
Abstract
Background
Distal hereditary motor neuropathy (dHMN) is a heterogeneous group of hereditary diseases caused by the gradual degeneration of the lower motor neuron. More than 30 genes associated with dHMN have been reported, while 70–80% of those with the condition are still unable to receive a genetic diagnosis.
Methods
A 26-year-old man experiencing gradual weakness in his lower limbs was referred to our hospital, and data on clinical features, laboratory tests, and electrophysiological tests were collected. To identify the disease-causing mutation, we conducted whole exome sequencing (WES) and then validated it through Sanger sequencing for the proband and his parents. Silico analysis was performed to predict the pathogenesis of the identified mutations. A literature review of all reported mutations of the related gene for the disease was performed.
Results
The patient presented with dHMN phenotype harboring a novel homozygous variant c.361G > C (p.Ala121Pro) in SORD, inherited from his parents, respectively. A121 is a highly conserved site and the mutation was categorized as “likely pathogenic” according to the criteria and guidelines of the American College of Medical Genetics and Genomics (ACMG). A total of 13 published articles including 101 patients reported 18 SORD variants. Almost all described cases have the homozygous deletion variant c.757delG (p.A253Qfs*27) or compound heterozygous state of a combination of c.757delG (p.A253Qfs*27) with another variant. The variant c.361G > C (p.Ala121Pro) detected in our patient was the second homozygous variant in SORD-associated hereditary neuropathy.
Conclusion
One novel homozygous variant c.361G > C (p.Ala121Pro) in SORD was identified in a Chinese patient with dHMN phenotype, which expands the mutation spectrum of SORD-associated hereditary neuropathy and underscores the significance of screening for SORD variants in patients with undiagnosed hereditary neuropathy patients.
Similar content being viewed by others

Introduction
Distal hereditary motor neuropathy (dHMN) is a heterogeneous group of hereditary diseases caused by the gradual degeneration of the lower motor neurons without notable sensory involvement [1]. The typical phenotype of dHMN is muscle weakness and atrophy, which starts in the distal lower limbs in childhood or adulthood [2]. The axonal type of Charcot-Marie-Tooth disease type 2 (CMT2) is characterized by limited sensory neuropathy, and dHMN sometimes share similar clinical features. Additionally, it overlaps with CMT2 in causative genes, including GARS, IGHMBP2, AARS, DYNC1H1, and HARS [3,4,5]. More than 30 genes associated with dHMN have been reported [6], while 70–80% of those with the condition are still unable to receive a genetic diagnosis [7]. In recent years, mutations in the sorbitol dehydrogenase (SORD) gene were found to be a prevalent cause of recessive dHMN, responsible for about 10% of undiagnosed cases of dHMN and CMT2 [8]. In this study, we reported a novel homozygous mutation c.361G > C (p.Ala121Pro) of SORD in a Chinese patient presented with dHMN and evaluated detailed clinical and electromyographic data. At last, a literature review of all reported variants of the SORD gene for the disease was performed.
Materials and methods
Subject
The proband, a 26-year-old man from Mianyang City in Sichuan Province, Southwest of China, was referred to our hospital because of progressive weakness in the lower limbs. The clinical presentation, laboratory data, and electrophysiological tests were collected.
Whole exome sequencing
Peripheral blood samples were collected from the patient and his parents, and genomic DNA was extracted by using a standard kit (Qiagen, Germany). At first, the 17p11.2 copy number variation was screened. Subsequently, whole-exome sequencing (WES) was conducted by using the Agilent SureSelectTM Human All Exome V6 kit (Agilent Technologies Inc, Canada) on an Illumina HiSeq2500 (Illumina, San Diego, CA, USA), and the variants were filtered (≤ 1%) according to their frequency in reference population databases.
Bioinformatics analysis
The variants were annotated by multiple databases, including HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) [9], OMIM (https://www.omim.org/) [10], gnomAD (http://gnomad.broadinstitute.org/) [11], ExAC (http://exac.broadinstitute.org/) [12], and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) [13], and variants with an allele frequency ≤ 1% were selected. To predict the potential pathogenicity of the variant, several in silico pathogenicity prediction tools, including PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) [14], NetGene2 (https://services.healthtech.dtu.dk/service.php?NetGene2-2.42) [15], and BDGP (http://www.fruitfly.org/seq_tools/splice.html) [16] were used. To predict the structural changes in protein by substitution of an amino acid, the Missense3D tool was used. (http://missense3d.bc.ic.ac.uk/~missense3d/) [17]. Finally, candidate variants were interpreted according to the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015) [18].
Sanger sequencing
Sanger sequencing was performed using an ABI 3730XL DNA analyzer (Applied Biosystems, Waltham, MA) following the manufacturer’s protocol to validate the filtered variants detected by WES and for segregation analysis.
