
Abstract
Real world evidence is now accepted by authorities charged with assessing the benefits and harms of new therapies. Clinical trials based on real world evidence are much less expensive than randomized clinical trials that do not rely on “real world evidence” such as contained in electronic health records (EHR). Consequently, we can expect an increase in the number of reports of these types of trials, which we identify here as ‘EHR-sourced trials.’ ‘In this selected literature review, we discuss the various designs and the ethical issues they raise. EHR-sourced trials have the potential to improve/increase common data elements and other aspects of the EHR and related systems. Caution is advised, however, in drawing causal inferences about the relationships among EHR variables. Nevertheless, we anticipate that EHR-CTs will play a central role in answering research and regulatory questions.
Similar content being viewed by others
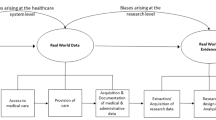


Introduction
Some who study randomized clinical trials broadly dichotomize them into explanatory (does this treatment work under ideal (or what Schwartz and Lellouch identified as “laboratory” conditions) [1] and efficacious (does this treatment work in the real world.
(what Schwartz and Lellouch identified as “normal” conditions) [2], while others prefer to see them as a continuum along this scale [1, 3, 4]. The ideal conditions needed for explanatory trials, however, are expensive, and do not allow inferences about generalizability of the findings. On the other hand, the normal conditions providing real world data are inexpensive, and do allow inferences about generalizability of the findings [5]. During the half-century since the ‘real world’ concept was introduced, 38 definitions of real-word data (RWD) have been offered with most approximating “data collected in a non-randomized controlled trial setting” [6].
Multiple forces are contributing to a rapid expansion of interest in real world data for clinical trials. These forces include the increasing availability of electronic data and the acceptability of real world evidence in support of applications for approval of medical products for marketing. We begin this selected review of the literature with a description of these recent developments driving the focus on this topic. We follow this with a discussion of the designs of studies intended to evaluate therapies and interventions using real world evidence, including the advantages and limitations of each, the inferences that can be drawn from study results, and issues related to recruiting/enrollment and ethics. We conclude with comments about the future.
Increasing availability of electronic data
The increasing availability of large integrated data sources [7,8,9] bodes well for clinical trials that rely on real world data, including the contents of the electronic health record. So does the availability of “learning health systems that use routine data from service delivery and patient care to generate knowledge to continuously improve healthcare” [10]. Groups of learning health systems are coming together to form networks, such as the nine Clinical Research Networks and the two Health Plan Research Networks that are part of Cornet [11]. The creation of an electronic health record data aggregation platform by one of the large vendors is likely to result in more and/or larger networks that will be able to function as integrated learning health systems [12].
More recently, the United States Centers for Medicare & Medicaid Services proposed rule (CMS 0057-P) is intended to advance interoperability among health information exchanges (entities such as hospitals, and public health agencies in states or regions that help share information among groups that have a legitimate need for it) [13]. To exchange data with health information exchanges, hospitals and networks of hospitals are expected to use application programming interfaces that can gather information from multiple sources and aggregate it in one place [14]. This would allow access specific pieces of information, rather than having to sort through pages of unnecessary or unhelpful records. The likely result is expected to be many more clinical trials based on real world data than in the recent past.
Government encouragement
The European Medicines Agency, the United States Food and Drug Administration, and Health Canada now accept real world evidence (RWE) in applications for approval of medical products for marketing [15].
The Food and Drug Administration defines RWD as “data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources” and defines RWE as “clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD,” regardless of the type of study design [16]. In the announced framework for its RWE program for drug and biological products, Food and Drug Administration acknowledged that clinical trials that use EHR data are acceptable, as are administrative claims data, or registry data [17],
The perceived need for transparency in applications to FDA prompted one group to create “The Structured Pre-Approval and post-approval Comparative study design framework to generate valid and transparent real-world Evidence (SPACE)” for identifying design elements, feasibility and validity concerns, and for documenting decisions [18]. An extension to SPACE, identified with another acronym, SPIFD for “Structured Process to Identify Fit-For-Purpose Data tool,” provides a guide to identify fit-for-purpose data required for the Food and Drug Administration’s (FDA) real-world-evidence (RWE) program [19].
Increasingly, routinely collected data are seen as an attractive source for post-marketing surveillance, complementing established spontaneous report mechanism [20, 21].
Here we focus on clinical trials that rely on data provided by EHRs, and what others have come to identify as "EHR-sourced" trials [22].
Designs: clinical effectiveness and implementation (Table 1)
EHR-sourced trials can be evaluated along a spectrum anchored at one end by those intended to assess only the clinical effectiveness of the intervention and anchored at the other end by those that assess only the adoption/uptake/acceptance of the intervention by groups of providers and institutions.
Increasingly, the word ‘hybrid’ is associated with EHR-sourced trials that try to evaluate (to varying degrees) both clinical effectiveness and acceptance of the intervention [23,24,25]. The three types of hybrid designs are:
-
(a)
Type 1: tests the clinical effectiveness of the intervention (reducing elevated blood pressure or blood glucose level) while observing and gathering information on the implementation,
-
(b)
Type 2: dual testing of clinical effectiveness and implementation interventions/strategies, and
-
(c)
Type 3: evaluates the adoption of an implementation strategy while observing and gathering information on its clinical effectiveness.
In the absence of obvious boundaries, some investigators avoid use of these labels (at least in the title). Only one report applied the term ‘continuum’ to the spectrum along the Type 1 to Type 3 classification [26]. More information about implementation strategies is available elsewhere [27, 28], as are examples of implementing self-management strategies for people with diabetes [29, 30], with asthma [31, 32], with hypertension [33, 34], and with epilepsy [35,36,37,38].
Justification for EHR based clinical trials
“The often stringent inclusion and exclusion criteria destined to provide for homogeneous study populations reduce the generalizability of randomized clinical trial results” [39]. Findings reported from observational studies, have, on important occasions, not been confirmed in randomized clinical trials [40,41,42,43,44,45,46,47,48,49,50,51,52,53]. This has been emphasized with the finding that in one-sixth of all comparisons between randomized clinical trials and observational studies, “there was a significant difference and the estimates pointed in opposite directions” [54].
The findings of EHR-sourced trials, especially those conducted in populations that are likely to benefit from the results of the study, tend to be more informative about what works in the real world than are the findings of explanatory trials (RCTs) [40, 55]. EHR-sourced trials are most suited to answer the question, “Will this intervention work in this population?” Investigators are encouraged to ‘embed’ randomization in learning health systems and networks [56].
Designs: randomized trials
The randomized trial design of an EHR-sourced trial can be identical to that of a randomized clinical trial in the most restrictive set of subjects. Two of the more common modifications of the conventional randomized clinical trial are conventional cluster randomized design and stepped-wedge cluster randomized design.
Cluster randomized trials compare the results of different interventions among groups of people whose members have an identifiable feature in common [57, 58]. They are most appropriate when.
-
the intervention evaluated is likely to be implemented subsequently among other patients who have the randomized groups’ characteristics.
-
the intervention carries a high risk of “contamination” (i.e., individuals randomized to different comparison groups are in frequent contact with one another and thus may be influenced (‘contaminated’) by recipients of the alternative treatment).
-
they have practical advantages over individual randomization (because of lower implementation costs, or administrative convenience).
