
Abstract
Background
Hereditary sensory and autonomic neuropathy type 2 (HSAN2) is an autosomal recessive disorder with predominant sensory dysfunction and severe complications such as limb destruction. There are different subtypes of HSAN2, including HSAN2A, which is caused by mutations in WNK1/HSN2 gene.
Methods
An Iranian family with four siblings and autosomal recessive inheritance pattern whom initially diagnosed with HSAN2 underwent whole exome sequencing (WES) followed by segregation analysis.
Results
According to the filtering criteria of the WES data, a novel candidate variation, c.3718C > A in WNK1/HSN2 gene that causes p.Tyr1025* was identified. This variation results in a truncated protein with 1025 amino acids instead of the wild-type product with 2645 amino acids. Sanger sequencing revealed that the mutation segregates with disease status in the pedigree.
Conclusions
The identified novel nonsense mutation in WNK1/HSN2 in an Iranian HSAN2 pedigree presents allelic heterogeneity of this gene in different populations. The result of current study expands the spectrum of mutations of the HSN2 gene as the genetic background of HSAN2A as well as further supports the hypothesis that HSN2 is a causative gene for HSAN2A. However, it seems that more research is required to determine the exact effects of this product in the nervous system.
Similar content being viewed by others

Background
Hereditary sensory and autonomic neuropathies (HSANs) are inherited group of neurodegenerative disorders of the peripheral nervous system associated with sensory dysfunction [1]. HSAN is characterized by the multimodal loss of sensation with or without presentation of autonomic disturbances [2]. In this group of neuropathies, motor neurons are relatively or entirely spared [3]. Based on clinical features and pattern of inheritance, HSAN has been categorized into seven types with additional entities related to mutations in different genes [4].
HSAN type 2 (HSAN2) was first described in 1973 by Ohta M. et al. [5], but its genetic etiology was initially demonstrated from Canadian patients in 2004, and causative mutation was reported in an intron within the WNK1 gene, referred to as HSN2 [6]. HSAN2 occurs sporadically or with autosomal recessive inheritance pattern, which is usually diagnosed during the first two decades of life [7]. Predominantly, HSAN2 patients present sensory deficit in distal lower limbs more severely than the upper ones with possible motor involvement and variable autonomic disturbances. With the passing of time, ulcero-mutilating complications will be revealed and, the disease can become more complicated by osteomyelitis and painless fractures [2, 8]. This disorder doesn’t have a sex preference or particular ethnic distribution [9].
The recommended approach taken to the diagnosis of HSAN2 is based on detailed family history, clinical and paraclinical findings comprising neurological examinations in order to determine the extent of sensory loss and involvement of autonomic and motor nervous system, electrophysiology, histopathological evaluation of sural nerve and molecular genetic testing of candidate genes [8]. Nowadays, the genetic methods such as next-generation sequencing (NGS) based methods, are good opportunities to reduce the requirement of invasive diagnostic tests such as histological evaluation [10].
HSAN type 2 is classified into four subtypes including HSAN2A, HSAN2B, HSAN2C and HSAN2D that respectively are ascribed to genes HSN2, FAM134B, KIF1A and SCN9A in which several mutations are known to develop the diseases [11,12,13].
HSN2 (NM_213655) is a single-exon gene located within intron 8 of WNK1 (WNK lysine deficient protein kinase 1). WNK1/HSN2 appears to be expressed in satellite and Schwann cells, and sensory neurons [14]. Various HSN2 mutations have been reported from patients with different ethnicity causing HSAN2 [1, 6, 14, 15]. Herein, we report a novel nonsense mutation in WNK1/HSN2 associated with the clinical presentations of HSAN2 in four siblings of an Iranian family.
Subjects and methods
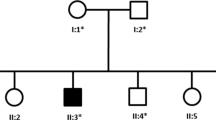
An HSAN2 pedigree with four affected and one unaffected sibling was recruited (Fig. 1a). The HSAN2 diagnosis was made based on clinical criteria, the results of electrophysiological evaluations, audiometry and tympanometry tests and laboratory assessments. The clinical features of the patients were recorded and summarized in Table 1.

