
Abstract
Background
Citrobacter species are Gram-negative opportunistic pathogens commonly reported in nosocomial-acquired infections. This study characterised four Citrobacter species that were isolated from surface water in the North West Province, South Africa.
Results
Phenotypic antimicrobial susceptibility profiles of the isolates demonstrated their ability to produce the extended-spectrum β-lactamase (ESBL). Whole genomes were sequenced to profile antibiotic resistance and virulence genes, as well as mobile genetic elements. In silico taxonomic identification was conducted by using multi-locus sequence typing and average nucleotide identity. A pangenome was used to determine the phylogenomic landscape of the Citrobacter species by using 109 publicly available genomes. The strains S21 and S23 were identified as C. braakii, while strains S24 and S25 were C. murliniae and C. portucalensis, respectively. Comparative genomics and sequenced genomes of the ESBL-producing isolates consisted of n = 91; 83% Citrobacter species in which bla-CMY−101 (n = 19; 32,2%) and bla-CMY−59 (n = 12; 38,7%) were prevalent in C. braakii, and C. portucalensis strains, respectively. Macrolide (acrAB-TolC, and mdtG) and aminoglycoside (acrD) efflux pumps genes were identified in the four sequenced Citrobacter spp. isolates. The quinolone resistance gene, qnrB13, was exclusive to the C. portucalensis S25 strain. In silico analysis detected plasmid replicon types IncHI1A, IncP, and Col(VCM04) in C. murliniae S24 and C. portucalensis S25, respectively. These potentially facilitate the T4SS secretion system in Citrobacter species. In this study, the C. braakii genomes could be distinguished from C. murliniae and C. portucalensis on the basis of gene encoding for cell surface localisation of the CPS (vexC) and identification of genes involved in capsule polymer synthesis (tviB and tviE). A cluster for the salmochelin siderophore system (iro-BCDEN) was found in C. murliniae S24. This is important when it comes to the pathogenicity pathway that confers an advantage in colonisation.
Conclusions
The emerging and genomic landscapes of these ESBL-producing Citrobacter species are of significant concern due to their dissemination potential in freshwater systems. The presence of these ESBL and multidrug-resistant (MDR) pathogens in aquatic environments is of One Health importance, since they potentially impact the clinical domain, that is, in terms of human health and the agricultural domain, that is, in terms of animal health and food production as well as the environmental domain.
Similar content being viewed by others


Background
Citrobacter species are characterised as Gram-negative, facultative anaerobe non-spore-forming rods that utilise citrate as the sole carbon source, and they belong to the family Enterobacteriaceae [1]. They are present in many environments, including water, soil, agriculture and food [2]. However, most species have been isolated from urinary and respiratory tract infections [3]. These Citrobacter species have also shown resistance to the common antimicrobials that are used to treat individuals who suffer from these infections [4].
Moreover, Citrobacter species can colonise the intestinal tract of humans and animals [5, 6], as they consist of virulence factors such as the Vi capsule polysaccharide of Salmonella enterica subsp. enterica serovar Typhi str. In its turn, CT18 (Salmonella Typhi, for short) plays a role in pathogenicity. Citrobacter species can be identified in terms of different metabolic and virulence gene associations. These associations are evidenced by species such as C. korseri, where macromolecular secretion systems and pathogenic islands occur [4]. The pathogenic islands involved in the salmochelin siderophore system (iroBCDEN gene cluster) for colonisation can also be acquired by means of horizontal gene transfer in some members of the family of Enterobacteriaceae [7]. These genes confer a competitive advantage for colonisation during infections [8].
Based on microbiological phenotypic methods and DNA hybridisation technology, the genus Citrobacter has been classified into 11 species [9]. The genus was further expanded to a total of 16 species based on whole-genome sequencing technology [10, 11]. Most of the phenotypical features of Citrobacter species mimic Escherichia coli, so that they are readily misidentified when phenotypic microbiological tests are used [3, 12]. The use of 16 S ribosomal RNA (rRNA) sequences and multi-locus sequence analysis (MLSA) can discriminate among the genera of Enterobacteriaceae [13, 14]. However, the use of 16 S rRNA gene sequencing does not enable differentiation among species [15,16,17,18]. Therefore, whole-genome sequencing has become the preferred genotypic tool for surveillance and detection of possible outbreaks of infections by Citrobacter species in the ecosystem [19,20,21,22]. This tool provides a higher resolution of phylogenetic relationships, as it can trace common infections from a One Health perspective [23, 24].
Citrobacter species that are multi-drug resistant, namely C. freundii, C. koseri, C. braakii and C. youngae, were recently identified and reported in the Democratic Republic of the Congo, Benin, Nigeria, Algeria, Tunisia and South Africa [25]. In South Africa, particularly in the Mooi River system of the North West Province, studies have shown the presence of multi-drug resistant bacteria [26,27,28]. The system flows through a combination of rural and urban residential settlements and is mainly influenced by urban, mine and agricultural activities. The latter two drivers contribute 5% towards the global domestic product (GDP) of South Africa [29]. Livestock farming is believed to play a part in the distribution of Citrobacter spp. in river systems, possibly by means of faecal run-off [30, 31]. The health problems caused by waterborne pathogen bacteria belonging to the Enterobacteriaceae are aggravated by the rise of the antimicrobial-resistant bacteria phenomenon. This phenomenon has been identified as one of the most considerable health challenges globally [32,33,34].
Many Enterobacteriaceae have been characterised as displaying the ability to produce ESBL [35]. These ESBLs are enzymes that can inactivate and hydrolyse clinically relevant third generation antibiotics such as cephalosporins and penicillins [36]. A whole-genome sequencing approach facilitates genomic profiling of ARBs and a comprehensive investigation of their shared genes. The genetic diversity of Citrobacter species strains can also be inferred by means of this approach. Environmental samples are often overlooked and could be the main drivers for AR that can be transmitted to animals and humans [37,38,39]. The present study characterised ESBL-producing Citrobacter species obtained from surface river water in the Mooi River, North West Province, by using antibiotic susceptibility testing, screening for ESBL production and whole-genome sequencing.
Results
Microbiological tests and antimicrobial resistance profiles
Based on their inability to ferment sorbitol, the four isolates were presumptively identified as pathogenic E. coli species on selective differential SMAC-CT agar. All isolates were characterised as ESBL-producing species upon observing sub-culturing on CHROMagar ESBL media. The metallic blue colonies with reddish halo formation indicated the presence of Klebsiella, Enterobacter or Citrobacter species. Furthermore, the Enteropluri results showed similar biochemical reactions between isolates S21 and S24. S25 was the only isolate that was able to produce hydrogen sulphide. All four isolates were able to utilise citrate as a carbon source. On the basis of a disc diffusion assay (DDA), the antimicrobial patterns reflected in Table 1 showed isolates’ resistance to ampicillin, amoxicillin and erythromycin, except for C. murliniae strain 24. All the isolates examined in the present project, apart from C. murliniae, were classified as multi-drug resistant (MDR), since they were resistant to beta-lactams, macrolides and aminoglycosides.