Cluster randomized trials, however, pose potential ethical concerns (v.i., Ethics section).
In a stepped wedge design, an intervention, an intervention is rolled out sequentially to the trial participants (either as individuals or clusters of individuals) [59, 60]. The order in which the different individuals or clusters receive the intervention is determined at random and, by the end of the random allocation, all individuals or groups will have received the intervention.
Among the reasons investigators chose the stepped wedge design in preference to others are:
-
The stepped wedge design avoids the logistical barriers that accompany efforts to implement the intervention simultaneously in many clusters,
-
a lack of equipoise for the intervention made the investigators feel it would be unethical to deny the intervention to some groups,
-
a desire to avoid the ‘disappointment effects’ possible in a parallel trial that follow from colleagues at some clusters who decide to drop out of the study when randomised to the control arm,
-
higher statistical power associated with clusters functioning as their own controls
-
the ability to adjust for time trends in outcomes,.
-
logistical, practical, or financial constraints require that the intervention be implemented in stages [61,62,63].
In such circumstances, determining the order in which participants receive the intervention at random is likely to be both morally and politically acceptable and may also be beneficial for trial recruitment.
The step wedge design was chosen for a study that evaluated if the presence of a seizure dog in the home reduced seizure and injury frequency [64]. “This design was chosen because it allows for rollout of the intervention to all participants. … The current capacity of the assistance dog schools participating in the EPISODE study would not permit simultaneous rollout of the required number of seizure dogs to all participants.” In addition, “blinding of the participants would be impossible.”
Group sequential design
Some patients exposed to an intervention that might be a source of potential benefit or harm can be monitored sequentially for either of these possibilities [65]. Group sequential design, also known as interim analysis offers an opportunity to make decisions along the way about whether or not future patients should be so exposed [66, 67].
Inferences
Randomized clinical trials (RCTs) are considered the "gold standard" for evaluating the safety and efficacy of new therapeutic agents because of their high quality data and strict inclusion and exclusion criteria [68, 69]. In contrast, however, EHR-sourced trials are fraught with potential biases and limited quality of the data in the EHRs [70,71,72,73,74,75,76,77,78,79]. These biases and limitations include selection bias [80,81,82,83], protopathic bias, [84,85,86], missingness and other data quality limitations, [78, 87,88,89,90,91,92,93,94], time-orientation challenges [88, 95], and potential confounding [96, 97]. These biases and limitations have the potential to limit severely the causal inferences that can be drawn from the contents of EHRs [98,99,100,101,102,103].
Although efforts are underway to address some of them, structural limitations, such as the paucity of common data elements recommended for research purposes by the National Institutes of Health [104] will limit how much bias can be reduced soon [77, 91, 105,106,107,108,109,110,111,112,113]. Nevertheless, planned EHR-sourced trials of technological care advances and changes in practice have the potential to enhance data quality in the EHR by requiring participating organizations to include selected common data elements, or by providing other data details (i.e., granularity) not previously included in the EHR. Data quality is also likely to be improved by the apparently increasing use of home monitoring devices, wearable devices, templated smartforms for documentation, automated transcription, artificial intelligence and natural language processing (to extract salient information from the EHR) [114, 115].
The Cochrane risk-of-bias groups have created tools for assessing “risk of bias in non-randomized studies of interventions” (ROBINS-I) [116], assessing risk of bias in randomised trials (RoB 2) [117], and even in cluster-randomized trials [118], as well as assessing risk of missing evidence (ROB-ME) [119, 120].
Recruiting/enrollment
Learning healthcare systems, with their large collections of EHRs, are likely to include a broad patient population with characteristics as close as possible to patients in routine clinical practice, whose responses to interventions will maximize the generalizability and applicability of trial results [121]. Data about such patients can help design suitable EHR-sourced trials [122]. EHR query tools can identify potential candidates for these clinical trials when the selection criteria are expressed in structured digital format [123, 124]. Once identified, eligible subjects can be enrolled electronically, either by email or via the patient portal [125, 126], which is recommended because of its security [127]. Doing so, however, will not eliminate bias [127,128,129,130,131].
Because recruitment for a randomized clinical trial can take a long time, alternative arrangements have been sought. One option is the ‘trial within cohorts’ design, which uses the infrastructure of an observational cohort study to identify possible participants for a randomized trial [132]. Upon cohort enrollment, all participants provide consent for being randomized in future studies without being informed. When a new treatment becomes available for evaluation, those randomized to the treatment arm are offered the new treatment, which they can choose to refuse. Those randomized to the standard of care arm are not informed about the trial and continue to receive standard of care as part of the cohort study. Patients do not appear to have ethical objections to serve as control without further notice [133, 134].
Ethics
The main ethical issue associated with EHR-sourced trials is when and how consent is obtained. Of 1988 EHR-sourced trial reports published during the years 2014 to 2019, 7% did not include a statement about participant consent and only 7.0% reported a waiver [135]. Cluster randomization studies were more likely than others to obtain consent or provide details about consent.
The role of institutional review boards
Given their complex coordination across multiple sites, EHR-sourced trials, in general, pose a challenge for research oversight mechanisms, including ethical review by multiple institutional review boards [136]. The Common Rule regulations allow waiver of consent by an institutional review board when the research poses no more than minimal risk, doing so does not adversely affect the rights or welfare of the participant, and obtaining direct consent is impracticable [137]. When a waiver is granted, subjects can/should still be informed of the nature of the study and how it might affect them, and perhaps even offered an opt-out option [138]. Minimal-risk determinations are the provenance of each institutional review board [139], but considerations can/should include “clinical equipoise, practice variation, research methods such as cluster randomization, and patients' perspectives” [140]. Yes, patients’ perspective! In one survey, three quarters of subjects approved obtaining post-randomization the consent of only those assigned to the treatment arm [141].
Nevertheless, suggested options “to make sure” everyone is on board, include investigators consulting with the institutional review board, [142] obtaining written informed consent for all participants before randomization, [143] and opportunities to ‘opt-out’ [144,145,146]. More than two-thirds of a national sample of US adults “reported interest in notification of research using their identified health information, and 40% reported interest in notification if the health information was deidentified [147].
Cluster randomized studies
In cluster randomized trials the unit of randomization might be a hospital or network of hospitals that might ask physicians to treat all patients with disease X alike and collect data from patients. The benchmark ethical recommendations about who needs to consent are in “The Ottawa statement on the ethical design and conduct of cluster randomized trials” [148] and in the 2016 Council for International Organizations of Medical Sciences (CIOMS) ethical guidelines [149]. Additional guidance has come from the FDA [150] and from “a guide for the perplexed” [151].
Stepped-wedge cluster randomized studies
Stepped-wedge cluster randomized trial designs pose additional challenges [143]. In contrast to parallel cluster randomized trials in which clusters are randomized at the outset to either intervention or control arms, stepped-wedge cluster randomized trials offer the intervention sequentially to each cluster so that each cluster begins the trial as its own control, and then receives the intervention [59, 152, 153]. This approach is thought to avoid ethical concerns about the denial of a desired intervention to participants in control groups [154,155,156].