The pedigree and clinical appearance of the studied family. a Iranian HSAN2A pedigree with a mutation in WNK1/HSN2 gene. Genotypes of studied individuals are presented. Filled circles and squares, affected individuals; unfilled circles and squares, unaffected members; Arrow shows proband. M, mutated allele; N, normal allele; B, C. Chronic ulcers as well as the amputated and mutilated sites on upper and lower extremities of the HSAN2-IV: 11 (b) and HSAN2-IV: 12 (c). D, E. Fingers deformity and Charcot joint in the left foot of the patients HSAN2-IV: 13 (d) and HSAN2-IV: 14 (e)
Genetics evaluations
According to the disease inheritance pattern, patients’ clinical history, and presentations, which complied with HSAN2, we initiated the molecular genetics evaluations with focused effort on this disease. Genomic DNA of all family members was extracted from blood samples using salting out protocol. The DNA fragments of genes WNK1, KIF1A, FAM134B, and SCN9A in which the prior mutations have been reported to be responsible for HSAN types 2 were respectively amplified by polymerase chain reaction (PCR). The amplicons were sequenced by Sanger method (Applied Biosystems, Foster City, CA). Afterwards, the extracted data were compared with relevant human reference sequences accessible in NCBI.
Whole exome sequencing
Whole exome sequencing (WES), Illumina HiSeq®2000 system (Illumina) was performed on the DNAs of two affected individuals HSAN- III:6 and HSAN- IV:13. Sequence alignment and variant calling were performed against human reference genome UCSC NCBI37/hg19. WANNOVAR (http://wannovar.wglab.org/) and ENSEMBL (http://asia.ensembl.org) are used to annotate the functional consequences of genetic variations. Based on the autosomal recessive inheritance of the disease and consanguinity of the parents, all heterozygous variations in father (HSAN- III:6) and homozygous variations in the proband (HSAN- IV:13) were selected. Subsequently, with the assumption that HSAN2 is a rare disease, SNPs with a reported minimal allele frequency of > 0.01 in the dbSNP database (http://www.ncbi.nlm.nih.gov/), the 1000 Genome project (http://www.internationalgenome.org/), or the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), GnomAD (http://gnomad.broadinstitute.org/) and Iranome (http://www.iranome.com/) were removed. In the next step, variants did not affect splicing, or amino acid change (e.g. synonymous, 3UTR, 5UTR variations) were filtered out which was followed by the removal of variations with mild or moderated effect (Fig. 2).

Flow diagram of WES data filtering process
Mutation screening
Using specific primers (fwd: 5’-ATTTCCCAGCGGCGTAAG-3′ and rev: 5’-CATTGAGACGTCAGAGCCA-3′), 984 bp of the HSN2 exon of WNK1 containing the candidate variation -c.3718C > A- was amplified, subsequently genotyped by Sanger sequencing (Applied Biosystems, Foster City, CA). WNK1 reference sequences used were NC_000012.12, NM_001184985, and NP_ 001171914.
Results
Clinical evaluations
HSAN2- iv: 11
A 36-year-old girl with below knee-amputated limbs and the mutilation of all distal phalanges of her upper extremities. She reported consecutive ulcers on the lower extremities from the age of six months old. Following deep ulcer infections and osteomyelitis occurrence, below knee amputations, were preceded on the left at age nine years old and on the right 13 years later (Fig. 1b).
HSAN2-iv: 12
A 32-year-old male subject, whose limb ulcerations and infections were initiated in the second year of his life. Bilateral transtibial amputations were done when he was 12 years old. The finger mutilation of his hands is shown in Fig. 1c.
HSAN2-IV: 13 and HSAN2-IV: 14
The third and fourth daughters of the family are aged 30 and 27 years old. They presented the first symptoms of consecutive limb ulcerations and infections, in seven and 10 years old, respectively. Over the time, the finger mutilation and deformity of upper and lower limbs occurred with prominent Charcot neuropathic arthropathies in their left feet (Fig. 1d and e). After profound infections around the ages between 10 and 15, the left third toe of HSAN2-IV: 13, the right second toe of HSAN2-IV: 14, and in both cases, the fifth finger of left lower extremities were amputated.