Genomic features and whole genome in silico taxonomic analysis
The scaffolds engendered by the sequenced Citrobacter genomes showed between 62 and 144 contigs (Table 2). Furthermore, pubMLST showed that genome strains S21 and S23 were C. braakii, while strains S24 and S25 were C. murliniae and C. portucalensis, respectively. The use of ANI showed that S21 and S23 were C. braakii (ANI > 98%). Strain S24 had a 99.26% ANI when the C. murliniae strain P080C CL was used as a reference. Strain S25 was identified by means of pubMLST as C. portucalensis and was found to share 90% of its identity with most of the C. freundii genomes. ANI classified this strain as C. portucalensis when C. portucalensis A60T was used as a reference.
The sequenced C. braakii strains S21 and S23 have genomes that are 4.98 Mb and 4.80 Mb in size, respectively. Different plasmids replicon types were present in the C. murliniae strain S24 and C. portucalensis strain S25. These genomes were larger (> 5.2 Mb) than the C. braakii genomes sequenced in this study. This was confirmed by the high number of coding sequences (CDS) in these two strains. The GC content of Citrobacter genomes ranged between 50% and 52%. The GC content of sequenced C. braakii genomes was approximately 52%, which was the same as the reference strain C. braakii ATCC 8090T and C. braakii strain DY2019 of the present study. The GC of the C. murliniae strain S24 was 50.62%, which was the same as the C. murliniae strain PC080C CL as isolated from human faeces available in GenBank. However, the genome size of C. murliniae strain PC080C CL was 5.0 Mb, which was slightly lower than that of C. murliniae strain S24 as sequenced in the present study. The genome size of the C. portucalensis strain S25 was 51.49%, which was similar to the C. portucalensis A60T reference strain.
Pangenomics and placement of the four sequenced Citrobacter species isolates
By using core and accessory genes, a pan-genome of 109 Citrobacter spp. was constructed to assess genetic diversity (Fig. 1). The sequenced and compared global Citrobacter species evidenced genetic diversity which facilitated the clustering of the Citrobacter species into distinct species. In this study, 58 924 genes were determined across the Citrobacter spp. and sequenced Citrobacter isolates genomes that were compared. The latter were used to construct a phylogenetic tree. A total of 1 790 core genes of the Citrobacter spp. Were identified, whereas the shell and cloud genes respectively totalled 2 976 and 53 243.

A pangenome analysis of the global 109 Citrobacter species strains, including the four Citrobacter isolates, highlighted by means of high font size on the phylogenetic tree. A maximum likelihood was used to construct the phylogenetic tree (unrooted), as based on the core and accessory genes. Citrobacter species are colour coded in terms of their different respective clusters. The first, inner circle represents the sources of the strains, and the second, outer circler the beta-lactamase genes (bla-CMY) identified in this study
The sequenced C. braakii strains S23 and S21 clustered together with C. braakii genome strains CB00164 and CB00130, respectively, as isolated from humans in the United States. The present study found that gene clusters distinguished C. braakii from the other Citrobacter species. Examples of these genes include chitoporin, sapF, yiaB, aminopeptidase ypdF, urease accessory protein UreD, putative protein YmdA, putative nucleoside permease nupX, streptomycin-3’-adenyl transferase, cfaA fimbrial subunit E and putative Type II secretion system proteins E and G, among others. The sequenced C. murliniae strain S24 grouped closely with the C. gillenii genomes that had mostly been isolated from wastewater treatment plants in the United Kingdom.
These two species, along with the unassigned Citrobacter species, lack most of the core genes when compared to the other assigned Citrobacter species. The core genes that are absent in the latter group include idcA, murein tetrapeptide carboxypeptidase, ppaC, putative manganese-dependent inorganic pyrophosphate, sutR, HTH-type transcriptional regulator, bioC, malonyl-acyl;-carrier, protein-O-methyltransferase, pdxK, pyridoxamine and protein FdrA. In the present study, strain S25 grouped with C. portucalensis strain CB00203 as isolated from a human rectal swab in the United States as well as from animal sample strains PBIO1950 and PBIO1938 as isolated from a fly (Musca domestica) in Rwanda.
Population structure of ESBL-producing Citrobacter species
Through a de novo assembly-based pangenome study, the evolutionary relationships among 109 ESBL-Citrobacter genomes were deduced. Except for the Citrobacter spp. cluster, which could not be assigned with species identification, strains consistently grouped in accordance with their MLST profiles. MLST and pangenome analysis allocated the following five species to their respective clusters: C. braakii (n = 59), C. gillenii (n = 6), C. europaeus (n = 7), C. murliniae (n = 1), C. portucalensis (n = 31) and Citrobacter spp. (n = 4). The pangenome phylogenetic tree is represented by samples from animals (n = 21), humans (n = 44), the environment (n = 35) and plants (n = 11). Each Citrobacter species cluster consisted of human and animal isolates, except for the clusters of C. murliniae and C. gillenii.
The vast majority of Citrobacter species (n = 91; 83%) were classified as ESBL-producing isolates comprising one of the 18 identified bla-CMY- genes: that is, bla-CMY-, bla-CMY−26, bla-CMY−39, bla-CMY−46, bla-CMY−59, bla-CMY−66, bla-CMY−70, bla-CMY−65, bla-CMY−71, bla-CMY−74, bla-CMY−77, bla-CMY−82, bla-CMY−83, bla-CMY−86, bla-CMY−93, bla-CMY−100, bla-CMY−101 and bla-CMY−104. No CMY genes were present in the C. europaeus and C. gillenii clustered strains. The bla-CMY−101 (n = 19; 32,2%) and bla-CMY−82 (n = 18; 30,5%) were prevalent in the genomes of C. braakii, whereas bla-CMY−59 (n = 12; 38,7%) and bla-CMY−77 (n = 10; 32,3%) occurred in the genomes of C. portucalensis.
Population structure of the quinolone-resistant Citrobacter species
The quinolone resistance genes (qnrB) were found in 42 of the 109 genomes of the Citrobacter species examined (Fig. 2). Gene coding for flouroquinolone resistance were found in only three genomes of Citrobacter spp.: C. europaeus and C. portucalensis showed thirteen quinolone resistance genes that were significant, namely qnrB10, qnrB13, qnrB17, qnrB18, qnrB21, qnrB27, qnrB33, qnrB44, qnrB49, qnrB57, qnrB58, qnrB6 and qnrB69. Each of the unnamed strains of Citrobacter species had the qnrB21 profile. The qnr27 gene was present in 83,3% (n = 5) of the C. europaeus strains that had been isolated from humans (n = 6), except for strain CB00169. The qnrB13 gene was present in the genome of the sequenced C. portucalensis strain S25 as isolated from the environment: however, this gene was also detected in human and animal samples. This strain clusters with the C. portucalensis genomes CB00079, PBIO1950 and PBIO1938 strains, which are qnrB17-positive. C. portucalensis appears to contain a variety of qnrB genes: eleven of these were determined in this cluster.