The Consolidated Standards of Reporting Trials has an extension for stepped-wedge cluster randomized trial that includes whether or not consent was obtained, the purpose of consent, when consent was sought (i.e., before or after randomization), and the forms of consent (differences between intervention and control clusters) [157]. A review of the stepped-wedge cluster randomized trials in the National Institutes of Health's Health Care Systems Research Collaboratory found that decisions to use the stepped-wedge cluster design were more often justified by practical and epistemic reasons than by ethical ones [143].
Limitations of this report
The main limitation of this report is that it is not a comprehensive overview of the literature. We specifically chose studies to show the breadth of the perspectives they offer about the designs most suitable for using EHR data, and to emphasize the data limitations, potential biases and other challenges that might hamper drawing causal inferences based on EHR contents.
The future
We see many phenomena contributing to large increases in the use of EHR data for clinical trials. First, the acceptance of real world evidence by authorities charged with assessing the benefits and harms of new therapies [158, 159], is highly likely to provide a strong impetus for EHR-sourced trials in preference to randomized clinical trials [160, 161]. Consequently, we can expect continued increase in the number of reports of EHR-sourced trials as well as efforts to have EHR data become “regulatory-grade) [69, 162].
Second, efforts to encourage an expanded set of common data elements for EHRs appear promising [105, 163, 164]. These efforts have the potential to lead to improvements have the potential to increase the validity of future EHR-sourced trials [165, 166].
Third, advances in artificial intelligence and machine learning bode well for significant improvement in the quality and quantity of information contained in the EHR [91, 115, 167, 168]. Although some will undoubtedly apply to natural language processing, we have little knowledge of the extraordinary achievements we can expect [169].
In light of these phenomena, EHR-sourced trials have a very bright future. Nevertheless, we are less optimistic that EHR data will be useful any time soon for providing valid information for drawing causal inferences about the relationships between EHR variables.
Conclusions
EHR-sourced trials have conceptual and logistic properties that make them especially attractive for future studies of what does and what does not work among those with the characteristics of the participants.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutical trials. J Chronic Dis. 1967;20(8):637–48.
Revicki DA, Frank L. Pharmacoeconomic evaluation in the real world. Effectiveness versus efficacy studies. Pharmacoeconomics. 1999;15(5):423–34.
Thorpe KE, Zwarenstein M, Oxman AD, Treweek S, Furberg CD, Altman DG, Tunis S, Bergel E, Harvey I, Magid DJ, et al. A pragmatic-explanatory continuum indicator summary (PRECIS): a tool to help trial designers. J Clin Epidemiol. 2009;62(5):464–75.
Zwarenstein M, Treweek S, Gagnier JJ, Altman DG, Tunis S, Haynes B, Oxman AD, Moher D. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ. 2008;337:a2390.
Weinfurt KP, Hernandez AF, Coronado GD, DeBar LL, Dember LM, Green BB, Heagerty PJ, Huang SS, James KT, Jarvik JG, et al. Pragmatic clinical trials embedded in healthcare systems: generalizable lessons from the NIH Collaboratory. BMC Med Res Methodol. 2017;17(1):144.
Makady A, de Boer A, Hillege H, Klungel O, Goettsch W. What is real-World Data? A review of definitions based on literature and stakeholder interviews. Value Health. 2017;20(7):858–65.
Friedman C, Rubin J, Brown J, Buntin M, Corn M, Etheredge L, Gunter C, Musen M, Platt R, Stead W, et al. Toward a science of learning systems: a research agenda for the high-functioning Learning Health System. J Am Med Inf Association. 2015;22(1):43–50.
Platt JE, Raj M, Wienroth M. An analysis of the Learning Health System in its first decade in practice: scoping review. J Med Internet Res. 2020;22(3):e17026.
Pomare C, Mahmoud Z, Vedovi A, Ellis LA, Knaggs G, Smith CL, Zurynski Y, Braithwaite J. Learning health systems: a review of key topic areas and bibliometric trends. Learn Health Syst. 2022;6(1):e10265.
Enticott J, Braaf S, Johnson A, Jones A, Teede HJ. Leaders' perspectives on learning health systems: a qualitative study. BMC Health Serv Res. 2020;20(1):1087.
Forrest CB, McTigue KM, Hernandez AF, Cohen LW, Cruz H, Haynes K, Kaushal R, Kho AN, Marsolo KA, Nair VP, et al. PCORnet(R) 2020: current state, accomplishments, and future directions. J Clin Epidemiol. 2021;129:60–7.
Tarabichi Y, Frees A, Honeywell S, Huang C, Naidech AM, Moore JH, Kaelber DC. The Cosmos Collaborative: a vendor-facilitated Electronic Health Record Data Aggregation platform. ACI open. 2021;5(1):e36–46.
Policies and Technology for Interoperability and, Reduction B. [https://www.cms.gov/regulations-and-guidance/guidance/interoperability/index]].
A Turning Point in Electronic Health Information Progress. [https://www.healthit.gov/buzz-blog/health-it-policy/a-turning-point-in-electronic-health-information-progress].
Lau C, Jamali F, Loebenberg R. Health Canada Usage of Real World Evidence (RWE) in Regulatory decision making compared with FDA/EMA usage based on publicly available information. J Pharm Pharm Sci. 2022;25:227–36.
Concato J, Corrigan-Curay J. Real-world evidence - where are we now? N Engl J Med. 2022;386(18):1680–2.
Real-World Evidence. Food and Drug Administation. [https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence].
Gatto NM, Reynolds RF, Campbell UB. A structured preapproval and Postapproval Comparative Study Design Framework to Generate Valid and Transparent Real-World evidence for Regulatory decisions. Clin Pharmacol Ther. 2019;106(1):103–15.
Gatto NM, Campbell UB, Rubinstein E, Jaksa A, Mattox P, Mo J, Reynolds RF. The structured process to identify fit-for-purpose data: A Data Feasibility Assessment Framework. Clin Pharmacol Ther. 2022;111(1):122–34.
Coste A, Wong A, Bokern M, Bate A, Douglas IJ. Methods for drug safety signal detection using routinely collected observational electronic health care data: a systematic review. Pharmacoepidemiol Drug Saf. 2023;32(1):28–43.
Nie X, Jia L, Peng X, Zhao H, Yu Y, Chen Z, Zhang L, Cheng X, Lyu Y, Cao W, et al. Detection of Drug-Induced Thrombocytopenia signals in children using Routine Electronic Medical records. Front Pharmacol. 2021;12:756207.
Raman SR, Qualls LG, Hammill BG, Nelson AJ, Nilles EK, Marsolo K, O’Brien EC. Optimizing data integration in trials that use EHR data: lessons learned from a multi-center randomized clinical trial. Trials. 2023;24(1):566.
Curran GM, Bauer M, Mittman B, Pyne JM, Stetler C. Effectiveness-implementation hybrid designs: combining elements of clinical effectiveness and implementation research to enhance public health impact. Med Care. 2012;50(3):217–26.
Wolfenden L, Williams CM, Wiggers J, Nathan N, Yoong SL. Improving the translation of health promotion interventions using effectiveness-implementation hybrid designs in program evaluations. Health Promot J Austr. 2016;27(3):204–7.
Root HJ, Lininger MR, DiStefano LJ. Hybrid effectiveness-implementation study designs in sports injury prevention research. Front Sports Act Living. 2022;4: 981656.
Green BB, Coronado GD, Schwartz M, Coury J, Baldwin LM. Using a continuum of hybrid effectiveness-implementation studies to put research-tested colorectal screening interventions into practice. Implement Sci. 2019;14(1):53.
