
Abstract
Background
Gut microbes can contribute to their hosts in food digestion, nutrient absorption, and inhibiting the growth of pathogens. However, only limited studies have focused on the gut microbiota of freshwater snails. Pomacea canaliculata is considered one of the worst invasive alien species in the world. Elucidating the diversity and composition of the microbiota in the gut of P. canaliculata snails may be helpful for better understanding the widespread invasion of this snail species. In this study, the buccal masses, stomachs, and intestines were isolated from seven P. canaliculata snails. The diversity and composition of the microbiota in the three gut sections were then investigated based on high-throughput Illumina sequencing targeting the V3-V4 regions of the 16S rRNA gene.
Results
The diversity of the microbiota was highest in the intestine but lowest in the buccal mass. A total of 29 phyla and 111 genera of bacteria were identified in all of the samples. In general, Ochrobactrum, a genus of putative cellulose-degrading bacteria, was the most abundant (overall relative abundance: 13.6%), followed by Sediminibacterium (9.7%), Desulfovibrio (7.8%), an unclassified genus in the family Aeromonadaceae (5.4%), and Cloacibacterium (5.4%). The composition of the microbiota was diverse among the different gut sections. Ochrobactrum (relative abundance: 23.15% ± 7.92%) and Sediminibacterium (16.95 ± 5.70%) were most abundant in the stomach, an unclassified genus in the family Porphyromonadaceae (14.28 ± 7.29%) and Leptotrichia (8.70 ± 4.46%) were highest in the buccal mass, and two genera in the families Aeromonadaceae (7.55 ± 4.53%) and Mollicutes (13.47 ± 13.03%) were highest in the intestine.
Conclusions
The diversity and composition of the microbiome vary among different gut sections of P. canaliculata snails. Putative cellulose-degrading bacteria are enriched in the gut of P. canaliculata.
Similar content being viewed by others
Background
Pomacea canaliculata, also known as the golden apple snail, is a large species of freshwater snail originating from South America. Because of its high adaptability, strong fecundity, diverse diet and lack of efficient predators, P. canaliculata is widely distributed in tropical and subtropical areas worldwide [1]. Therefore, this species is now considered one of the most invasive alien species in the world and causes serious damage to agriculture and the ecological environment [2]. It is also an intermediate host of Angiostrongylus cantonensis, the etiological agent of angiostrongyliasis [3].
Animals sometimes coevolve with the bacteria residing in their gut. Many studies have successfully correlated gut microbiota to host physiology. For example, gut microbiota may play roles in food digestion, absorption and metabolism in humans and other animals [4, 5]; microbiota-derived lactate can activate the production of reactive oxygen species and shorten the lifespan of Drosophila [6]; and microbiota can regulate midgut homeostasis to prevent the systemic infection of mosquitoes by inducing the peritrophic matrix [7]. Although the composition and function of gut microbiota have been well studied in humans, several mammals and insects, only limited studies have focused on the gut microbiota of freshwater snails. So far, the microbial community in the guts of P. canaliculata snails has not been systematically characterized. Understanding the gut microbiota of P. canaliculata might provide insight into the behavior of the host and might be helpful for better understanding the widespread invasion of this snail species. In this study, we investigated the diversity and composition of the microbiota in different gut sections of P. canaliculata snails using high-throughput Illumina sequencing targeting the V3-V4 regions of the 16S rRNA gene.
Results
Bacterial complexity of the microbiome in the three gut sections of P. canaliculata snails
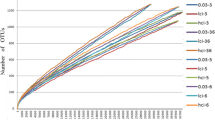
All 21 snail DNA samples were amplified successfully and sequenced. However, the extraction products of the blank control failed to be amplified by PCR under the same conditions as the snail samples. A total of 1,075,200 valid sequences were acquired from the 21 snail samples, yielding 23,151 valid OTUs at 97% identity. After removing the OTUs with relative abundance less than 0.001%, 2234 OTUs remained and were included in further analysis. The rarefaction curve of observed species reached asymptote (Additional file 1: Fig. S1), which indicated that the sequencing depth was sufficient to represent the majority of species richness in each sample.