Pangenome phylogenetic tree showing clustering of the quinolone resistance Citrobacter species strains (n = 42) as based on a maximum-likelihood phylogenetic tree. Approximately 13 qnrB genes were determined and clustering was based on MLST genome identification of the Citrobacter species
Antibiotic resistance genes on the four Citrobacter genomes
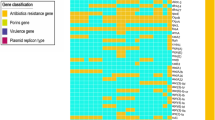
The sequences of the Citrobacter species were analysed for antibiotic resistance genes on the four sequenced Citrobacter genomes. Forty antibiotic resistance genes were identified by the Comprehensive Antibiotic Resistance Database (CARD) across the four Citrobacter species (Fig. 3). The acrAB-TolC complex which encode for the intermembrane tripartite multidrug for macrolides, including transcriptional activator marA were the common identified ARGs determined in the four sequenced Citrobacter genomes. This genotypic profile of each of the four isolates is well supported by the phenotypic profile (Table 1) observed by using disc diffusion assay (DDA). The Citrobacter spp. S21, S23, S24 and S25 strains appear to have distinctive beta-lactamase genes such as bla-CMY−83, bla-CMY−74, bla-CMY−59 and bla-CMY−77, respectively (Fig. 3).

Heatmap reflecting the presence and absence of virulence genes (65 in total) and antibiotic resistant genes (40 in total) across the four sequenced Citrobacter spp. isolates
The acrD efflux pump gene and the regulon baeR that were identified in these genomes corroborated the finding of phenotypic resistance to aminoglycosides, as was demonstrated by all four Citrobacter spp. The C. portucalensis strain S25 consisted of the quinolone-resistant gene qnrB13; which is confirmed by phenotypic susceptibility to nalidixic acid. The quinolone resistance genes such as emrAB/TolC, patA, and mdtHK efflux complexes were detected in the sequenced Citrobacter genomes. Most of these variables seemed to be connected to multidrug efflux pump processes, which included genes such as mdtABCGHK. The heatmap depicts the fact that C. braakii S21 and S23 strains were similar in their ARG profile to a greater degree than the others (Fig. 3). This was confirmed by the presence of multidrug resistance mdtF gene in these two genomes.
The phenotypic features that were identified by using DDA affirmed the presence of the extended beta-lactamase (bla-CMY) genes in all four sequenced genomes. The pmrE gene, which affects the peptide antibiotics class by means of antibiotic target alteration, was the only AR gene found in C. braakii strain S21 (Fig. 3). The inner membrane transporter gene, acrF, which is known as a multi drug resistant gene, was only found in the C. murliniae S24 strain. However, this gene is related to the acrB gene, which was also identified in the four Citrobacter genomes in this study. A quinolone resistance gene qnrB13 was present in the genome of C. portucalensis strain S25. Pangenomic analysis of the C. portucalensis strains showed that the quinolone gene (qnrB) differentiates this species from the other three genomes strains that were compared in this study.
Determination of the virulence assays and genes on the four Citrobacter genomes
All four isolates showed a similar extracellular enzyme production profile, except for isolate S24, which did not produce catalase and gelatinase. Overall, though, lecithinase, gelatinase, lipase, DNase, proteinase and catalase were produced. A heatmap was generated of the virulence gene profile across the four sequenced genomes so as to identify the main pathogenicity genes in C. braakii, C. murliniae, and C. portucalensis (Fig. 3). The present project discovered primary virulence factors linked to adhesion, biofilm formation, iron intake, the movement of siderophores, environmental stress response and cell surface localisation in the associated strains (Fig. 3). It was found that gene-encoding cell surface localisation of the CPS (vexABCDE), and genes involved in capsule polysaccharide biosynthesis (tviBCDE) could be exclusively used to differentiate the C. braakii genomes from those of C. murliniae and C. portucalensis. The C. murliniae S24 strain exhibited a virulence profile that was distinct from the other genomes that had been compared. This genome consisted of the iroABCDEN gene cluster that encodes a heme receptor protein shuA for the iron-salmochelin siderophore system. Moreover, the iron uptake gene, chuY, was exclusive to this strain.
Mobile genetic elements and plasmids
Table 3 reflects the fact that all the Citrobacter genomes of this study, excluding C. braakii S23, indicated the presence of mobile genetic elements such as insertion sequences (ISs) and transposons (Tns). The three genomes that had insertion sequences shared a commonly identified element, IS5075. Mobile genetic elements such as ISEsa1 and ISEsa2 were shared among C. murliniae and C. portucalensis genomes. The unique profile of IS elements in C. portucalensis strain S25 were found in IS903, ISSpu2 and ISSKox1. In contrast, ISEc33, ISKpn24 and ISKpn26 were present in C. murliniae strain S24. Except for the C. braakii strain S23 genome, only the transposase Tn5403 was found in the sequenced Citrobacter genomes in this study. Type IV coupling proteins (T4CPs) were present in all the sequenced Citrobacter spp. genomes, except in C. braakii strain S21.
Genomes of Citrobacter spp. consisted of a diverse category of translocation systems known as the bacterial Type IV secretion systems (T4SSs), except in the genome of C. braakii strain S23. Genome C. murliniae strain S24 presented a large number of T4SSs, which merits further investigation. The TraD, TraF, TraH and TraG Type IV secretion were found to be common among C. murliniae S23 and C. portucalensis strain S25. Except for C. braakii S23, all the other sequenced Citrobacter species carried plasmids, while C. murliniae consisted of different plasmids (Table 3 and Supplementary Table 1). Plasmid replicons and incompatibility types were detected on the draft of assembled plasmids. Four draft plasmids were identified in the C. murliniae strain, which is associated with the T4SS clusters (Vir-Tra-Trb). The plasmid AA543 of C. murliniae strain S25 had a VirD4-TraABCDEFGH-TrbABCDEFGHIJKLV gene cluster, while plasmid AC978 had a TraX_F-TrbBCDEFGHIJLN gene cluster. Citrobacter portucalensis strain S25 carried four plasmids, and plasmid AB130 consisted of the TraABCEFGHIKLNVW gene cluster.
Typing analysis of this plasmid AC978 demonstrated that it was related to the C. freundii (CP037735) (Supplementary Table 1). Plasmids AA543 and AC978 of C. murliniae strain S24 were classified as conjugative plasmids and were closely related to Klebsiella oxytoca strain CAV1374 plasmid pKPC (CP011635) and Escherichia coli JJ1886 (CP006788), respectively. None of the plasmids discovered in the reference strain P080C_CL and sequenced C. murliniae strain S24 were comparable. Sequence typing of the C. portucalensis strain S25 plasmid AB130 and AF578 were identified to be closely related to C. freundii (CP037735) and Salmonella enterica subsp. enterica serovar Cubana str. CFSAN002050 (NC_021819), respectively. Meanwhile, plasmids AF384 and AF578 of this strain were found to be closely related to Enterobacter roggenkampii (CP019840) and Pelobacter propionicus DSM 2379 (CP000484), respectively.