Perry CK, Damschroder LJ, Hemler JR, Woodson TT, Ono SS, Cohen DJ. Specifying and comparing implementation strategies across seven large implementation interventions: a practical application of theory. Implement Sci. 2019;14(1):32.
Proctor EK, Powell BJ, McMillen JC. Implementation strategies: recommendations for specifying and reporting. Implement Sci. 2013;8:139.
Pamungkas RA, Usman AM, Chamroonsawasdi K, Abdurrasyid. A smartphone application of Diabetes coaching intervention to prevent the onset of Complications and to improve Diabetes self-management: a randomized control trial. Diabetes Metab Syndr. 2022;16(7):102537.
Woodard L, Amspoker AB, Hundt NE, Gordon HS, Hertz B, Odom E, Utech A, Razjouyan J, Rajan SS, Kamdar N, et al. Comparison of collaborative goal setting with enhanced education for managing Diabetes-Associated distress and hemoglobin A1c levels: a Randomized Clinical Trial. JAMA Netw Open. 2022;5(5):e229975.
MacBride-Stewart S, Marwick C, Ryan M, Guthrie B. Feedback of actionable individual patient prescription data to improve Asthma prescribing: pragmatic cluster randomised trial in 233 UK general practices. Br J Gen Pract. 2022;72(722):e627-33.
Romani ED, Siddharthan T, Lovaton N, Alvitez-Luna CC, Flores-Flores O, Pollard SL. Implementation of an intervention to improve the adoption of Asthma self-management practices in Peru: Asthma implementation research (AIRE) randomized trial study protocol. Trials. 2020;21(1):377.
Lobo EH, Karmakar C, Abdelrazek M, Abawajy J, Chow CK, Zhang Y, Kabir MA, Daryabeygi R, Maddison R, Islam SMS. Design and development of a smartphone app for Hypertension management: an intervention mapping approach. Front Public Health. 2023;11:1092755.
Valerio-Shewmaker MA, Heredia NI, Pulicken C, Mathews PD, Chenier R, Swoboda TL, Garza ER, Velasco-Huerta F, Fernandez ME. Using implementation mapping for the adoption and implementation of target:BP in community health centers. Front Public Health. 2022;10:928148.
Pandey DK, Dasgupta R, Levy J, Wang H, Serafini A, Habibi M, Song W, Shafer PO, Loeb JA. Enhancing Epilepsy self-management and quality of life for adults with Epilepsy with varying social and educational backgrounds using PAUSE to learn your Epilepsy. Epilepsy Behav. 2020;111:107228.
Schougaard LMV, Mejdahl CT, Christensen J, Lomborg K, Maindal HT, de Thurah A, Hjollund NH. Patient-initiated versus fixed-interval patient-reported outcome-based follow-up in outpatients with Epilepsy: a pragmatic randomized controlled trial. J Patient Rep Outcomes. 2019;3(1):61.
Ridsdale L, Wojewodka G, Robinson EJ, Noble AJ, Morgan M, Taylor SJC, McCrone P, Richardson MP, Baker G, Landau S, et al. The effectiveness of a group self-management education course for adults with poorly controlled Epilepsy, SMILE (UK): a randomized controlled trial. Epilepsia. 2018;59(5):1048–61.
Ghearing GR, Briggs F, Cassidy K, Privitera M, Blixen C, Sajatovic M. A randomized controlled trial of self-management for people with Epilepsy and a history of negative health events (SMART) targeting rural and underserved people with Epilepsy: a methodologic report. Trials. 2021;22(1):821.
Moore N, Blin P, Droz C. Pharmacoepidemiology. Handb Exp Pharmacol. 2019;260:433–51.
Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342(25):1887–92.
Odgaard-Jensen J, Vist GE, Timmer A, Kunz R, Akl EA, Schunemann H, Briel M, Nordmann AJ, Pregno S, Oxman AD. Randomisation to protect against selection bias in healthcare trials. Cochrane Database Syst Rev. 2011;4:MR000012.
Ankarfeldt MZ, Adalsteinsson E, Groenwold RH, Ali MS, Klungel OH. A systematic literature review on the efficacy-effectiveness gap: comparison of randomized controlled trials and observational studies of glucose-lowering Drugs. Clin Epidemiol. 2017;9:41–51.
Tonin FS, Steimbach LM, Leonart LP, Ferreira VL, Borba HH, Piazza T, Araujo AG, Fernandez-Llimos F, Pontarolo R, Wiens A. Discontinuation of non-anti-TNF Drugs for rheumatoid arthritis in interventional versus observational studies: a systematic review and meta-analysis. Eur J Clin Pharmacol. 2018;74(11):1513–21.
Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med. 2000;342(25):1878–86.
Coscia C, Jaureguizar A, Quezada CA, Muriel A, Monreal M, Villen T, Barbero E, Chiluiza D, Yusen RD, Jimenez D. Comparison of all-cause Mortality following VTE Treatment between Propensity score-adjusted observational studies and matched randomized controlled trials: Meta-epidemiologic study. Chest. 2019;155(4):689–98.
Huynh T, Perron S, O’Loughlin J, Joseph L, Labrecque M, Tu JV, Theroux P. Comparison of primary percutaneous coronary intervention and fibrinolytic therapy in ST-segment-elevation Myocardial Infarction: bayesian hierarchical meta-analyses of randomized controlled trials and observational studies. Circulation. 2009;119(24):3101–9.
Li G, Holbrook A, Jin Y, Zhang Y, Levine MA, Mbuagbaw L, Witt DM, Crowther M, Connolly S, Chai-Adisaksopha C, et al. Comparison of treatment effect estimates of non-vitamin K antagonist oral anticoagulants versus warfarin between observational studies using propensity score methods and randomized controlled trials. Eur J Epidemiol. 2016;31(6):541–61.
Liu HY, Han Y, Chen XS, Bai L, Guo SP, Li L, Wu P, Yin YP. Comparison of efficacy of treatments for early Syphilis: a systematic review and network meta-analysis of randomized controlled trials and observational studies. PLoS One. 2017;12(6):e0180001.
Naudet F, Maria AS, Falissard B. Antidepressant response in major depressive disorder: a meta-regression comparison of randomized controlled trials and observational studies. PLoS One. 2011;6(6):e20811.
Shikata S, Nakayama T, Noguchi Y, Taji Y, Yamagishi H. Comparison of effects in randomized controlled trials with observational studies in digestive Surgery. Ann Surg. 2006;244(5):668–76.
Smeeing DPJ, van der Ven DJC, Hietbrink F, Timmers TK, van Heijl M, Kruyt MC, Groenwold RHH, van der Meijden OAJ, Houwert RM. Surgical Versus Nonsurgical Treatment for Midshaft Clavicle Fractures in patients aged 16 years and older: a systematic review, Meta-analysis, and comparison of Randomized controlled trials and observational studies. Am J Sports Med. 2017;45(8):1937–45.
Zhai C, Cong H, Hou K, Hu Y, Zhang J, Zhang Y. Clinical outcome comparison of percutaneous coronary intervention and bypass Surgery in diabetic patients with coronary artery Disease: a meta-analysis of randomized controlled trials and observational studies. Diabetol Metab Syndr. 2019;11:110.