When analyzed by group, the number of OTUs was higher in the intestine samples (1049.4 ± 184.3) than in the buccal mass (719.0 ± 81.7) and stomach (808.6 ± 189.3) samples (Table 1). Among the 2234 OTUs, 786 (35.5%) were shared by all three groups; 163 (7.0%), 215 (9.6%), and 314 (14.1%) were unique in the buccal mass, stomach, and intestine samples, respectively (Additional file 2: Fig. S2).
The alpha diversity of the gut microbiome was different among the three tissues of P. canaliculata. In general, the bacterial diversity was highest in the intestine samples but lowest in the buccal mass samples as assessed by Chao1, ACE, and Shannon indices (Table 1).
Taxonomic composition of the microbiome in the three gut sections of P. canaliculata snails
Among the 2234 OTUs, 99.8% were assigned to the family level, and 48.7% were assigned to the genus level. Finally, 29 phyla and 111 genera were identified from all 21 samples.
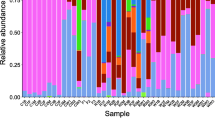
At the phylum level, Proteobacteria and Bacteroidetes were the two most dominant phyla in the gut of P. canaliculata snails (Fig. 1a), with overall relative abundances of 51.6 and 23.6%, respectively. At the genus level, Ochrobactrum was the most dominant genus (overall relative abundance: 13.6%), followed by Sediminibacterium (9.7%), Desulfovibrio (7.8%), an unclassified genus in the family Aeromonadaceae (5.4%), and Cloacibacterium (5.4%, Fig. 1b).

Composition of the bacterial community in the gut of Pomacea canaliculata snails (a) at the phylum level; (b) at the genus level. C1 – C7: buccal mass samples; S1 – S7: stomach samples; I1 – I7: intestine samples
The taxonomic compositions of the microbiome among samples from the different gut sections were diverse. At the phylum level, the relative abundance of Bacteroidetes (mean ± standard error: 34.10% ± 14.72%) and Fusobacteria (9.23% ± 4.76%) was higher in the buccal mass, the abundance of Cyanobacteria (1.55% ± 0.50%) was higher in the stomach, and the abundances of Tenericutes (14.67% ± 13.14%) and Spirochaetes (2.02% ± 1.28%) were higher in the intestine (Additional file 3: Table S1). At the genus level, the relative abundances of an unclassified genus in the family Porphyromonadaceae (14.28 ± 7.29%) and Leptotrichia (8.70 ± 4.46%) were highest in the buccal mass, Ochrobactrum (23.15 ± 7.92%) and Sediminibacterium (16.95 ± 5.70%) were highest in the stomach, and two unclassified genera in the families Aeromonadaceae (7.55 ± 4.53%) and Mollicutes (13.47 ± 13.03%) were highest in the intestine (Additional file 4: Table S2). Interestingly, the structure of the microbiome in the gut of P. canaliculata snails was quite different from that in the water samples (Fig. 1b).
Similarity of the bacterial community in the three gut sections of P. canaliculata snails
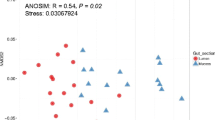
According to the results of the unweighted UniFrac distance-based NMS analysis, the intergroup distance was higher than the intragroup distance (Fig. 2a, Additional file 5: Table S3). Moreover, a similar pattern was confirmed by weighted UniFrac distance-based NMDS analysis (Fig. 2b, Additional file 6: Table S4). The results of ANOSIM also suggested that the intragroup similarity of the gut microbiome was different from the intergroup similarity (R = 0.5623, P = 0.001 for unweighted UniFrac distance; R = 0.4893, P = 0.001 for weighted UniFrac distance).