Review of the literature
We reviewed all the previously reported cases of CMT or dHMN associated with SORD mutations, with particular emphasis on clinical, electrophysiologic, pathologic, and genetic data. The search strategy included the following: PubMed search with “SORD,” “Charcot-Marie-Tooth” or “Distal hereditary motor neuropathies” used as keywords for articles.
Results
Clinical information
The male proband developed progressive weakness and atrophy in the lower limbs at the age of 11 and had a duration of 15 years. He had normal developmental history. He also complained of numbness besides weakness. His parents were not consanguineous and did not have any neurological disorders. A neurological examination was performed on the patient in May 2022, when he was 26 years old. The examination revealed bilateral distal weakness, in the upper limbs (graded as 4+) and lower limbs (graded as 4). Apparent muscle atrophy of lower limbs and pes cavus were noticed. Deep tendon reflexes showed the brisk knee jerk and absence of ankle reflex and Babinski’s sign. The pinprick and temperature sensation were normal. Mild fasciculations were observed in the thighs.
Laboratory tests revealed that blood routine, routine urine, liver and kidney function, and creatine kinase were normal. There were no abnormalities in fasting blood glucose, glycosylated hemoglobin, thyroid function, antibodies, tumor markers, ANA/ENA/ANCA antibody profiles, vitamin B12, and intrinsic factor antibodies. Hepatitis B markers, HIV, and TPPA, were negative. Head MRI showed no remarkable abnormalities. Nerve conduction studies showed reduced compound muscle action potential (CMAP) amplitude in both lower limbs. Motor and sensory nerve conduction velocity and sensory nerve action potentials were in the normal range. Needle electromyography revealed chronic denervation in the proximal and distal muscles (Table 1).
Genetic data
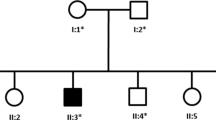
Whole exome sequencing of the proband detected the homozygous variant c.361G > C (p.Ala121Pro) in the SORD gene. The variant is a missense mutation in the coding region of the SORD gene (NM_003104.6), which was not reported in the 1000 Genomes Project, Exome Aggregation Consortium (ExAC), and the Genome Aggregation Database (gnomAD). The pedigree of the family is shown in Fig. 1. Sanger sequencing and co-segregation were further conducted on the family, including the proband and unaffected parents (Fig. 2). Conserved sequence analysis suggested the variant is located in a highly conserved sequences across various mammalian species (Fig. 3). The variant c.361G > C was predicted to be possibly damaging with a score of 0.822 by PolyPhen-2. The MutationTaster was used to predict the pathogenicity of the mutation, and it showed that the variation was disease-causing, with a PhyloP score of 5.277 and a PhastCons score of 1. The impact of the p.Ala121Pro mutation on 3D structure of the protein was detected by the Missense3D tool, which showed structural damage of buried proline. The buried proline was predicated to lead to secondary structure changes. The protein structure of SORD wild-type and mutant-type were constructed by the Missense 3D tool (Fig. 4). As a result, the variant was categorized as “likely pathogenic” according to the criteria and guidelines of the American College of Medical Genetics and Genomics (ACMG) (PM1, PM2, PP3, PP4).

Pedigree of the Chinese family with dHMN. The arrow indicates the proband

Chromatograms of the SORD gene. The patient carried the homozygous c.361G > C mutation and his parents harbored the heterozygous c.361G > C mutation (arrows)

Analysis of conserved sequences showed a total of 75 amino acids surrounding the mutation position (marked with a black box)

Three-dimensional(3D) structures of the SORD wild-type protein (A) and the mutant protein (B). The blue represents the ALA residue. The red represents the Pro residue
Here, we also summarized all the SORD mutations reported in previous literatures (Table 2). A total of 101cases with 18 mutations were reported [4, 8, 19,20,21,22,23,24,25,26,27,28,29]. Among these, 73 patients carried the homozygous deletion variant c.757delG (p.A253Qfs*27), 27 cases in a compound heterozygous state of combination of c.757delG with another variant, one patient harboring a compound heterozygous variants of c.404 A > G and c.908 + 1G > C. Most of the variants in SORD are frameshift or splicing variants. Surprisingly, a case of a 24-year-old man diagnosed with juvenile amyotrophic lateral sclerosis (JALS) was documented. The patient was found to carry the homozygous c.757delG variant in the SORD gene [29]. The novel homozygous variant c.361G > C (p.Ala121Pro) detected in our patient was the second homozygous variant identified in the SORD gene related dHMN.