Janiaud P, Agarwal A, Tzoulaki I, Theodoratou E, Tsilidis KK, Evangelou E, Ioannidis JPA. Validity of observational evidence on putative risk and protective factors: appraisal of 3744 meta-analyses on 57 topics. BMC Med. 2021;19(1):157.
Hong YD, Jansen JP, Guerino J, Berger ML, Crown W, Goettsch WG, Mullins CD, Willke RJ, Orsini LS. Comparative effectiveness and safety of pharmaceuticals assessed in observational studies compared with randomized controlled trials. BMC Med. 2021;19(1):307.
Usman MS, Van Spall HGC, Greene SJ, Pandey A, McGuire DK, Ali ZA, Mentz RJ, Fonarow GC, Spertus JA, Anker SD, et al. The need for increased Pragmatism in cardiovascular clinical trials. Nat Reviews Cardiol. 2022;19(11):737–50.
Pencina MJ, Rockhold FW, D’Agostino RB. Deriving real-world insights from real-World Data: Biostatistics to the rescue. Ann Intern Med. 2018;169(6):401–2.
Moberg J, Kramer M. A brief history of the cluster randomized trial design. In: JLL (James Lind Library) Bulletin: Commentaries on the history of treatment evaluation. 2015.
Hallett J, Feng D, McCormick A, Allen S, Inouye J, Schure M, Holder S, Medicine LO, Held S. Improving chronic Illness self-management with the Apsaalooke Nation: the Baa Nnilah Project, a cluster randomized trial protocol. Contemp Clin Trials. 2022;119:106835.
Hemming K, Haines TP, Chilton PJ, Girling AJ, Lilford RJ. The stepped wedge cluster randomised trial: rationale, design, analysis, and reporting. BMJ. 2015;350:h391.
Brown CA, Lilford RJ. The stepped wedge trial design: a systematic review. BMC Med Res Methodol. 2006;6:54.
Nguyen AM, Cleland CM, Dickinson LM, Barry MP, Cykert S, Duffy FD, Kuzel AJ, Lindner SR, Parchman ML, Shelley DR, et al. Considerations before selecting a stepped-Wedge Cluster Randomized Trial Design for a practice improvement study. Ann Fam Med. 2022;20(3):255–61.
Li YH, Mullette E, Brant JM. The stepped-Wedge Trial Design: paving the way for Cancer Care Delivery Research. J Adv Pract Oncol. 2018;9(7):722–7.
Beard E, Lewis JJ, Copas A, Davey C, Osrin D, Baio G, Thompson JA, Fielding KL, Omar RZ, Ononge S, et al. Stepped wedge randomised controlled trials: systematic review of studies published between 2010 and 2014. Trials. 2015;16:353.
Wester V, de Groot S, Kanters T, Wagner L, Ardesch J, Corro Ramos I, Enders-Slegers MJ, de Ruiter M, le Cessie S, Los J, et al. Evaluating the effectiveness and cost-effectiveness of seizure dogs in persons with medically refractory Epilepsy in the Netherlands: study protocol for a stepped Wedge Randomized Controlled Trial (EPISODE). Front Neurol. 2020;11:3.
Galbraith S, Marschner IC. Interim analysis of continuous long-term endpoints in clinical trials with longitudinal outcomes. Stat Med. 2003;22(11):1787–805.
Parsons NR, Stallard N, Parsons H, Haque A, Underwood M, Mason J, Khan I, Costa ML, Griffin DR, Griffin J, et al. Group sequential designs in pragmatic trials: feasibility and assessment of utility using data from a number of recent surgical RCTs. BMC Med Res Methodol. 2022;22(1):256.
Group sequential design. [https://toolbox.eupati.eu/glossary/group-sequential-design/].
Blonde L, Khunti K, Harris SB, Meizinger C, Skolnik NS. Interpretation and impact of real-world Clinical Data for the practicing clinician. Adv Ther. 2018;35(11):1763–74.
Liu F, Panagiotakos D. Real-world data: a brief review of the methods, applications, challenges and opportunities. BMC Med Res Methodol. 2022;22(1):287.
Agniel D, Kohane IS, Weber GM. Biases in electronic health record data due to processes within the healthcare system: retrospective observational study. BMJ. 2018;361:k1479.
Kohane IS, Aronow BJ, Avillach P, Beaulieu-Jones BK, Bellazzi R, Bradford RL, Brat GA, Cannataro M, Cimino JJ, García-Barrio N, et al. What every reader should know about studies using Electronic Health Record Data but May be afraid to ask. J Med Internet Res. 2021;23(3):e22219.
Gokhale M, Stürmer T, Buse JB. Real-world evidence: the devil is in the detail. Diabetologia. 2020;63(9):1694–705.
von Lucadou M, Ganslandt T, Prokosch HU, Toddenroth D. Feasibility analysis of conducting observational studies with the electronic health record. BMC Med Inf Decis Mak. 2019;19(1):202.
Beesley LJ, Salvatore M, Fritsche LG, Pandit A, Rao A, Brummett C, Willer CJ, Lisabeth LD, Mukherjee B. The emerging landscape of health research based on biobanks linked to electronic health records: existing resources, statistical challenges, and potential opportunities. Stat Med. 2020;39(6):773–800.
Callahan A, Shah NH, Chen JH. Research and Reporting Considerations for Observational Studies Using Electronic Health Record Data. Ann Intern Med. 2020;172(11 Suppl):79-S84.
Holmes JH, Beinlich J, Boland MR, Bowles KH, Chen Y, Cook TS, Demiris G, Draugelis M, Fluharty L, Gabriel PE, et al. Why is the Electronic Health Record so challenging for Research and Clinical Care? Methods Inf Med. 2021;60(1–02):32–48.
Acton EK, Willis AW, Hennessy S. Core concepts in pharmacoepidemiology: key biases arising in pharmacoepidemiologic studies. Pharmacoepidemiol Drug Saf. 2023;32(1):9–18.
Lewis AE, Weiskopf N, Abrams ZB, Foraker R, Lai AM, Payne PRO, Gupta A. Electronic health record data quality assessment and tools: a systematic review. J Am Med Inf Association. 2023;30(10):1730–40.
Bykov K, Patorno E, D’Andrea E, He M, Lee H, Graff JS, Franklin JM. Prevalence of Avoidable and Bias-Inflicting Methodological pitfalls in Real-World studies of Medication Safety and Effectiveness. Clin Pharmacol Ther. 2022;111(1):209–17.
Xue TM, Pan W, Tsumura H, Wei S, Lee C, McConnell ES. Impact of Dementia on long-term hip fracture Surgery outcomes: an Electronic Health Record Analysis. J Am Med Dir Assoc. 2023;24(2):235-241e232.
Li F, Tian Z, Bobb J, Papadogeorgou G, Li F. Clarifying selection bias in cluster randomized trials. Clin Trials. 2022;19(1):33–41.
Goldstein ND, Kahal D, Testa K, Burstyn I. Inverse probability weighting for selection bias in a Delaware community health center electronic medical record study of community deprivation and Hepatitis C prevalence. Ann Epidemiol. 2021;60:1–7.
Beesley LJ, Mukherjee B. Statistical inference for association studies using electronic health records: handling both selection bias and outcome misclassification. Biometrics. 2022;78(1):214–26.