Two-dimensional distribution of samples according to (a) unweighted (b) weighted UniFrac distance-based NMS analysis
Discussion
The gut microbiota of animals can play roles in the food ingestion, digestion and nutrient absorption of the host [8]. In snails, food is scraped by radula and mixed with the secretions of the salivary gland after being ingested by the buccal mass and digested in the stomach; nutrients are then absorbed in the intestine [9]. Little is known about the spatial structure of the gut microbiota in P. canaliculata snails. In this study, we assumed that the different gut sections, the buccal mass, stomach, and intestine, could be unique microenvironments and harbor distinct bacterial communities. To our knowledge, this is the first study to investigate the diversity and composition of bacterial communities in different gut sections of P. canaliculata snails. Our finding that the bacterial diversity was higher in the intestine (Table 1) is in agreement with reports from other animals [10]. This might be attributed to the characteristics that make the intestine more hospitable to bacteria than other regions of the gut [11].
Only a few studies have investigated the gut microbiota of snails. Moreover, most of these studies were not based on high-throughput sequencing. For example, Van and colleagues investigated intestinal bacterial communities in three species of planorbid snails (Biomphalaria pfeifferi, Bulinus africanus, and Helisoma duryi) via PCR amplification and sequencing of nearly full-length 16S rRNA genes. They revealed that six bacterial taxa (Aeromonas, Bacilli, Chryseobacterium, Chloroacidobacterium, Comamonadaceae and Verrucomicrobiae) were present in at least one of the three snail species [12]. Kiran et al. studied cellulolytic bacteria from Achatina fulica using both culture and PCR product clones and sequencing of the 16S rRNA gene. They found that Citrobacter, Escherichia, Klebsiella, Salmonella, Raoultella, Stenotrophomonas, Enterobacter, and Ochrobactrum were present in the gastrointestinal tract [13].
Hu and colleagues investigated the gut microbiota of Radix auricularia via high-throughput sequencing of the 16S rRNA gene. The results showed that unclassified genera of Mycoplasmataceae and Chloroflexaceae, Paracoccus, Microcoleus, Pleurocapsa, etc. were the most abundant genera [14]. In contrast, our study found that Ochrobactrum, Sediminibacterium, Desulfovibrio, an unclassified genus of Aeromonadaceae, and Cloacibacterium were dominant bacteria in the gut of P. canaliculata snails. The composition of the gut microbiota reflects natural selection between the bacteria and host, which promotes functional stability of this complex ecosystem [15]. The diverse composition of the gut microbiota in distinct snail species may be attributed to the differences in environments, habits, physiological states, genetic characteristics of the host, or the methods used for studying the bacterial community [16].
Bacteria with the potential to degrade plant components are common in the gut of snails. The present study showed that Ochrobactrum was most abundant in the stomach. Ochrobactrum bacteria are putative cellulose-degrading bacteria, which may play important roles in plant fiber digestion in herbivores [17]. They have also been reported in various ecological niches, including soil, water, plants, and animals [18], and have been reported in the human stomach [19] and the gut of A. fulica [13, 20]. Some researchers have suggested that the exogenous bacteria entering the intestinal tract of the snail with food have enzymatic activities that improve digestive processes [21]. In this study, Ochrobactrum was detected in all three gut sections of P. canaliculata snails, and it was the most abundant bacterium in the stomach. Moreover, another common bacterial composition from the gut of P. canaliculata snails, bacteria of the family Comamonadaceae, are also putative cellulose-degrading bacteria [12]. Koch et al. reported that P. canaliculata can survive 56 days on a cellulose-rich diet and suggested that bacterial endoglucanases could help the snail to utilize cellulose polymers [2]. However, whether the gut bacteria can degrade cellulose for the host cannot be inferred from the sequencing data [22]. More studies are still needed to further investigate the roles of putative cellulose-degrading bacteria in the gut of P. canaliculata.