Discussion
Four Citrobacter species that carried ESBL genes were identified during the present study, which assessed the characteristics of Enterobacteriaceae bacteria that circulate in the Mooi River system in the North West Province, South Africa. The prevalence of ESBL-producing Enterobacteriaceae is receiving increasing attention as a threat to global health [40]. Environmental surface water is frequently disregarded in these studies, though. In the present study, the genomes of the four Citrobacter species were first assumed to be E. coli on the basis of conventional phenotypic microbiological tests. However, by utilising high-resolution taxonomic categorisation that included ANI and pangenome analysis, these isolates were verified as belonging to the Citrobacter species. The use of traditional methods for Citrobacter species identification is not recommended, because they are frequently flawed [3, 17].
The present project dispersed the environmental strains of the Citrobacter species among its various genetic clusters, which raises concerns about the spread of antibiotic-resistance genes in humans and animals as well as the acquisition of such genes. Additionally, AR genes, virulence genes and mobile genetic elements were investigated. In the case of river systems especially, the population genetic structure of Citrobacter genomes has not been fully documented. The first Citrobacter species genomes to be isolated from surface water in South Africa exhibit heterologous unique structures that are closely related to the human and animal-associated Citrobacter spp. genomes. These Citrobacter species were successfully identified in the present study as C. braakii (n = 2), C. murliniae (n = 1) and C. portucalensis (n = 1). Thus far, only one genome was sequenced and documented from a human feaces sample in Norwich, United Kingdom [41] for C. murliniae: it is the only current genome that is available in GenBank. C. portucalensis has been well characterised by means of various biochemical tests [11] However, a test differentiation among Citrobacter species such as C. braakii, C. freundii, and C. europaeus is lacking.
Antibiotic resistance remains a public health concern [42]. Some of the antibiotics that screened for antibiotic susceptibility in the four Citrobacter species belong to the critically essential antibiotic classes [43]. Resistance against fluoroquinolones, aminoglycosides, β-lactams, and macrolides is problematic, particularly when it comes to treating Enterobacteriaceae infections [40]. The sequenced Citrobacter spp. genomes of the present study displayed resistance against these critically and highly important antibiotic classes, the latter as described by the WHO [42]. The phenotypic presentation of the classes of antibiotics referred to in the present study included aminoglycosides, β-lactams and macrolides.
A comprehensive antimicrobial profile of the four Citrobacter genomes was explored by means of WGS. We found that all species had genes that encoded diverse types of antibiotic efflux pumps, including resistance-nodulation-cell division (RND) types, major facilitator superfamily (MFS) types, ATP-binding cassette (ABC) types, all of which confer resistance to aminoglycoside antibiotics, fluoroquinolone and macrolides (Fig. 3). If resistant bacteria enter a freshwater stream, their presence could be associated with the potential risk of transporting resistant genes from harmless bacteria to pathogenic ones that end up in humans who interact with aquatic environments [44].
Over the last decade, Citrobacter spp. have also been reported to be resistant to the most generally used antibiotics, including ampicillin, cefotaxime, aminoglycoside and tetracyclines [45]. The present study established that the four sequenced Citrobacter spp. and global strains harboured genes that encode resistance to a critically important antimicrobial such as beta-lactamase. The phylogenomic landscape of the ESBL-producing Citrobacter spp. was employed to infer the prevalence of CMY genes. The vast majority of Citrobacter spp. (n = 91; 83%) were classified as ESBL-producing isolates consisting of one of the 18 identified bla-CMY− genes, including bla-CMY−26, bla-CMY−39, bla-CMY−46, bla-CMY−59, bla-CMY−66, bla-CMY−70, bla-CMY−65, bla-CMY−71, bla-CMY−74, bla-CMY−77, bla-CMY−82, bla-CMY−83, bla-CMY−86, bla-CMY−93, bla-CMY−100, bla-CMY−101 and bla-CMY−104. Most ESBL-producing isolates have been reported in Enterobacteriaceae [36, 40]. In the present case, the bla-CMY−101 (n = 19, 32,2%) and bla-CMY−82 (n = 18; 30,5%) were prevalent in the C. braakii genomes. The bla-CMY−101 and bla-CMY−82 genes have been reported from C. braakii strains that had been isolated from a river in China [46].
The present study suggests that the beta-lactamase genes bla-CMY−101 and bla-CMY−82, which contain isolates, are circulating in the river system, However, they are not limited to this environment, as some of bla-CMY−101 and bla-CMY−82 were isolated from plants and humans (Fig. 1). The widespread use of broad-spectrum antibiotics might be the key cause of the emergence and dissemination of elevated levels of antimicrobial-resistant strains in the freshwater of the Mooi River [28]. The present study depicts the spectrum of Citrobacter in the freshwater of the Mooi River and found that Citrobacter is most often associated with multidrug resistance. This resistance might be due to the acquisition of resistance genes such as β-lactamase, efflux pumps or alternation in porins that act synergistically as channels for drug entry so as to confer resistance among Citrobacter isolates [4].
A fluoroquinolone resistance indicator, qnrB13, was found in C. portucalensis strain S25 and was compared with other C. portucalensis genomes. The chromosomal antibiotic resistance qnrB gene has also been reported in other Citrobacter species [12, 47], which suggests that Citrobacter spp. may be the origin of chromosomal antibiotic resistance. The hypothesis was based on species distribution (> 60% in Citrobacter spp.) [47,48,49] and the fact that the qnrB gene is prevalent in C. freundii strains isolated from human clinical specimens [50]. Other studies support the notion that the genus Citrobacter is the origin of qnrB, as this gene is distributed in species such as C. freundii, C. braakii, C. youngae, and C. werkmanii, However, allele variation specific to each species does exist [12, 49].
The present study demonstrated that the C. braakii genomes can be distinguished from C. murliniae and C. portucalensis on the basis of gene encoding for cell surface localisation of the CPS (vexC) as well as genes involved in capsule polymer synthesis (tviB and tviE). Extant studies also report these genes in this kind of context [4]. The present study found that C. murliniae strain S24 could not produce catalase, which was confirmed by the absence of the katB gene. A gene cluster of the salmochelin siderophore system (iroBCDEN) was found in C. murliniae S24: it is important for the pathogenicity pathway that confers an advantage in colonisation. This gene cluster was first described in Salmonella enterica [51], and orthologous genes were also reported in some E. coli strains, which suggests acquisition through horizontal gene transfer [52]. The present study is the first report that iron-BCDEN gene clusters were identified in Citrobacter species, particularly in the C. murliniae genome.