Hayakawa T, Nagashima T, Akimoto H, Minagawa K, Takahashi Y, Asai S. Benzodiazepine-related Dementia risks and protopathic biases revealed by multiple-kernel learning with electronic medical records. Digit Health. 2023;9:20552076231178576.
Rafiq M, Abel G, Renzi C, Lyratzopoulos G. Steroid prescribing in primary care increases prior to Hodgkin Lymphoma diagnosis: a UK nationwide case-control study. Cancer Epidemiol. 2022;81: 102284.
Schuemie MJ, Coloma PM, Straatman H, Herings RM, Trifiro G, Matthews JN, Prieto-Merino D, Molokhia M, Pedersen L, Gini R, et al. Using electronic health care records for drug safety signal detection: a comparative evaluation of statistical methods. Med Care. 2012;50(10):890–7.
Groenwold RHH. Informative missingness in electronic health record systems: the curse of knowing. Diagn Progn Res. 2020;4:8.
Perez-Lebel A, Varoquaux G, Le Morvan M, Josse J, Poline JB. Benchmarking missing-values approaches for predictive models on health databases. Gigascience. 2022;11:11.
Patino CM, Ferreira JC. Internal and external validity: can you apply research study results to your patients? J Bras Pneumol. 2018;44(3):183.
Fawcett N, Young B, Peto L, Quan TP, Gillott R, Wu J, Middlemass C, Weston S, Crook DW, Peto TEA, et al. Caveat emptor’: the cautionary tale of endocarditis and the potential pitfalls of clinical coding data-an electronic health records study. BMC Med. 2019;17(1):169.
Weinstein EJ, Ritchey ME, Lo Re V 3rd. Core concepts in pharmacoepidemiology: validation of health outcomes of interest within real-world healthcare databases. Pharmacoepidemiol Drug Saf. 2023;32(1):1–8.
Cook LA, Sachs J, Weiskopf NG. The quality of social determinants data in the electronic health record: a systematic review. J Am Med Inf Association. 2021;29(1):187–96.
Goldstein ND, Kahal D, Testa K, Gracely EJ, Burstyn I. Data Quality in Electronic Health Record Research: an Approach for Validation and Quantitative Bias Analysis for Imperfectly Ascertained Health outcomes Via Diagnostic codes. Harv Data Sci Rev. 2022;4(2). https://doi.org/10.1162/99608f92.cbe67e91.
Vest JR, Adler-Milstein J, Gottlieb LM, Bian J, Campion TR Jr, Cohen GR, Donnelly N, Harper J, Huerta TR, Kansky JP, et al. Assessment of structured data elements for social risk factors. Am J Manag Care. 2022;28(1):e14–23.
Saez C, Gutierrez-Sacristan A, Kohane I, Garcia-Gomez JM, Avillach P. EHR temporal variability: delineating temporal data-set shifts in electronic health records. Gigascience. 2020;9(8):giaa079.
Zeng J, Gensheimer MF, Rubin DL, Athey S, Shachter RD. Uncovering interpretable potential confounders in electronic medical records. Nat Commun. 2022;13(1):1014.
Zhang L, Wang Y, Schuemie MJ, Blei DM, Hripcsak G. Adjusting for indirectly measured confounding using large-scale propensity score. J Biomed Inform. 2022;134:104204.
Zang H, Kim HJ, Huang B, Szczesniak R. Bayesian causal inference for observational studies with missingness in covariates and outcomes. Biometrics. 2023. https://doi.org/10.1111/biom.13918. Epub ahead of print.
Xiong R, Koenecke A, Powell M, Shen Z, Vogelstein JT, Athey S. Federated causal inference in heterogeneous observational data. Stat Med. 2023;42(24):4418–39.
Steinberg E, Ignatiadis N, Yadlowsky S, Xu Y, Shah N. Using public clinical trial reports to probe non-experimental causal inference methods. BMC Med Res Methodol. 2023;23(1):204.
Michoel T, Zhang JD. Causal inference in drug discovery and development. Drug Discov Today. 2023;28(10):103737.
Lane M, Berlin NL, Chung KC, Waljee JF. Strengthening Association through Causal Inference. Plast Reconstr Surg. 2023;152(4):899–907.
Hufstedler H, Mauer N, Yeboah E, Carr S, Rahman S, Danzer AM, Debray TPA, Jong VMT, Campbell H, Gustafson P, et al. Application of causal inference methods to pooled Longitudinal non- Randomized studies: a methodological systematic review. Res Sq. 2023:rs.3.rs-3282208.
Common Data Elements. : Increasing FAIR Data Sharing. https://nexus.od.nih.gov/all/2021/06/24/common-data-elements-increasing-fair-data-sharing/.
Grinspan ZM, Patel AD, Shellhaas RA, Berg AT, Axeen ET, Bolton J, Clarke DF, Coryell J, Gaillard WD, Goodkin HP, et al. Design and implementation of electronic health record common data elements for pediatric Epilepsy: foundations for a learning health care system. Epilepsia. 2021;62(1):198–216.
Wyles CC, Fu S, Odum SL, Rowe T, Habet NA, Berry DJ, Lewallen DG, Maradit-Kremers H, Sohn S, Springer BD. External Validation of Natural Language Processing Algorithms to Extract Common Data Elements in THA Operative Notes. J Arthroplasty. 2023;38(10):2081-4.
Wang L, Fu S, Wen A, Ruan X, He H, Liu S, Moon S, Mai M, Riaz IB, Wang N, et al. Assessment of Electronic Health Record for Cancer Research and Patient Care through a scoping review of Cancer Natural Language Processing. JCO Clin Cancer Inform. 2022;6:e2200006.
Lyketsos CG, Roberts SB, Swift EK, Quina A, Moon G, Kremer I, Tariot P, Fillit H, Bovenkamp DE, Zandi PP, et al. Standardizing Electronic Health Record Data on AD/ADRD to Accelerate Health Equity in Prevention, Detection, and treatment. J Prev Alzheimers Dis. 2022;9(3):556–60.
Bradwell KR, Wooldridge JT, Amor B, Bennett TD, Anand A, Bremer C, Yoo YJ, Qian Z, Johnson SG, Pfaff ER, et al. Harmonizing units and values of quantitative data elements in a very large nationally pooled electronic health record (EHR) dataset. J Am Med Inf Association. 2022;29(7):1172–82.
Fitzgerald MP, Kaufman MC, Massey SL, Fridinger S, Prelack M, Ellis C, Ortiz-Gonzalez X, Fried LE, DiGiovine MP, Collaborative CPEP, et al. Assessing seizure burden in pediatric Epilepsy using an electronic medical record-based tool through a common data element approach. Epilepsia. 2021;62(7):1617–28.
Thandi M, Brown S, Wong ST. Mapping frailty concepts to SNOMED CT. Int J Med Inform. 2021;149:104409.
Hurst JH, Liu Y, Maxson PJ, Permar SR, Boulware LE, Goldstein BA. Development of an electronic health records datamart to support clinical and population health research. J Clin Transl Sci. 2020;5(1):e13.
Hammond WE, Bent B, West VL. Goodbye Electronic Health Record? Stud Health Technol Inform. 2022;298:107–11.
Clermont G. The Learning Electronic Health Record. Crit Care Clin. 2023;39(4):689–700.