Our study showed that the relative abundance of Sediminibacterium was higher in the stomach than in the buccal mass and intestine. Sediminibacterium is a common reagent contaminant [23]. However, the extraction products of the blank control failed to be amplified by PCR in this study. Therefore, it is reasonable to consider that the microbes reported in this study may not come from laboratory reagents. In previous studies, the genus Sediminibacterium was isolated from aquatic environments such as environmental water samples and sediments [24]. Similarly, Sediminibacterium was also detected from water samples (Fig. 1b). Therefore, Sediminibacterium in the gut was probably derived from the habitat of the snails.
Snails usually use copper for the formation of hemocyanin. Desulfovibrio spp. are sulfate-reducing bacteria and can chelate metals such as Cu, Fe, Ni, and Zn to enhance their absorption. Desulfovibrio has been reported in Helix aspersa crops [25]. In our study, Desulfovibrio was identified in all gut tissues. More studies are warranted to investigate whether Desulfovibrio plays a role in the metabolism of trace elements in P. canaliculata snails.
The relative abundance of Leptotrichia and bacteria from the family Porphyromonadaceae was significantly higher in the buccal mass than in the stomach and intestine of P. canaliculata snails. Leptotrichia is a facultative anaerobic bacterium and has been found mostly in the oral cavity and some other parts of the human body, in animals, and even in ocean sediments [26]. Leptotrichia species can ferment carbohydrates and produce lactic acid and might be associated with lactic acid metabolism in the buccal masses of snails [27]. Bacteria of the families Aeromonadaceae and Mollicute have been reported in various tissues of A. fulica and H. pomatia, respectively [16, 28]. Both of these bacterial taxa were more abundant in the intestine than in the buccal mass and stomach of P. canaliculata snails. Cloacibacterium was first described in 2006 and is usually found in wastewater [29]. It has also been isolated from sediment of freshwater lake [30] and the gut of abalone [31]. However, how these gut bacteria affect snail hosts remains to be investigated.
As a preliminary study on the gut microbiome of P. canaliculata snails, there are several limitations to the present study. First, bacterial DNA is ubiquitous in reagents and can cause problems when samples have a low microbial biomass [23, 32]. The extraction products of the blank control failed to be amplified by PCR in this study. Therefore, it is reasonable to consider that the microbes reported in this study may not come from laboratory reagents. However, contaminants from the environment cannot be excluded completely by surface sterilization of the shell.
Second, only three gut sections of seven P. canaliculata snails from one location were sequenced, and other snail species from the same ecological niche or the actual diet of the snails were not analyzed. Therefore, it is difficult to determine whether the bacteria detected from the snails are inherent or derived from the environment. In fact, most dominant bacteria detected in the gut of P. canaliculata snails existed in water samples at quite a low abundance (Fig. 1b) in this study. In another study, the composition and abundance of intestinal microbiota was found to be quite different in aquatic invertebrates collected from a single small pond [33]. These results suggested that certain microorganisms derived from the environment might be selectively colonized and established in the gut of the host. However, the gut microbiome of snails from different locations and other snail species needs to be analyzed in the future.
Third, P. canaliculata snails were dissected after starvation for 24 h in this study. Some researchers suggest that the bacterial community in the digestive tract of snails will be reduced to stable members after starvation [20]. However, it could have the opposite effect, for example, starvation may promote the growth of transient bacteria by inducing metabolic or immunological changes in the host. Therefore, the influence of starvation on the gut microbiome could not be determined. Since external factors, including diet, can largely affect the gut microbiota of the host [34], further investigation to compare the bacterial structure of snails that have and have not been starved is recommended. Moreover, the importance of the gut microbiota for the biology of the P. canaliculata snail cannot be inferred from the sequencing data and also requires further investigation.
Conclusions
This study first describes the spatial structure of the microbiota in the gut of P. canaliculata snails using high-throughput sequencing. The results demonstrate that the diversity and composition of the microbiome vary among different gut sections of P. canaliculata snails. Putative cellulose-degrading bacteria, including Ochrobactrum, were abundant in the gut of P. canaliculata. More studies are required to better understand the interaction between the gut microbiota and its snail host, including P. canaliculata.