Other virulence factors identified in all Citrobacter spp. strains were associated with iron uptake (chu and ent). Pathogenic bacteria need iron, which they must acquire during infection, if they are to thrive in a mammalian host [53]. However, C. murliniae contained an additional iron update gene, namely chuF. This gene is an anearobilin reductase that exhibits kinetic cooperativity during the process of biochemical degradation in E. coli O157:H7 [54]. Among members of the genus Citrobacter., especially C. koseri, extant studies have found strains that consisted of this iron gene cluster [4]. However, in the present study, this gene cluster was found in the four sequenced genomes.
The identified plasmids, as determined in the present study, were mostly associated with T4SSs. The latter is a broad distribution group that secretes macromolecules in Gram-negative and positive bacteria as well as other eukaryotic cells [55]. They play a crucial role in the interactions between bacteria and their surrounding environments by secreting many virulence factors, particularly in the case of pathogenic bacteria [56]. The Type IV secretory genes are not fully exploited: however, they have been reported in most genomes of the Citrobacter species [4]. The present study established that bacterial T4SSs were present among the sequenced Citrobacter genomes strains, except in the case of C. braakii strain S23. This was most probably due to the fact that the plasmid was absent from the strain.
We detected an incompatibility group of plasmids replicon genes, including IncP and IncH. The IncP plasmid replicon type plays important roles around the transfer of antibiotic resistance genes and other types of genetic information among bacteria [57]. They also contain genes that produce virulence factors, engendering bacteria that carry IncP plasmids that are more dangerous to humans than other organisms. The IncP plasmids are sometimes referred to as wild-type ones due to their ability to transfer themselves among many different bacterial species [57]. The colicinogenic (or Col) factors, such as plasmid Col(VCM04), determine the production of proteins called colicins, which can kill other bacteria. Given certain environmental conditions, plasmids can provide many different types of selective advantages, including antibiotic resistance, resistance to pollutants or UV and biofilm formation [58].
The sequenced C. Murliniae strain S24 consisted of three distinct types of T4SS gene clusters (Vir, Tra and Trb) that were encoded in the plasmid AA543: VirD4, TraABCDEFGH, and TrbABCDEFGHIJKLV. Meanwhile, the plasmid AC978 encoded the TraXF and Trb gene clusters TraXF and TrbBCDEFGHIJLN. It is well known that some T4SS components, such as the tra-operon involved in conjugation, exhibit significant sequence and structural homology and have identical protein activities that are associated with pathogenicity [59, 60]. Therefore, the T4SS is not limited to certain bacterial species, as it has been reported in other pathogenic species, such as Pseudomonas aeruginosa, Vibrio cholerae, enteroaggregative Escherichia coli, Burkholderia thailandensis, Serratia marcescens, Burkholderia malle, and Salmonella enterica, which are also involved in conjugative transfer and plasmid replication [61].
Conclusion
Our findings suggest that C. portucalensis, C. murliniae, and C. braakii are multi-drug resistant pathogens with intrinsic genes that encode forESBL. The use of whole-genome sequencing identified these species and the phylogenomic structure of the four Citrobacter species that circulate in surface water in the Mooi River of the North West Province, South Africa. The C. murliniae strain showed a unique profile with an iron-BCDEN gene cluster of salmochelin siderophore system. Fluoroquinolone resistance (qnrB13) was only detected in C. portucalensis, which was closely related to animal and human C. portucalensis.
These genomes represent important reference points. They act as crucial pieces of detailed genetic information and phenotypic data that will be used as models for the characterisation of antibiotic resistance mechanisms and interactions. Of further importance is that the study, as centred on ESBL-producing Citrobacter species, is that all four genomes carried the bla-CMY-gene, thus presenting a threat in its transmission potential for the environment and possibly other sectors, the latter as described in the One Health model.
Materials and methods
Sample collection and isolation
Samples were collected from a Mooi River site (26° 42’ 29.3” S 027° 06’ 20.6” E), as described in earlier part of the present project [62]. However, in the original study, the aim was to characterise Enterobacteriaceae and their potential antibiotic resistance within surface water in the Mooi River system. In summary of that study, water samples (100 ml) were processed by the Colilert-18 (IDEXX, USA) procedure. Selective media of Sorbitol MacConkey agar (Oxoid, UK) infused with antibiotics (0.05 mg/L Cefixime trihydrate (Sigma Aldrich, India) and 2.5 mg/L Potassium Tellurite (Sigma Aldrich, Japan) (SMAC-CT agar)) were further used to isolate presumptive pathogenic Enterobacteriaceae species incubated at 37 °C for 24 h.
The isolates were further subjected to Rainbow agar O157 (Biolog, USA) infused with Novobiocin sodium salt (Sigma Aldrich), and were subsequently incubated at the same conditions as those of the SMAC-CT agar. The reference strains that were used in this study were non-pathogenic E. coli ATCC 10,536, pathogenic E. coli O157:H7 ATCC 700,728 and ESBL-producing E. coli ATCC 35,218 (Microbiologics, USA). ESBL-producing Citrobacter isolates were confirmed phenotypically on CHROMagar™ ESBL (CHROMagar, France). The agar differentiates between ESBL E. coli (red colony presentation) and ESBL Klebsiella, Enterobacter and Citrobacter spp. (metallic blue, with or without a red halo). The selection of these four isolates was based on their ability to produce the ESBL enzymes.
These ESBL-producing Enterobacteriaceae were analysed according to the manufacturer’s instructions on the Enteropluri-Test strip (Liofilchem, Italy). A set of 13 biochemical reactions was detected, which included glucose fermentation, gas production, hydrogen sulphide production, lysine decarboxylation, ornithine decarboxylation, adonitol fermentation, lactose fermentation, arabinose fermentation, sorbitol fermentation, dulcitol fermentation, phenylalanine deamination, urea hydrolysis and citrate utilisation [63]. Furthermore, seven in-vitro virulence assays were conducted, which included catalase, proteinase, DNase, lipase, gelatinase and lecithinase [63].
Antimicrobial susceptivity testing
Pure cultures of Citrobacter isolates (n = 4) were sub-cultured onto Mueller-Hinton agar (Biolabs, USA) by using the Kirby-Bauer disc diffusion method [64] for a set of 12 antibiotics. The antibiotics included ampicillin (10 µg), amoxicillin (10 µg), cefazolin (30 µg), erythromycin (15 µg), gentamycin (10 µg), kanamycin (30 µg), streptomycin (10 µg), tetracycline (30 µg), ciprofloxacin (5 µg), chloramphenicol (30 µg), neomycin (30 µg) and nalidixic (30 µg) (Oxoid, UK). The results were interpreted in accordance with the M100 CLSI guideline for Antimicrobial Susceptibility Testing (2017) (https://clsi.org/standards/products/free-resources/access-our-free-resources/) and the clinical breakpoint for bacteria (v12.0) EUCAST guidelines (https://www.eucast.org/clinical_breakpoints/).