Discussion
Even after conducting screenings for all known genetic mutations, a significant number of patients with hereditary neuropathies remain without a confirmed genetic diagnosis. In 2020, Cortese et al. reported that SORD was identified as a novel causative gene of recessive forms of dHMN and CMT2, with an estimated frequency of up to ∼ 10% in undiagnosed dHMN and CMT2 cases [8]. Our systemic literature review found 18 mutations from 101 affected individuals among different populations were identified to carry biallelic variants in SORD. Almost all described patients had the homozygous deletion variant c.757delG (p.A253Qfs*27) or a compound heterozygous state of a combination of c.757delG with another variant, besides one patient harboring a compound heterozygous for c.404 A > G and c.908 + 1G > C [19]. In the present study, the novel homozygous mutation c.361G > C (p.Ala121Pro) of SORD identified in our patient with dHMN was the second homozygous variant in SORD gene related dHMN.
The clinical manifestation of the patient was childhood-onset and symmetrical distal peripheral neuropathy. Physical examination showed brisk knee jerk and mild fasciculations in the thighs. Electrophysiological studies, including MNC, SNC, and EMG, revealed reduced compound muscle action potential (CMAP) amplitude in both lower limbs, normal sensory conductions, and chronic denervation in the upper and lower limbs. The characteristics of SORD-related CMT have been typically described as a length-dependent neuropathy, characterized by distal hereditary motor neuropathy (dHMN) or a predominantly motor presentation resembling CMT2. Unusual features were also observed, including mild hearing loss, dermographism, brisk reflexes, denervation, conduction block, and small fiber impairment on EMG [8, 26]. This may be attributed to diabetic mononeuropathy caused by high sorbitol levels involved in the pathogenesis.
SORD encodes a 357 amino acid protein and is widely expressed in mammalian tissues. SORD protein is sorbitol dehydrogenase involved in a two-step of polyol pathway. The initial step is the conversion of glucose to sorbitol by aldose reductase, followed by sorbitol dehydrogenase catalyzes sorbitol into fructose, which has been implicated in the pathophysiology of preclinical models of diabetic neuropathy [30, 31]. In the present study, a homozygous variant c.361G > C (p.Ala121Pro) of SORD gene were identified in a dHMN patient using whole exome sequencing. In silico pathogenicity prediction tools revealed that the mutation was categorized as “likely pathogenic” according to the criteria and guidelines of ACMG. Previous study found that patients with SORD mutations exhibited elevated fasting serum sorbitol levels, which resulted from decreased SORD enzyme levels and impaired enzyme function. The absence of SORD orthologues in Drosophila caused synaptic degeneration and motor impairment. It is suggested that loss-of-function of the enzyme and subsequent sorbitol accumulation was the mechanism of SORD-associated neuropathy [8]. A previous study demonstrated that aldose reductase inhibitors effectively reduced sorbitol accumulation in mouse models of diabetes and in humans [31]. This represents a promising therapeutic intervention for SORD-associated patients.
In conclusion, we reported one novel homozygous variant c.361G > C (p.Ala121Pro) of SORD identified in a Chinese patient with dHMN phenotype. This study expands the clinical and mutational spectrum of SORD-associated hereditary neuropathy. Further, these findings provide support for SORD being a disease-causing gene in dHMN, underscoring the significance of screening for SORD variants in patients with undiagnosed hereditary neuropathy patients. Enzyme therapy of the sorbitol metabolic pathway may be a promising therapeutic intervention for SORD-related neuropathy in the future.
Data availability
The datasets analyzed during the current study are available in the ClinVar database (https://submit.ncbi.nlm.nih.gov/subs/variation_clinvar/SUB14261784/.
Submission ID: SUB14261784).
References
Frasquet M, Sevilla T. Hereditary motor neuropathies. Curr Opin Neurol. 2022;35(5):562–70.
2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II) and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT) 26–28 September 1997, Naarden, The Netherlands. Neuromuscular disorders: NMD. 1998;8(6):426–431.
Becker LL, Dafsari HS, Schallner J, Abdin D, Seifert M, Petit F, Smol T, Bok L, Rodan L, Krapels I, et al. The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification. J Hum Genet. 2020;65(11):1003–17.
Xie Y, Lin Z, Pakhrin PS, Li X, Wang B, Liu L, Huang S, Zhao H, Cao W, Hu Z, et al. Genetic and clinical features in 24 Chinese distal Hereditary Motor Neuropathy families. Front Neurol. 2020;11:603003.
Liu X, Duan X, Zhang Y, Sun A, Fan D. Molecular analysis and clinical diversity of distal hereditary motor neuropathy. Eur J Neurol. 2020;27(7):1319–26.
Bansagi B, Griffin H, Whittaker RG, Antoniadi T, Evangelista T, Miller J, Greenslade M, Forester N, Duff J, Bradshaw A, et al. Genetic heterogeneity of motor neuropathies. Neurology. 2017;88(13):1226–34.
Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6–14.
Cortese A, Zhu Y, Rebelo AP, Negri S, Courel S, Abreu L, Bacon CJ, Bai Y, Bis-Brewer DM, Bugiardini E, et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet. 2020;52(5):473–81.