Methods
Sample collection and tissue processing
P. canaliculata snails were collected from Nanheng River (31.052649°N, 120.99297°E) in Rentun village, Qingpu district, Shanghai, PR China, in July 2018. Prior to dissection, the snails were starved for 24 h to minimize the amount of partially digested food in the gut [12]. Simultaneously, three water samples were collected from the habitat of the snails using sterilized bottles and transferred to the laboratory immediately. Water samples were concentrated using 0.22 μm polyether sulfone membrane filters (Millipore, Darmstadt, Germany). The filtration volume was one liter per sample [35]. The membranes coated with microbes from the water samples were used for DNA extraction. The membranes were cut into small pieces and homogenized in SLX-Mlus Buffer (Omega, Norcross, United States) using a Tissuelyser (Jingxin Industrial development Co., Ltd., Shanghai, China). DNA was extracted from the homogenate using the Mag-Bind Soil DNA Kit M5635–02 (Omega, Norcross, U.S.A.) following the manufacturer’s protocol.
Seven female P. canaliculata snails weighing 9–10 g were selected for dissection. The shell was removed from each snail after wiping the shell with 70% ethanol three times and rinsing it twice in distilled water. Dissection was performed on ice in sterile Petri dishes using flame-sterilized tools. The buccal mass, stomach, and intestine of each snail were isolated (Fig. 3) and homogenized separately in centrifuge tubes with a Tissuelyser. DNA was extracted from the homogenized tissue using the Mag-Bind Soil DNA Kit M5635–02 as described above. To exclude contaminants from reagents, three blank controls were extracted simultaneously using the same DNA extraction kit.

The simplified anatomic diagram of Pomacea canaliculata (www.applesnail.net, by Dr. Stijn Ghesquiere. We thank Dr. Stijn Ghesquiere for permission to use the diagram)
Sequencing of the microbial 16S rRNA genes
The variable V3-V4 region of the 16S rRNA gene was amplified by PCR using universal bacterial primers (338F: 5′-ACTCCTACGGGAGGCAGCA-3′, 806R: 5′-GGACTACHVGGGTWTCTAAT-3′). PCR amplification was performed with an ABI 2720 thermal cycler (Applied Biosystems, Foster City, United States) in a total volume of 25 μL containing 8.75 μL of ddH2O, 5 μL of 5× reaction buffer, 5 μL of 5× GC buffer, 2 μL of dNTPs (2.5 mM), 1 μL of each primer (10 μM), 2 μL of DNA template, and 0.25 μL of Q5 High-Fidelity DNA Polymerase (NEB, Ipswich, UK). The thermal cycling conditions were an initial denaturation at 98 °C for 5 min, followed by 25 cycles of 98 °C for 30 s, 55 °C for 45 s and 72 °C for 30 s, and a final extension at 72 °C for 5 min. The PCR products were detected by 2% agarose gel electrophoresis and purified with an AxyPrep DNA Gel Extraction Kit (Axygen, New York, United States). The purified PCR amplicons were used to construct paired-end DNA libraries using the TruSeq Nano DNA LT Library Prep Kit (Illumina, San Diego, United States). Each PCR product was tagged with an index sequence at the 5′ end of the forward primer and then sequenced on the Illumina MiSeq platform (300 bp paired-end reads) by Personal Biotechnology Co., Ltd. (Shanghai, China).