In all patients, there is a history of non-progressive symmetrical reduced multimodal sensory function in distal areas of upper and lower extremities. In the clinical examinations, pain, temperature, vibration, position, pressure, fine and crude touch perception had decreased in distal half of lower limbs in cases IV:11 and IV:12 as well as in distal one-third of lower extremities in cases IV:13 and IV:14, with the distal end of upper limbs in all patients. The sensory impairment was more profound in pain, touch and temperature sensation with more severity in cases IV: 11 and IV: 12. Moreover, deep tendon reflexes were diminished in all patients. The clinical examinations of motor neurons, autonomic systems and cranial nerves, had no abnormal findings. All patients were in normal cognitive status. They had normal tickling sensation and sexual activity. Skin exam showed hyperkeratosis in the limbs and the lingual fungiform papillae were observed in the oral examination. Besides, there was history of neither hypertension nor recurrent fever episodes in the family (Table 1).
Electrophysiological evaluations including sensory and motor conduction studies, F wave and H reflex analysis, electromyography and the sympathetic skin response test which have been done in cases IV: 13, 14 revealed symmetric peripheral sensory neuropathy, axonal type, with the impaired skin sympathetic response. The audiometry and tympanometry tests confirmed normal hearing. Laboratory assessments of fasting blood sugar (FBS), liver function tests (LFT), electrolytes, blood urea nitrogen, creatinine, and urine analysis were normal in all patients.
Genetics evaluations
Sanger sequencing of PCR products containing previously mutations in WNK1, KIF1A, FAM134B, and SCN9A genes showed wild type alleles; hence, we decided to perform the WES to have a better outline of genetic variations. According to the filtering criteria of the WES data a novel candidate variation, c.3718C > A in WNK1/HSN2 gene that causes p.Tyr1025* was identified. This variation results in a truncated protein with 1025 amino acids instead of the wild-type product with 2645 amino acids; p.Tyr1025* (Fig. 3a) and segregated with disease status in the pedigree. All affected members were in the homozygous state while unaffected parents and sibling were in the heterozygous state (Fig. 3b). Also, c.3718C > A was not observed in the Iranome (http://www.iranome.ir/) and other public databases. Given that, variations in this gene have been reported as a cause of HSAN2A, so, it was assessed as the likely cause of the neurologic disorder in this family.

a The schematic diagram of WNK/HSN2 gene. The identified nonsense mutation c.3718C > A in HSN2 exon of WNK1 transcript alters the tyrosine codon TAC to the stop codon TAA which leads to a truncated protein. b Direct sequencing of the HSN2 amplicon containing the mutation in all the members of the affected family. Healthy subjects are in heterozygous status (III:5, III:6, and IV:10) but affected members (IV:11, IV:12, IV:13, and IV:14) are homozygous for this mutation
Discussion
With-no-lysine (K) kinase 1 (WNK1), lysine deficient protein kinase 1, a gene located on 12p13.33, consists of 28 coding exons and encodes multiple isoforms of serine/threonine-protein kinase WNK1. WNK1 proteins regulate the activity of other proteins by attaching the phosphate group to specific positions. Due to the protein kinase properties, WNK1 isoforms can control the fluxes of sodium and chloride ions and have been considered as one of the primary regulators of blood pressure. Accordingly, several mutations resulted in WNK1 over-expression have been reported as the causative mutations of an autosomal dominant disorder, pseudohypoaldosteronism type II, which is characterized by hypertension, hyperkalemia, and renal tubular acidosis [8, 16]. In summary, it has been suggested that WNK1 proteins in cooperation with the other WNK kinases and with mediation of kinases SPAK and OSR1 have role in renal epithelial transport, maintaining cell volume homeostasis, GABA signaling, immune function and cell migration through the effect on cation-Cl− co-transporters [17].
HSN2 is an alternatively spliced exon in WNK1 providing the nervous system specific isoform of WNK1 transcript, namely WNK1/HSN2 [14]. It has been hypothesized that WNK1/HSN2 product involves the control of neuronal ion transportation and affect membrane excitability. Furthermore, several lines of studies have been shown this protein regulates neurite extension via its effect on Nogo signaling during neuronal differentiation [18, 19]. To date, 20 homozygous mutations in WNK1/HSN2 have been reported and almost all are in the HSN2 exon leading to truncating and loss-of-function [15] and WNK1/HSN2 protein leads to HSAN2A disease [6, 20,21,22,23]. Analyzing the results of genetics evaluations including Sanger sequencing of previously reported mutations as well as exome sequencing showed that none of the previously reported variations for HSAN2 were present in this family, but a novel nonsense mutation c.3718C > A in HSN2 exon of WNK1 transcript was found that resulted in alteration of the tyrosine codon TAC to the stop codon TAA and a truncated protein with 1025 amino acids. This result has been confirmed by subsequent segregation analysis and introduced c.3718C > A as a novel mutation for HSAN2A.