Juhn Y, Liu H. Artificial intelligence approaches using natural language processing to advance EHR-based clinical research. J Allergy Clin Immunol. 2020;145(2):463–9.
Sterne JA, Hernan MA, Reeves BC, Savovic J, Berkman ND, Viswanathan M, Henry D, Altman DG, Ansari MT, Boutron I, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. 2016;355:i4919.
Sterne JAC, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, Cates CJ, Cheng HY, Corbett MS, Eldridge SM, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898.
Revised Cochrane risk of bias tool for randomized trials (RoB 2.) Additional considerations for cluster-randomized trials (RoB 2 CRT) [https://drive.google.com/file/d/1yDQtDkrp68_8kJiIUdbongK99sx7RFI-/view.
Page MJ, Higgins JPT, Sterne JAC, et al. Chap. 13: Assessing risk of bias due to missing results in a synthesis. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al., editors. Cochrane Handbook for Systematic Reviews of Interventions. 3rd ed. Chichester: John Wiley and Sons; 2019.
Risk Of Bias due to Missing Evidence (ROB-ME). : a new tool for assessing risk of non-reporting biases in evidence syntheses. [https://drive.google.com/file/d/1BaF3lZ6j1ZIx208gsoYab8uGzk6zlqvw/view].
Leather DA, Jones R, Woodcock A, Vestbo J, Jacques L, Thomas M. Real-World Data and Randomised controlled trials: the Salford Lung Study. Adv Ther. 2020;37(3):977–97.
Shortreed SM, Rutter CM, Cook AJ, Simon GE. Improving pragmatic clinical trial design using real-world data. Clin Trials. 2019;16(3):273–82.
Laaksonen N, Varjonen JM, Blomster M, Palomaki A, Vasankari T, Airaksinen J, Huupponen R, Scheinin M, Juuso B. Assessing an Electronic Health Record research platform for identification of clinical trial participants. Contemp Clin Trials Commun. 2021;21:100692.
Claerhout B, Kalra D, Mueller C, Singh G, Ammour N, Meloni L, Blomster J, Hopley M, Kafatos G, Garvey A, et al. Federated electronic health records research technology to support clinical trial protocol optimization: evidence from EHR4CR and the InSite platform. J Biomed Inform. 2019;90:103090.
Conley S, O’Connell M, Linsky S, Moemeka L, Darden JWt, Gaiser EC, Jacoby D, Yaggi H, Redeker NS. Evaluating recruitment strategies for a Randomized Clinical Trial with Heart Failure patients. West J Nurs Res. 2021;43(8):785–90.
Bennett WL, Bramante CT, Rothenberger SD, Kraschnewski JL, Herring SJ, Lent MR, Clark JM, Conroy MB, Lehmann H, Cappella N, et al. Patient recruitment into a Multicenter Clinical Cohort Linking Electronic Health Records from 5 Health Systems: cross-sectional analysis. J Med Internet Res. 2021;23(5):e24003.
Pfaff E, Lee A, Bradford R, Pae J, Potter C, Blue P, Knoepp P, Thompson K, Roumie CL, Crenshaw D, et al. Recruiting for a pragmatic trial using the electronic health record and patient portal: successes and lessons learned. J Am Med Inf Association. 2019;26(1):44–9.
Plante TB, Gleason KT, Miller HN, Charleston J, McArthur K, Himmelfarb CD, Lazo M, Ford DE, Miller ER 3rd, Appel LJ, et al. Recruitment of trial participants through electronic medical record patient portal messaging: a pilot study. Clin Trials. 2020;17(1):30–8.
Tabriz AA, Fleming PJ, Shin Y, Resnicow K, Jones RM, Flocke SA, Shires DA, Hawley ST, Willens D, Lafata JE. Challenges and opportunities using online portals to recruit diverse patients to behavioral trials. J Am Med Inf Association. 2019;26(12):1637–44.
Kannan V, Wilkinson KE, Varghese M, Lynch-Medick S, Willett DL, Bosler TA, Chu L, Gates SI, Holbein MEB, Willett MM, et al. Count me in: using a patient portal to minimize implicit bias in clinical research recruitment. J Am Med Inf Assoc. 2019;26(8–9):703–13.
Gehtland LM, Paquin RS, Andrews SM, Lee AM, Gwaltney A, Duparc M, Pfaff ER, Bailey DB Jr. Using a patient Portal to increase enrollment in a newborn Screening Research Study: Observational Study. JMIR Pediatr Parent. 2022;5(1):e30941.
Kim SY, Flory J, Relton C. Ethics and practice of trials within cohorts: an emerging pragmatic trial design. Clin Trials. 2018;15(1):9–16.
Verweij ME, Gal R, Burbach JPM, Young-Afat DA, van der Velden JM, van der Graaf R, May AM, Relton C, Intven MPW, Verkooijen HM. Most patients reported positively or neutrally of having served as controls in the trials within cohorts design. J Clin Epidemiol. 2022;148:39–47.
Young-Afat DA, Gal R, Gerlich S, Burbach JPM, van der Velden JM, van den Bongard D, Intven MPW, Kasperts N, May AM, van der Graaf R, et al. Oncology patients were found to understand and accept the trials within cohorts design. J Clin Epidemiol. 2021;130:135–42.
Zhang JZ, Nicholls SG, Carroll K, Nix HP, Goldstein CE, Hey SP, Brehaut JC, McLean PC, Weijer C, Fergusson DA, Taljaard M. Informed consent in pragmatic trials: results from a survey of trials published 2014-2019. J Med Ethics. 2021:medethics-2021-107765.
O’Rourke PP, Carrithers J, Patrick-Lake B, Rice TW, Corsmo J, Hart R, Drezner MK, Lantos JD. Harmonization and streamlining of research oversight for pragmatic clinical trials. Clin Trials. 2015;12(5):449–56.
Code of Federal Regulations. 45 CFR 46.116(d). Protection of Human Subjects. General Requirements for Informed Consent. [https://www.hhs.gov/ohrp/regulations-and-policy/regulations/45-cfr-46/index.html#46.116].
McKinney RE Jr, Beskow LM, Ford DE, Lantos JD, McCall J, Patrick-Lake B, Pletcher MJ, Rath B, Schmidt H, Weinfurt K. Use of altered informed consent in pragmatic clinical research. Clin Trials. 2015;12(5):494–502.
Kim SY, Kimmelman J. Practical steps to identifying the research risk of pragmatic trials. Clin Trials. 2022;19(2):211–6.
Lantos JD, Wendler D, Septimus E, Wahba S, Madigan R, Bliss G. Considerations in the evaluation and determination of minimal risk in pragmatic clinical trials. Clin Trials. 2015;12(5):485–93.
Miller DG, Kim SYH, Li X, Dickert NW, Flory J, Runge CP, Relton C. Ethical acceptability of postrandomization consent in pragmatic clinical trials. JAMA Netw Open. 2018;1(8):e186149.
Marquis-Gravel G, Robertson H, Jones WS, Riley D, Ford DE, Crenshaw D, Joosten YA, Rudov L, Hernandez AF, Hess R. Streamlining the institutional review board process in pragmatic randomized clinical trials: challenges and lessons learned from the aspirin dosing: a patient-centric trial assessing benefits and long-term effectiveness (ADAPTABLE) trial. Trials. 2021;22(1):90.