Sequencing data analysis
Quantitative Insights Into Microbial Ecology (QIIME) software (v1.8.0) was used to process the raw sequences. Reads containing any ambiguous bases, sequences shorter than 150 bp, or chimeric sequences were removed. All of the trimmed sequences were normalized to the same sequencing depth using the Mothur software package (v.1.31.2) [36]. The operational taxonomic units (OTUs) were clustered at 97% identity using the UCLUST tool of QIIME software [37]. The sequence with the highest abundance was selected as a representative sequence of each OTU. The taxonomy of each representative sequence was assigned according to the Greengenes 13.8 database [38]. The original OTU abundance matrix usually contains a large number of OTUs with very low abundance, which often occurs occasionally in a small number of samples (i.e., low frequency), while the number of OTUs with high abundance is relatively small. Those rare OTUs with very low abundance and frequency can greatly increase the complexity of data analysis. Removing these rare OTUs has little effect on the diversity of the bacterial community but can significantly improve the efficiency of data analysis. Therefore, OTUs with relative abundance less than 0.001% of all OTUs were removed prior to analysis [39].
A rarefaction curve was drawn to determine whether the sequencing depth was sufficient to represent the bacterial diversity of each sample using the Mothur software package [40]. Alpha diversity indices of the gut microbiome, including the ACE, Chao1, Shannon and Simpson indices, were estimated using QIIME.
To estimate the beta diversity or similarity of the gut microbiome among tissues, nonmetric multidimensional scaling (NMDS) analysis was performed to visualize the pairwise UniFrac distances among samples using R software based on unweighted and abundance weighted UniFrac distance [41]. The tests of significance between intragroup and intergroup UniFrac distance were performed using the Monte Carlo permutation test with QIIME software. Analysis of similarity (ANOSIM) was further conducted to analyze the differences between the intragroup and intergroup distances.
SPSS 19.0 software (IBM, Armonk, USA) was used for statistical analysis of the alpha diversity, number of OTUs, and relative abundance of bacterial taxa among groups using one-way ANOVA. A P-value less than 0.05 was considered statistically significant.
Availability of data and materials
The sequences generated in the present study were deposited in the NCBI Sequence Read Archive (nos. SRR9166284 - SRR9166304).
Abbreviations
- ANOSIM:
-
Analysis of similarity
- ANOVA:
-
Analysis of variance
- LEfSe:
-
Linear discriminant analysis effect size
- NMS:
-
Nonmetric multidimensional scaling
- OTU:
-
Operational taxonomic unit
References
Carey MP, Wahl DH. Native fish diversity alters the effects of an invasive species on food webs. Ecology. 2010;91(10):2965–74.
Koch E, Lozada M, Dionisi H, Castro-Vazquez A. Uric acid-degrading bacteria in the gut of the invading apple snail Pomacea canaliculata and their possible symbiotic significance. Symbiosis. 2014;63(3):149–55.
Lv S, Zhang Y, Chen SR, Wang LB, Fang W, Chen F, Jiang JY, Li YL, Du ZW, Zhou XN. Human angiostrongyliasis outbreak in Dali, China. PLoS Negl Trop Dis. 2009;3(9):e520.
Mitev K, Taleski V. Association between the gut microbiota and obesity. Open Access Maced J Med Sci. 2019;7(12):2050–6.
Yadav S, Jha R. Strategies to modulate the intestinal microbiota and their effects on nutrient utilization, performance, and health of poultry. J Anim Sci Biotechnol. 2019;10:2.
Iatsenko I, Boquete JP, Lemaitre B. Microbiota-derived lactate activates production of reactive oxygen species by the intestinal NADPH oxidase Nox and shortens Drosophila lifespan. Immunity. 2018;49(5):929–42.
Rodgers FH, Gendrin M, Wyer CAS, Christophides GK. Microbiota-induced peritrophic matrix regulates midgut homeostasis and prevents systemic infection of malaria vector mosquitoes. PLoS Pathog. 2017;13(5):e1006391.
Pawar KD, Banskar S, Rane SD, Charan SS, Kulkarni GJ, Sawant SS, Ghate HV, Patole MS, Shouche YS. Bacterial diversity in different regions of gastrointestinal tract of Giant African snail (Achatina fulica). Microbiologyopen. 2012;1(4):415–26.
Dar MA, Pawar KD, Pandit RS. Gut microbiome analysis of snails: a biotechnological approach. In: Organismal and Molecular Malacology. Edited by Ray S; 2017: 189–217.