Clinical symptoms of the prior reported HSAN2A patients characterized within first two decades of life with predominant sensory polyneuropathy manifested most profound distally in a glove-stocking distribution. Sensory neuropathy was reported in lower limbs more severely than the upper limbs, although the trunk was involving in some patients. In addition, the previously reported patients presented with decreased or absent of DTRs, limb ulceration and mutilation with or without muscle weakness and variable autonomic disturbances [6, 15, 21, 23]. Our studied patients’ clinical characteristics include reduced perception to pain, touch, sense of temperature, and vibration and position sensation in distal areas of upper and lower extremities with more severity in the lower limbs. They express reduced DTRs, limb mutilation and amputation, with unremarkable autonomic clinical examination without motor dysfunction (Table 1) which are consistent to the clinical presentations of previously reported data in patients with HSAN2A [8].
Clinical presentations, inheritance pattern, and genetic analyses in our reported patients confirmed the HSAN2A diagnosis by identification of a novel mutation. It is worth mentioning there are several reports of HSAN2 patients from Iran but none of them conducted genetic investigations [24,25,26,27]; thus the current study is the first report of HSAN2A in Iranian patients with complementary genetic analyses. Detection of this new mutation in HSN2 presents allelic heterogeneity of this gene in different populations.
Consistent with previously reported mutations in HSN2 exon, the novel mutation identified in the current Iranian HSAN2 pedigree also leads to a truncating loss-of-function mutation. Therefore, the result of present study expands the spectrum of mutations of the HSN2 gene as the genetic background of HSAN2A as well as further support the hypothesis that HSN2 is a causative gene for HSAN2A. However, more research is required to determine the exact effects of this mutation in the nervous system. Moreover, the relevance of WNK1/HSN2 transcript and its pathogenic mutations to HSAN needs more deciphering.
References
Rotthier A, Baets J, Vriendt ED, Jacobs A, Auer-Grumbach M, Lévy N, et al. Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain. 2009;132:2699–711.
Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM. Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg psychiatry. NIH public. Access. 2012;83:119–20.
Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179–81.
Axelrod FB, Kaufmann H. Hereditary sensory and autonomic neuropathies. Neuromuscul Disord Infancy, Childhood: Adolesc A Clin Approach; 2014. p. 340–52
Ohta M, Ellefson R, Lambert E. Hereditary sensory neuropathy, type II: clinical, electrophysiologic, histologic, and biochemical studies of a Quebec kinship. Arch. 1973.
Lafreniere RG, MacDonald MLE, Dube M-P, MacFarlane J, O’Driscoll M, Brais B, et al. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the study of Canadian genetic isolates. Am J Hum Genet. 2004;74:1064–73.
Bercier V, Brustein E, Liao M, Dion PA, Lafrenière RG, Rouleau GA, et al. WNK1/HSN2 mutation in human peripheral neuropathy deregulates KCC2 expression and posterior lateral line development in zebrafish (Danio rerio). PLoS Genet. 2013;9:e1003124.
Kurth I. In: Pagon RA, Adam MP, Al AHH, editors. Hereditary Sensory and Autonomic Neuropathy Type II. 2010 Nov 23 [Updated 2015 Feb 19]. Washington: GeneReviews®. Seattle Univ; 2015. p. 1993–2016.
Axelrod FB, Gold-von SG. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis. 2007;2:39.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11.
Roddier K, Thomas T, Marleau G, Gagnon A, Dicaire M, St-Denis A, et al. Two mutations in the HSN2 gene explain the high prevalence of HSAN2 in French Canadians. Neurology. 2005;64:1762-7.
Rivire JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011;89:219–301.
Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, et al. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. 2013;80:1641–9.
Shekarabi M, Girard N, Rivière JB, Dion P, Houle M, Toulouse A, et al. Mutations in the nervous system-specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest. 2008;118:2496–505.