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–81.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43(Database issue):D789–798.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, Hoffman D, Jang W, Kaur K, Liu C, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835–44.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics 2013;Chap. 7:Unit7.20.
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24(17):3439–52.
Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. The Berkeley Drosophila Genome Project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics. 1999;153(1):135–77.
Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJE. Can predicted protein 3D structures provide Reliable insights into whether missense variants are Disease Associated? J Mol Biol. 2019;431(11):2197–212.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Medicine: Official J Am Coll Med Genet. 2015;17(5):405–24.
Dong HL, Li JQ, Liu GL, Yu H, Wu ZY. Biallelic SORD pathogenic variants cause Chinese patients with distal hereditary motor neuropathy. NPJ Genomic Med. 2021;6(1):1.
Frasquet M, Rojas-García R, Argente-Escrig H, Vázquez-Costa JF, Muelas N, Vílchez JJ, Sivera R, Millet E, Barreiro M, Díaz-Manera J, et al. Distal hereditary motor neuropathies: mutation spectrum and genotype-phenotype correlation. Eur J Neurol. 2021;28(4):1334–43.
Laššuthová P, Mazanec R, Staněk D, Sedláčková L, Plevová B, Haberlová J, Seeman P. Biallelic variants in the SORD gene are one of the most common causes of hereditary neuropathy among Czech patients. Sci Rep. 2021;11(1):8443.
Liu X, He J, Yilihamu M, Duan X, Fan D. Clinical and genetic features of biallelic mutations in SORD in a series of Chinese patients with Charcot-Marie-tooth and distal Hereditary Motor Neuropathy. Front Neurol. 2021;12:733926.
Yuan RY, Ye ZL, Zhang XR, Xu LQ, He J. Evaluation of SORD mutations as a novel cause of Charcot-Marie-tooth disease. Ann Clin Transl Neurol. 2021;8(1):266–70.
Alluqmani M, Basit S. Association of SORD mutation with autosomal recessive asymmetric distal hereditary motor neuropathy. BMC Med Genom. 2022;15(1):88.
Grosz BR, Stevanovski I, Negri S, Ellis M, Barnes S, Reddel S, Vucic S, Nicholson GA, Cortese A, Kumar KR, et al. Long read sequencing overcomes challenges in the diagnosis of SORD neuropathy. J Peripheral Nerv System: JPNS. 2022;27(2):120–6.
Record CJ, Pipis M, Blake J, Curro R, Lunn MP, Rossor AM, Laura M, Cortese A, Reilly MM. Unusual upper limb features in SORD neuropathy. J Peripheral Nerv System: JPNS. 2022;27(2):175–7.
Wu C, Xiang H, Chen R, Zheng Y, Zhu M, Chen S, Yu Y, Peng Y, Yu Y, Deng J, et al. Genetic spectrum in a cohort of patients with distal hereditary motor neuropathy. Ann Clin Transl Neurol. 2022;9(5):633–43.
Chen B, Zhang Z, Zhang C, Niu S, Pan H, Dong G, Wang X. Clinical and pathological study of SORD-related distal motor neuropathy caused by novel compound heterozygous mutations in a Chinese patient. Clin Neurol Neurosurg. 2022;213:107118.
Bernard E, Pegat A, Vallet AE, Leblanc P, Lumbroso S, Mouzat K, Latour P. Juvenile amyotrophic lateral sclerosis associated with biallelic c.757delG mutation of sorbitol dehydrogenase gene. Amyotroph Lateral Scler Frontotemporal Degeneration. 2022;23(5–6):473–5.
Ng TF, Lee FK, Song ZT, Calcutt NA, Lee AY, Chung SS, Chung SK. Effects of sorbitol dehydrogenase deficiency on nerve conduction in experimental diabetic mice. Diabetes. 1998;47(6):961–6.
Grewal AS, Bhardwaj S, Pandita D, Lather V, Sekhon BS. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and non-diabetic diseases. Mini Rev Med Chem. 2016;16(2):120–62.
Acknowledgements
The authors thank the patient and his family members for their participation in the study.
Funding
The study thank the financial support from Mianyang Central Hospital and Sichuan Medical Association (No. Q17045).
Author information
Authors and Affiliations
Contributions
Data analysis and writing original draft: XQ Y. Project administration: SS Z. Manuscript review and editing: HF S. Supervision: YF Tang.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Mianyang Central Hospital, School of Medicine, University of Electronic Science and Technology of China. Written informed consents were obtained from all the individuals included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, X., Zhang, S., Shang, H. et al. A novel mutation in SORD gene associated with distal hereditary motor neuropathies. BMC Med Genomics 17, 169 (2024). https://doi.org/10.1186/s12920-024-01940-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01940-5