Genomic DNA extraction and whole-genome sequencing
Overnight broth cultures of the bacterial isolates were centrifuged and pelleted for DNA extraction. The Chemagic kit (Perkin Elmer, Germany) was used to extract the total genomic DNA of the Citrobacter isolates while following the manufacturer’s protocol. The concentration and quality of the DNA were determined by using the Qubit 2.0 fluorometer (Thermofisher-Scientific, USA). The DNA integrity was monitored on a 2% agarose gel pre-stained with ethidium bromide and visualised on a UV transilluminator. Paired end libraries were generated by using the NEBNext kit (BioLabs, England) according to the NEBNext for Illumina protocol. Cluster generation and sequencing were performed by means of the MiSeq Reagent Kit V3 (2 × 300 bp) on the MiSeq 2000 (Illumina, USA).
Genome assembly and annotation
Quality of the sequenced reads were assessed by using FastQC software 0:10.1 [65]. Trimmomatic [66] was used to remove the ambiguous nucleotide reads in terms of a threshold of reads > Q28. The paired end trimmed reads of Citrobacter spp. strains were assembled de novo by using the SPAdes v1.1.0 pipeline [67]. The minimum contig length was set to 500 bp, and kmer sizes 21, 33, 55, 77, 99 and 127 were used for the assembly. CheckM [68] was additionally used to assess potential contaminants in individual assembled genomes. Quast v 2.3 [69] was used to evaluate the draft genome assemblies of Citrobacter species. The assembled contigs were annotated by means of the NCBI prokaryotic genome automatic annotation pipeline (PGAAP) [70].
Whole genome in-silico taxonomic analysis and pangenomics
The genomic sequence and the pubMLST database of Citrobacter spp. on the website https://pubmlst.org/ were used to determine the species identity. FastANI version 1.3 [71] was used to estimate the isolate’s ANI against closely related genomes of Citrobacter species as references. Based on the ANI, a percentage identity of > 90% against the reference genome was considered to belong to the same species. For pangenome analysis, 109 Citrobacter publicly available genomes were retrieved from GenBank (Supplementary Table 2), and were selected on the basis of pubMLST match profiles of the sequence genomes. The inclusion was also based on the One Health concept, so as to determine relatedness to animal, human and environmental strains. This included the geographical region of Africa and worldwide genomes representation.
All the retrieved and sequenced Citrobacter strain genomes in this study were further annotated by using Prokka v.1.14.0 [72]. Similarity searches in terms of the coding domain sequences (CDSs) of assembled genomes were conducted by using pair-wise BLASTp [73] and the Markov Cluster Algorithm (MCL). Clusters were created by using paralogs of the genomes and were ordered with a view to the presence/ absence of orthologs [74]. Pangenome clusters were defined as follows: core genes present in all isolates; soft core genes present in at least 95% of isolates; shell genes present between 15 and 95% of isolates; cloud genes in less than 15% of isolates. The phylogenetic tree of the Citrobacter genomes was visualised by using ITOL [75].
Antibiotic resistance, virulence gene detection, mobile genetic elements, and accession numbers
The ABRicate pipeline, which was assessed on 25 January 2023, was employed to identify antibiotic resistance and virulence genes in the four genomes of the isolated Citrobacter species. Antimicrobial resistance determinants were identified in each assembled genome by using the ResFinder database (–db ResFinder) [76] with minimum identity and coverage thresholds of 75 (– minid 75) and 50% (–mincov 50), respectively. The Comprehensive Antibiotic Resistance Database (CARD) was also employed to determine the AR genes. ABRicate was further used to detect virulence factors in the sequenced genomes of Citrobacter spp. By using the Virulence Factor Database (VFDB; –db vfdb) [77, 78] in terms of minimum identity and coverage thresholds of 70 (–minid 70) and 50% (–mincov 50), respectively.
Mobile genetic elements were investigated on the sequenced genomes of the Citrobacter by means of Mobile Element Finder v1.0.3 (2023-01-20) on the Center for Genomic Epidemiology platform [79]. Plasmid replicons were identified by ABRicate on the sequenced genomes by using the PlasmidFinder database [80]. The MOB-Typer tool from MOB-Suite software v1.4.9 [81], and OriTfinder [82] was used to characterise plasmid sequences.
Data Availability
Citrobacter species isolates S21, S23, S24 and S25 genome sequences were deposited in NCBI GenBank under the accession numbers SRR23239838, SRR23239837, SRR23239836, and SRR23239835, respectively.
References
Frederiksen W. Citrobacter. Bergey’s Man Syst Archaea Bacteria 2015:1–23.
Murray PR, Holmes B, Aucken HM. Citrobacter, Enterobacter, Klebsiella, Plesiomonas, Serratia, and other members of the enterobacteriaceae. Topley & Wilson’s Microbiology and Microbial Infections[if a book, double check if city and place of publication are necessary] 2010.
Nayar R, Shukla I, Sultan A. Epidemiology, prevalence and identification of Citrobacter species in clinical specimens in a tertiary care hospital in India. Int J Sci Res Publications. 2014;4(4):1–6.
Yuan C, Yin Z, Wang J, Qian C, Wei Y, Zhang S, Jiang L, Liu B. Comparative genomic analysis of Citrobacter and key genes essential for the pathogenicity of Citrobacter koseri. Front Microbiol. 2019;10:2774.
Arens S, Verhaegen J, Verbist L. Differentiation and susceptibility of Citrobacter isolates from patients in a university hospital. Clin Microbiol Infect. 1997;3(1):53–7.
Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. Citrobacter rodentium: Infection, inflammation and the microbiota. Nat Rev Microbiol. 2014;12(9):612–23.
Magistro G, Hoffmann C, Schubert S. The salmochelin receptor IroN itself, but not salmochelin-mediated iron uptake promotes biofilm formation in extraintestinal pathogenic Escherichia coli (ExPEC). Int J Med Microbiol. 2015;305(4–5):435–45.
Balbontín R, Villagra N, Pardos de la Gándara M, Mora G, Figueroa-Bossi N, Bossi L. Expression of IroN, the salmochelin siderophore receptor, requires mRNA activation by RyhB small RNA homologues. Mol Microbiol. 2016;100(1):139–55.
Pilar AVC, Petronella N, Dussault FM, Verster AJ, Bekal S, Levesque RC, Goodridge L, Tamber S. Similar yet different: phylogenomic analysis to delineate Salmonella and Citrobacter species boundaries. BMC Genomics. 2020;21:1–13.
Ribeiro TG, Izdebski R, Urbanowicz P, Carmeli Y, Gniadkowski M, Peixe L. Citrobacter telavivum sp. nov. with chromosomal mcr-9 from hospitalized patients. Eur J Clin Microbiol Infect Dis. 2021;40:123–31.