Federico CA, Heagerty PJ, Lantos J, O’Rourke P, Rahimzadeh V, Sugarman J, Weinfurt K, Wendler D, Wilfond BS, Magnus D. Ethical and epistemic issues in the design and conduct of pragmatic stepped-wedge cluster randomized clinical trials. Contemp Clin Trials. 2022;115:106703.
Lakin JR, Brannen EN, Tulsky JA, Paasche-Orlow MK, Lindvall C, Chang Y, Gundersen DA, El-Jawahri A, Volandes A, Investigators A-P. Advance Care Planning: promoting effective and aligned communication in the Elderly (ACP-PEACE): the study protocol for a pragmatic stepped-wedge trial of older patients with cancer. BMJ Open. 2020;10(7):e040999.
van Oostveen RB, Romero-Palacios A, Whitlock R, Lee SF, Connolly S, Carignan A, Mazer CD, Loeb M, Mertz D. Prevention of Infections in Cardiac Surgery study (PICS): study protocol for a pragmatic cluster-randomized factorial crossover pilot trial. Trials. 2018;19(1):688.
Dember LM, Lacson E Jr, Brunelli SM, Hsu JY, Cheung AK, Daugirdas JT, Greene T, Kovesdy CP, Miskulin DC, Thadhani RI, et al. The TiME trial: a fully embedded, Cluster-Randomized, pragmatic trial of Hemodialysis Session Duration. J Am Soc Nephrol. 2019;30(5):890–903.
Spector-Bagdady K, Trinidad G, Kardia S, Krenz CD, Nong P, Raj M, Platt JE. Reported interest in notification regarding use of Health Information and Biospecimens. JAMA. 2022;328(5):474–6.
Weijer C, Grimshaw JM, Eccles MP, McRae AD, White A, Brehaut JC, Taljaard M. The Ottawa Statement on the ethical Design and Conduct of Cluster Randomized trials. PLoS Med. 2012;9(11):e1001346.
International Ethical Guidelines for Health-related Research Involving Humans, Fourth Edition. Geneva. Council for International Organizations of Medical Sciences (CIOMS); 2016.
IRB waiver or. Alteration of informed consent for clinical investigations involving no more than minimal risk to human subjects: guidance for sponsors, investigators and institutional review boards. https://www.fda.gov/media/106587/download.
Nix HP, Weijer C, Brehaut JC, Forster D, Goldstein CE, Taljaard M. Informed consent in cluster randomised trials: a guide for the perplexed. BMJ Open. 2021;11(9):e054213.
Copas AJ, Lewis JJ, Thompson JA, Davey C, Baio G, Hargreaves JR. Designing a stepped wedge trial: three main designs, carry-over effects and randomisation approaches. Trials. 2015;16:352.
Hemming K, Taljaard M. Reflection on modern methods: when is a stepped-wedge cluster randomized trial a good study design choice? Int J Epidemiol. 2020;49(3):1043–52.
Hemming K, Taljaard M, Grimshaw J. Introducing the new CONSORT extension for stepped-wedge cluster randomised trials. Trials. 2019;20(1):68.
Binik A. Delaying and withholding interventions: ethics and the stepped wedge trial. J Med Ethics. 2019;45(10):662–7.
Hughes JP, Granston TS, Heagerty PJ. Current issues in the design and analysis of stepped wedge trials. Contemp Clin Trials. 2015;45(Pt A):55–60.
Hemming K, Taljaard M, McKenzie JE, Hooper R, Copas A, Thompson JA, Dixon-Woods M, Aldcroft A, Doussau A, Grayling M, et al. Reporting of stepped wedge cluster randomised trials: extension of the CONSORT 2010 statement with explanation and elaboration. BMJ. 2018;363:k1614.
Eskola SM, Leufkens HGM, Bate A, De Bruin ML, Gardarsdottir H. Use of Real-World Data and evidence in Drug Development of Medicinal products centrally authorized in Europe in 2018–2019. Clin Pharmacol Ther. 2022;111(1):310–20.
Purpura CA, Garry EM, Honig N, Case A, Rassen JA. The role of real-world evidence in FDA-Approved New Drug and Biologics license applications. Clin Pharmacol Ther. 2022;111(1):135–44.
You SC, Krumholz HM. The evolution of evidence-based medicine: when the magic of the Randomized Clinical Trial meets real-World Data. Circulation. 2022;145(2):107–9.
Mackowiak JI, Mack CD, Irwin DE, Zura R. Randomized Clinical Trial or Real-World evidence: how historical events, public demand, and the resulting laws and regulations shaped the body of medical evidence. J Orthop Trauma. 2021;35(Suppl 1):17-S21.
Miksad RA, Abernethy AP. Harnessing the power of real-world evidence (RWE): a Checklist to Ensure Regulatory-Grade Data Quality. Clin Pharmacol Ther. 2018;103(2):202–5.
Sajatovic M, Wilson B, Shegog R, Escoffery FBSB, Jobst C, Johnson BC, Fraser EK, Quarells RT, Spruill RC. The managing Epilepsy Well (MEW) network database: lessons learned in refining and implementing an integrated data tool in service of a national U.S. Research Collaborative. Epilepsy Behav. 2021;115:107650.
Lhatoo SD, Bernasconi N, Blumcke I, Braun K, Buchhalter J, Denaxas S, Galanopoulou A, Josephson C, Kobow K, Lowenstein D, et al. Big data in Epilepsy: clinical and research considerations. Report from the Epilepsy Big Data Task Force of the International League against Epilepsy. Epilepsia. 2020;61(9):1869–83.
Karanatsios B, Prang KH, Verbunt E, Yeung JM, Kelaher M, Gibbs P. Defining key design elements of registry-based randomised controlled trials: a scoping review. Trials. 2020;21(1):552.
Concannon TW, Guise JM, Dolor RJ, Meissner P, Tunis S, Krishnan JA, Pace WD, Saltz J, Hersh WR, Michener L, et al. A national strategy to develop pragmatic clinical trials infrastructure. Clin Transl Sci. 2014;7(2):164–71.
Kumar K, Kumar P, Deb D, Unguresan ML, Muresan V. Artificial Intelligence and Machine Learning Based Intervention in Medical Infrastructure: a Review and Future trends. Healthc (Basel). 2023;11(2):207.
Sezgin E, Hussain SA, Rust S, Huang Y. Extracting Medical Information From Free-Text and Unstructured Patient-Generated Health Data Using Natural Language Processing Methods: Feasibility Study With Real-world Data. JMIR Form Res. 2023;7:e43014.
Negro-Calduch E, Azzopardi-Muscat N, Krishnamurthy RS, Novillo-Ortiz D. Technological progress in electronic health record system optimization: systematic review of systematic literature reviews. Int J Med Inform. 2021;152:104507.
Funding
Preparation of this report was supported by the Epilepsy Research Foundation. The sponsor had no involvement in the writing of this report or in the decision to submit it for publication.
Author information
Authors and Affiliations
Contributions
AL prepared the first draft; TL then provided multiple edits iteratively. All authors read and approved the final draft.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Leviton, A., Loddenkemper, T. Design, implementation, and inferential issues associated with clinical trials that rely on data in electronic medical records: a narrative review. BMC Med Res Methodol 23, 271 (2023). https://doi.org/10.1186/s12874-023-02102-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12874-023-02102-4