Godoy-Vitorino F, Goldfarb KC, Karaoz U, Leal S, Garcia-Amado MA, Hugenholtz P, Tringe SG, Brodie EL, Dominguez-Bello MG. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 2012;6(3):531–41.
Smith CC, Srygley RB, Healy F, Swaminath K, Mueller UG. Spatial structure of the Mormon cricket gut microbiome and its predicted contribution to nutrition and immune function. Front Microbiol. 2017;8:801.
Van Horn DJ, Garcia JR, Loker ES, Mitchell KR, Mkoji GM, Adema CM, Takacs-Vesbach CD. Complex intestinal bacterial communities in three species of planorbid snails. J Mollus Stud. 2011;78(1):74–80.
Pawar KD, Dar MA, Rajput BP, Kulkarni GJ. Enrichment and identification of cellulolytic bacteria from the gastrointestinal tract of Giant African snail, Achatina fulica. Appl Biochem Biotechnol. 2015;175(4):1971–80.
Hu Z, Chen X, Chang J, Yu J, Tong Q, Li S, Niu H. Compositional and predicted functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing. PeerJ. 2018;6:e5537.
O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–93.
Nicolai A, Rouland-Lefèvre C, Ansart A, Filser J, Lenz R, Pando A, Charrier M. Inter-population differences and seasonal dynamic of the bacterial gut Community in the Endangered Land Snail Helix pomatia (Gastropoda: Helicidae). Malacologia. 2015;59(1):177–90.
Morrison M, Pope PB, Denman SE, McSweeney CS. Plant biomass degradation by gut microbiomes: more of the same or something new? Curr Opin Biotechnol. 2009;20(3):358–63.
Kulkarni G, Gohil K, Misra V, Kakrani AL, Misra SP, Patole M, Shouche Y, Dharne M. Multilocus sequence typing of Ochrobactrum spp. isolated from gastric niche. J Infect Public Health. 2017;10(2):201–10.
Dharne MS, Misra SP, Misra V, Dwivedi M, Patole MS, Shouche YS. Isolation of urease-positive Ochrobactrum intermedium in the stomach of a non-ulcer dyspeptic patient from North India. J Microbiol Immunol Infect. 2008;41(2):183–6.
Cardoso AM, Cavalcante JJV, Vieira RP, Lima JL, Grieco MAB, Clementino MM, Vasconcelos ATR, Garcia ES, de Souza W, Albano RM, Martins OB. Gut bacterial communities in the Giant land snail Achatina fulica and their modification by sugarcane-based diet. PLoS One. 2012;7(3):e33440.
Koleva ZV, Kizheva YK, Tishkov SH, Dedov IK, Kirova EL, Stefanova PM, Moncheva PA, Hristova PK. Dynamics of bacterial community in the gut of Cornu aspersum. J Biosci Bioeng. 2015;4(3):263–9.
Hammer TJ, Sanders JG, Fierer N. Not all animals need a microbiome. FEMS Microbiol Lett. 2019;366(10).
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27(2):105–17.
Ingerslev HC, von Gersdorff JL, Lenz Strube M, Larsen N, Dalsgaard I, Boye M, Madsen L. The development of the gut microbiota in rainbow trout (Oncorhynchus mykiss) is affected by first feeding and diet type. Aquaculture. 2014;424-425:24–34.
Simkiss K. Prokaryote-eukaryote interactions in trace element metabolism. Desulfovibrio sp. in Helix aspersa. Experientia. 1985;41(9):1195–7.
Eribe ER, Olsen I. Leptotrichia species in human infections. Anaerobe. 2008;14(3):131–7.
Eribe ER, Olsen I. Leptotrichia species in human infections II. J Oral Microbiol. 2017;9(1):1368848.
Morocoima A, Rodríguez V, Rivas R, Coriano H, Rivero S. Achatina fulica Bowdich, 1822 (Mollusca, Gastropoda, Achatinidae) carrier of helminthes, protozoa and bacteria in Northeast Venezuela. Boletín De Malariología Y Salud Ambiental. 2014;54(2):174–85.