Ribeiro TG, Goncalves BR, da Silva MS, Novais A, Machado E, Carrico JA, Peixe L. Citrobacter portucalensis sp. nov., isolated from an aquatic sample. Int J Syst Evol MicroBiol. 2017;67(9):3513–7.
Ribeiro TG, Novais Â, Branquinho R, Machado E, Peixe L. Phylogeny and comparative genomics unveil Independent diversification trajectories of qnrB and genetic platforms within particular Citrobacter species. Antimicrob Agents Chemother. 2015;59(10):5951–8.
Clermont D, Motreff L, Passet V, Fernandez J-C, Bizet C, Brisse S. Multilocus sequence analysis of the genus Citrobacter and description of Citrobacter pasteurii sp. nov. Int J Syst Evol MicroBiol. 2015;65(Pt5):1486–90.
Warren JR, Farmer J III, Dewhirst FE, Birkhead K, Zembower T, Peterson LR, Sims L, Bhattacharya M. Outbreak of nosocomial Infections due to extended-spectrum β-lactamase-producing strains of enteric group 137, a new member of the family Enterobacteriaceae closely related to Citrobacter farmeri and Citrobacter amalonaticus. J Clin Microbiol. 2000;38(11):3946–52.
Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029.
Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45(9):2761–4.
Fox GE, Wisotzkey JD, Jurtshuk P Jr. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Evol MicroBiol. 1992;42(1):166–70.
Lekota KE, Hassim A, Mafofo J, Rees J, Muchadeyi FC, Van Heerden H, Madoroba E. Polyphasic characterization of Bacillus species from anthrax outbreaks in animals from South Africa and Lesotho. J Infect Developing Ctries. 2016;10(8):814–23.
Reuter S, Ellington MJ, Cartwright EJ, Köser CU, Török ME, Gouliouris T, Harris SR, Brown NM, Holden MT, Quail M. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern Med. 2013;173(15):1397–404.
Dallman TJ, Byrne L, Ashton PM, Cowley LA, Perry NT, Adak G, Petrovska L, Ellis RJ, Elson R, Underwood A. Whole-genome sequencing for national surveillance of Shiga toxin–producing Escherichia coli O157. Clin Infect Dis. 2015;61(3):305–12.
Park ST, Kim J. Trends in next-generation sequencing and a new era for whole genome sequencing. Int Neurourol J. 2016;20(Suppl 2):76.
Fono-Tamo EUK, Kamika I, Dewar JB, Lekota KE. Comparative Genomics revealed a potential threat of Aeromonas Rivipollensis G87 strain and its antibiotic resistance. Antibiot (Basel) 2023, 12(1).
Ashton PM, Nair S, Peters TM, Bale JA, Powell DG, Painset A, Tewolde R, Schaefer U, Jenkins C, Dallman TJ. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ. 2016;4:e1752.
Lekota KE, Hassim A, Madoroba E, Hefer CA, van Heerden H. Phylogenomic structure of Bacillus anthracis isolates in the Northern Cape Province, South Africa revealed novel single nucleotide polymorphisms. Infect Genet Evol. 2020;80:104146.
Ogunlaja A, Ogunlaja OO, Olukanni OD, Taylor GO, Olorunnisola CG, Dougnon VT, Mousse W, Fatta-Kassinos D, Msagati TAM, Unuabonah EI. Antibiotic resistomes and their chemical residues in aquatic environments in Africa. Environ Pollut. 2022;312:119783.
Carstens A, Bartie C, Dennis R, Bezuidenhout C. Antibiotic-resistant heterotrophic plate count bacteria and amoeba-resistant bacteria in aquifers of the Mooi River, North West Province, South Africa. [Smart-case journal titles, viz. important word takes capital, check the rest of the list for this]. J Water Health. 2014;12(4):835–45.
Molale L, Bezuidenhout CC. Antibiotic resistance, efflux pump genes and virulence determinants in Enterococcus spp. from surface water systems. Environ Sci Pollut Res. 2016;23:21501–10.
Bosch J, Bezuidenhout C, Coertze R, Molale-Tom L. Metal-and antibiotic-resistant heterotrophic plate count bacteria from a gold mine impacted river: the Mooi River system, South Africa. Environ Sci Pollut Res 2022:1–15.
Saayman M, Saayman A, Rhodes JA. Domestic tourist spending and economic development: the case of the North West Province. Dev South Afr. 2001;18(4):443–55.
McAllister T, Topp E. Role of livestock in microbiological contamination of water: commonly the blame, but not always the source. Anim Front. 2012;2(2):17–27.
Nag R, Nolan S, O’Flaherty V, Fenton O, Richards KG, Markey BK, Whyte P, Bolton D, Cummins E. Quantitative microbial human exposure model for faecal indicator bacteria and risk assessment of pathogenic Escherichia coli in surface runoff following application of dairy cattle slurry and co-digestate to grassland. J Environ Manage. 2021;299:113627.
Fouladkhah AC, Thompson B, Camp JS. The threat of antibiotic resistance in changing climate. In., vol. 8: MDPI; 2020: 748.
Huang F-Y, Chen Q-L, Zhang X, Neilson R, Su J-Q, Zhou S-Y-D. Dynamics of antibiotic resistance and its association with bacterial community in a drinking water treatment plant and the residential area. Environ Sci Pollut Res. 2021;28(39):55690–9.
Buelow E, Ploy M-C, Dagot C. Role of pollution on the selection of antibiotic resistance and bacterial pathogens in the environment. Curr Opin Microbiol. 2021;64:117–24.
Zurfluh K, Hächler H, Nüesch-Inderbinen M, Stephan R. Characteristics of extended-spectrum β-lactamase-and carbapenemase-producing Enterobacteriaceae isolates from rivers and lakes in Switzerland. Appl Environ Microbiol. 2013;79(9):3021–6.
Jaén-Luchoro D, Busquets A, Karlsson R, Salvà-Serra F, Åhrén C, Karami N, Moore ER. Genomic and proteomic characterization of the extended-spectrum β-lactamase (ESBL)-producing Escherichia coli strain CCUG 73778: a virulent, nosocomial outbreak strain. Microorganisms. 2020;8(6):893.
Dolejska M. Antibiotic-resistant bacteria in wildlife. Antibiotic Resist Environment: Worldw Overv 2020:19–70.
Hernando-Amado S, Coque TM, Baquero F, Martínez JL. Defining and combating antibiotic resistance from one health and Global Health perspectives. Nat Microbiol. 2019;4(9):1432–42.
Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious Diseases. Nature. 2004;430(6996):242–9.
Fadare FT, Okoh AI. Distribution and molecular characterization of ESBL, pAmpC β-lactamases, and non-β-lactam encoding genes in Enterobacteriaceae isolated from hospital wastewater in Eastern Cape Province, South Africa. PLoS ONE. 2021;16(7):e0254753.
