DVH. Design of the work: KH, SH, GK. Acquisition of data for the work: BL, JL-M, DRichards, GI, DZC, GK, PS, DRichel, CS, SC, GLF, TC, DVH, EC, KH. Analysis of data for the work: SH, JL, ABL, GK, DZC, DVH, TC. Interpretation of data for the work: BL, JL-M, KH, SH, JL, ABL, GK, EC, DZC, DVH, TC. Drafting of the manuscript: SH, JL, SC, TC. Critical revision of the manuscript for important intellectual content: BL, JL-M, DRichards, GI, DZC, GK, PS, DRichel, CS, GLF, KH, SH, ABL, DVH, EC.
Competing interests
P. Stella is employed by a for-profit health care company, Physician Resource Management, and is the president as well as part-owner and founder of the company. D. Von Hoff is currently a consultant for Eli Lilly and Company. K. Hurt, S. Hynes, J. Lin, and A. Bence Lin are employees of Eli Lilly and Company and own Eli Lilly and Company stock. All remaining authors have declared no conflicts of interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Prior to enrollment, the study protocol, patient informed consent, and any other written study documentation were approved by an ethics committee. These documents were approved in each country by the following ethical review boards: Ethik-kommission Des Fb Medizin Der J. W. Goethe-universität (Germany), Ospedale Le Torrette (Italy), Azienda Ospedaliera Universitaria Careggi (Italy), Istituto Scientifico Romagnolo - Studio E La Cura Dei Tumori (Italy), Academisch Medisch Centrum Metc (Netherlands), Wojskowy Instytut Medyczny (Poland), Comisia Nationala De Etica Pentru Studiul Clinic Al Medicame (Romania), Hospital Madrid Norte Sanchinarro (Spain), Hospital Duran I Reynals (Spain), Hospital Universitario 12 De Octubre (Spain), US Oncology (United States), Mayo Clinic (United States), Sanford Health Institutional Review Board (United States), Western Institutional Review Board - Wirb (United States). In addition, this trial was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization. Adult patients who had given written informed consent were eligible to participate in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Laquente, B., Lopez-Martin, J., Richards, D. et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 17, 137 (2017). https://doi.org/10.1186/s12885-017-3131-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3131-x