Yuan J-H, Hashiguchi A, Yoshimura A, Sakai N, Takahashi MP, Ueda T, et al. WNK1/HSN2 founder mutation in patients with hereditary sensory and autonomic neuropathy: a Japanese cohort study. Clin Genet. 2017;92:659–63.
Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Willams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science (80- ). 2001;293:1107–12.
Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017;25:285–99.
Zhang ZH, Li JJ, Wang QJ, Zhao WQ, Hong J, Jie LS, et al. WNK1 is involved in Nogo66 inhibition of OPC differentiation. Mol Cell Neurosci. 2015;65:135–42.
Krueger EM, Miranpuri GS, Resnick DK. Emerging role of WNK1 in pathologic central nervous system signaling. Ann. Neurosci. 2011;18:70–5.
Cho H-J, Kim BJ, Suh Y-L, An J-Y, Ki C-S. Novel mutation in the HSN2 gene in a Korean patient with hereditary sensory and autonomic neuropathy type 2. J Hum Genet. 2006;51:905–8.
Coen K, Pareyson D, Auer-Grumbach M, Buyse G, Goemans N, Claeys KG, et al. Novel mutations in the HSN2 gene causing hereditary sensory and autonomic neuropathy type II. Neurology. 2006;66:748–51.
Rivière JB, Verlaan DJ, Shekarabi M, Lafrenière RG, Bénard M, Der Kaloustian VM, et al. A mutation in the HSN2 gene causes sensory neuropathy type II in a lebanese family. Ann Neurol. 2004;56:572–5.
Pacheco-Cuellar G, González-Huerta LM, Valdés-Miranda JM, Peláez-González H, Zenteno-Bacheron S, Cazarin-Barrientos J, et al. Hereditary sensory and autonomic neuropathy II due to novel mutation in the HSN2 gene in Mexican families. J Neurol. 2011;258:1890–2.
Eslami H, Fakhrzadeh V, Golizadeh N. Synchronized occurrence of two rare genetic disorders: Amelogenesis imperfect and hereditary sensory autonomic neuropathy type II. Int J Curr Res Aca Rev. 2014;2:16–20.
Aghaei S, Journal KP-D. Online, 2006 U. three cousins with chronic foot ulcers resulting from late-onset hereditary sensory and autonomic neuropathies type 2 (HSAN2). Dermatol Online J. 2006;12:5.
Gharagozlou M, Zandieh F, Tabatabaei P, Zamani G. Congenital sensory neuropathy as a differential diagnosis for phagocytic immunodeficiency. Iran J Allergy, Asthma Immunol. 2006;5:35–7.
Eslamian F, Soleymanpour J. Case report: two case reports of hereditary sensory autonomic neuropathy type II in a family. Med J Tabriz Uni Med Sci. 2009;31:91–4.
Acknowledgements
The authors thank the family members for participation in this study. We would like to thank Ms. Fatemeh Sadat Rashidi for the technical support.
Funding
This research was supported by a grant from Neuroscience Research Center, Shahid Beheshti University of Medical Sciences, Iran.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
BR contributed in conception and design of the study, literature search, experimental studies, and drafting the manuscript; FF contributed in clinical data acquisition, manuscript editing and review. KA contributed in clinical data acquisition, manuscript editing and review; TA contributed in clinical data acquisition, manuscript editing and review; A Alavi contributed in exome sequencing analysis, manuscript editing and review, A Ahmadiani contributed in conception and design of the study, manuscript editing and review; SA contributed in conception and design of the study, literature search, experimental studies, manuscript editing and review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was performed in accordance with the Declaration of Helsinki and with the approval of Research Ethics Committee of Neuroscience Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran (IR.SBMU.PHNS.REC.1395.122). Participants gave written consent to participate after being informed of the nature of the research.
Consent for publication
Written informed consent was obtained directly from all members of the studied family for publication of personal and clinical information in this report and any accompanying images at the same time.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rahmani, B., Fekrmandi, F., Ahadi, K. et al. A novel nonsense mutation in WNK1/HSN2 associated with sensory neuropathy and limb destruction in four siblings of a large Iranian pedigree. BMC Neurol 18, 195 (2018). https://doi.org/10.1186/s12883-018-1201-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-018-1201-6