Chen Y, Brook TC, Alcon-Giner C, Clarke P, Hall LJ, Hoyles L. Draft genome sequences of Citrobacter freundii and Citrobacter murliniae strains isolated from the feces of Preterm infants. Microbiol Resour Announc 2019, 8(33).
Organization WH. Critically important antimicrobials for human medicine. 2019.
Organization WH. Critically important antimicrobials for human medicine: ranking of antimicrobial agents for risk management of antimicrobial resistance due to non-human use. 2017.
Costanzo SD, Murby J, Bates J. Ecosystem response to antibiotics entering the aquatic environment. Mar Pollut Bull. 2005;51(1–4):218–23.
Cizmas L, Sharma VK, Gray CM, McDonald TJ. Pharmaceuticals and personal care products in waters: occurrence, toxicity, and risk. Environ Chem Lett. 2015;13(4):381–94.
Qin J, Zhao Y, Wang A, Chi X, Wen P, Li S, Wu L, Bi S, Xu H. Comparative genomic characterization of multidrug-resistant Citrobacter spp. strains in Fennec fox imported to China. Gut Pathog. 2021;13(1):59.
Jacoby GA, Griffin CM, Hooper DC. Citrobacter spp. as a source of qnrB alleles. Antimicrob Agents Chemother. 2011;55(11):4979–84.
Kehrenberg C, Friederichs S, de Jong A, Schwarz S. Novel variant of the qnrB gene, qnrB12, in Citrobacter werkmanii. Antimicrob Agents Chemother. 2008;52(3):1206–7.
Saga T, Sabtcheva S, Mitsutake K, Ishii Y, Tateda K, Yamaguchi K, Kaku M. Characterization of qnrb-like genes in Citrobacter species of the American Type Culture Collection. Antimicrob Agents Chemother. 2013;57(6):2863–6.
Park YJ, Yu JK, Lee S, Oh EJ, Woo GJ. Prevalence and diversity of qnr alleles in AmpC-producing Enterobacter cloacae, Enterobacter aerogenes, Citrobacter freundii and Serratia marcescens: a multicentre study from Korea. J Antimicrob Chemother. 2007;60(4):868–71.
Baumler AJ, Tsolis RM, Ficht TA, Adams LG. Evolution of host adaptation in Salmonella enterica. Infect Immun. 1998;66(10):4579–87.
Sorsa LJ, Dufke S, Heesemann J, Schubert S. Characterization of an iroBCDEN gene cluster on a transmissible plasmid of uropathogenic Escherichia coli: evidence for horizontal transfer of a chromosomal virulence factor. Infect Immun. 2003;71(6):3285–93.
Carniel E. The Yersinia high-pathogenicity island: an iron-uptake island. Microbes Infect. 2001;3(7):561–9.
LaMattina JW, Delrossi M, Uy KG, Keul ND, Nix DB, Neelam AR, Lanzilotta WN. Anaerobic heme degradation: ChuY is an Anaerobilin reductase that exhibits kinetic cooperativity. Biochemistry-Us. 2017;56(6):845–55.
Grohmann E, Christie PJ, Waksman G, Backert S. Type IV secretion in Gram-negative and Gram-positive bacteria. Mol Microbiol. 2018;107(4):455–71.
Blasey N, Rehrmann D, Riebisch AK, Muhlen S. Targeting bacterial pathogenesis by inhibiting virulence-associated type III and type IV secretion systems. Front Cell Infect Microbiol. 2022;12:1065561.
Jechalke S, Dealtry S, Smalla K, Heuer H. Quantification of IncP-1 plasmid prevalence in environmental samples. Appl Environ Microbiol. 2013;79(4):1410–3.
Dionisio F, Conceicao IC, Marques AC, Fernandes L, Gordo I. The evolution of a conjugative plasmid and its ability to increase bacterial fitness. Biol Lett. 2005;1(2):250–2.
Fischer W, Haas R, Odenbreit S. Type IV secretion systems in pathogenic bacteria. Int J Med Microbiol. 2002;292(3–4):159–68.
Christie PJ, Cascales E. Structural and dynamic properties of bacterial type IV secretion systems. Mol Membr Biol. 2005;22(1–2):51–61.
Juhas M, Crook DW, Hood DW. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell Microbiol. 2008;10(12):2377–86.
Molale LG. Surface water quality of the North West Province based on physico-chemical properties and faecal streptococci levels. Citeseer; 2012.
Horn S, Pieters R, Bezuidenhout C. Pathogenic features of heterotrophic plate count bacteria from drinking-water boreholes. J Water Health. 2016;14(6):890–900.
Hudzicki J. Kirby-Bauer disk diffusion susceptibility test protocol. Am Soc Microbiol. 2009;15:55–63.
Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt K, Borodovsky M, Ostell J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614–24.
Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9(1):5114.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3.
Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–9.
Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, Tyson GH, Zhao S, Hsu C-H, McDermott PF. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. 2019;63(11):e00483–00419.
Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, Jin Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(suppl1):D325–8.
Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47(D1):D687–92.
Johansson MH, Bortolaia V, Tansirichaiya S, Aarestrup FM, Roberts AP, Petersen TN. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J Antimicrob Chemother. 2021;76(1):101–9.
Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, Hasman H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–903.
Robertson J, Nash JHE. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genomics 2018, 4(8).
Li XB, Xie YZ, Liu M, Tai C, Sung JY, Deng ZX, Ou HY. oriTfinder: a web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 2018;46(W1):W229–34.
Acknowledgements
The authors would like to thank Dr. Mzimkhulu Monapathi for assisting with the collection of samples, and Mr Thuto Magome for assisting in some of the bioinformatics analysis. The opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the funders.
Funding
Open access funding provided by North-West University. This study was supported in part by grants from the Water Research Commission (WRC) - ESKAPE [C2019/2020 − 00224] and the National Research Foundation (NRF) - WATNET [No. UID105825].
Open access funding provided by North-West University.
Author information
Authors and Affiliations
Contributions
The basic idea and the study design were from C.C.B. and K.E.L. The practical work was carried out by L.C. Analysis and interpretation of the results L.C., C.M., D.A.B.V, C.C.B and K.E.L. Sequencing and bioinformatics anlysis was carried out by D.A.B.V and K.E.L. The wrote of the original draft of the manuscript D.A.B.V., and K.E.L. All authors critically reviewed and edited the manuscript. Lastly, all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study was carried out in accordance with the guidelines and regulations of the Ethics Committee, Faculty of Natural and Agricultural Sciences, North-West University, South Africa (No: NWU-01728-20-A9).
Consent for publication
None-applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chenhaka, LH., Van Wyk, D., Mienie, C. et al. The phylogenomic landscape of extended-spectrum β-lactamase producing Citrobacter species isolated from surface water. BMC Genomics 24, 755 (2023). https://doi.org/10.1186/s12864-023-09867-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09867-4