Allen TD, Lawson PA, Collins MD, Falsen E, Tanner RS. Cloacibacterium normanense gen. Nov., sp. nov., a novel bacterium in the family Flavobacteriaceae isolated from municipal wastewater. Int J Syst Evol Microbiol. 2006;56(Pt 6):1311–6.
Cao SJ, Deng CP, Li BZ, Dong XQ, Yuan HL. Cloacibacterium rupense sp. nov., isolated from freshwater lake sediment. Int J Syst Evol Microbiol. 2010;60(Pt 9):2023–6.
Hyun DW, Shin NR, Kim MS, Kim JY, Kim PS, Oh SJ, Whon TW, Bae JW. Cloacibacterium haliotis sp. nov., isolated from the gut of an abalone, Haliotis discus hannai. Int J Syst Evol Microbiol. 2014;64(Pt 1):72–7.
de Goffau MC, Lager S, Salter SJ, Wagner J, Kronbichler A, Charnock-Jones DS, Peacock SJ, Smith GCS, Parkhill J. Recognizing the reagent microbiome. Nat Microbiol. 2018;3(8):851–3.
Lyra ML, Bletz MC, Haddad CFB, Vences M. The intestinal microbiota of tadpoles differs from those of Syntopic aquatic invertebrates. Microb Ecol. 2018;76(1):121–4.
Xiang Q, Zhu D, Chen QL, Delgado-Baquerizo M, Su JQ, Qiao M, Yang XR, Zhu YG. Effects of diet on gut microbiota of soil collembolans. Sci Total Environ. 2019;676:197–205.
Ling F, Whitaker R, LeChevallier MW, Liu WT. Drinking water microbiome assembly induced by water stagnation. ISME J. 2018;12(6):1520–31.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72.
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10(1):57–9.
Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol. 2004;47(2):161–77.
Ramette A. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 2007;62(2):142–60.
Acknowledgments
We thank Mr. Dan Zhu and Ms. He-Xiang Liu for their help with snail collection and dissection and Dr. Stijn Ghesquiere for permission to reprint the anatomic diagram of Pomacea canaliculata.
Funding
This research was partially supported by the Special Foundation of Basic Science and Technology Resources Survey of Ministry of Science and Technology of China (2017FY101203), the Shandong Provincial Natural Science Foundation (ZR2019MH093), National Natural Science Foundation of China (81902095), and the Open Project of Key Laboratory of Parasite and Vector Biology, China Ministry of Health (WSBKFKT-201804). The funding body had no role in the design of the study, data collection and analysis, interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
LHL designed the study, performed the experiments and drafted the manuscript. SL, YL, DQB, YHG, JTW, ZYY, GYM and ZXG discussed the study design and data analysis and revised the manuscript. YZ and YFT conceived the study and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures for snail treatment were approved by the Ethics Committee of Weifang Medical University. Collection of the P. canaliculata snails from Nanheng River was permitted by Qingpu Center for Disease Control and Prevention.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1.
The rarefaction curve of observed species in the samples.
Additional file 2: Fig. S2.
Venn diagram of shared OTUs among different gut sections of P. canaliculata.
Additional file 3: Table S1.
The relative abundance of the top 10 phyla of the gut microbiome in the three gut sections of P. canaliculata.
Additional file 4: Table S2.
The relative abundance of the top 10 genera of the gut microbiome in the three gut sections of P. canaliculata.
Additional file 5: Table S3.
Differences between intragroup and intergroup unweighted UniFrac distances.
Additional file 6: Table S4.
Differences between intragroup and intergroup weighted UniFrac distances.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Li, LH., Lv, S., Lu, Y. et al. Spatial structure of the microbiome in the gut of Pomacea canaliculata. BMC Microbiol 19, 273 (2019). https://doi.org/10.1186/s12866-019-1661-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1661-x