
Abstract
Background
Ophiorrhiza pumila (Rubiaceae) is capable of producing camptothecin (CPT), one monoterpene indole alkaloid extensively employed in the treatment of multiple cancers. Transcription factors (TFs) GATA are a group of transcription regulators involved in plant development and metabolism, and show the feature of binding to the GATA motif within the promoters of target genes. However, GATA TFs have not been characterized in O. pumila.
Result
In this study, a total of 18 GATA genes classified into four subfamilies were identified, which randomly distributed on 11 chromosomes of O. pumila. Synteny analysis of GATA genes between O. pumila and other plant species such as Arabidopsis thaliana, Oryza sativa, Glycine max, Solanum lycopersicum, Vitis vinifera, and Catharanthus roseus genomes were analyzed. Tissue expression pattern revealed that OpGATA1 and OpGATA18 were found to be correlated with ASA, MK, CPR and GPPS, which were highly expressed in leaves. OpGATA7, showed high expression in roots as most of the CPT biosynthetic pathway genes did, suggesting that these OpGATAs may be potential candidates regulating CPT biosynthesis in O. pumila.
Conclusions
In this study, we systematically analyzed the OpGATA TFs, and provided insights into the involvement of OpGATA TFs from O. pumila in CPT biosynthesis.
Similar content being viewed by others

Background
Ophiorrhiza pumila is a dicotyledonous plant classified into Rubiaceae family and remains as a sustainable source of camptothecin (CPT). CPT is a type of monoterpene indole alkaloids (MIAs) commonly used in treatment of cancers and was initially isolated from Camptotheca acuminate [1], and subsequently detected in Nothapodytes nimmoniana and other plants [2,3,4]. CPT inhibits tumor growth by blocking DNA topoisomerase I [5, 6]. Topotecan and irinotecan, two drugs developed by CPT derivatives, have been extensively employed in various cancers including lung, colorectal, cervical, and ovarian cancers [7]. The biosynthesis pathway of CPT is complex and remains not fully resolved [8]. Briefly, the terpene section of CPT is derived from the 2-C-methyl-d-erythritol 4-phosphate (MEP) and mevalonate (MVA) pathways. The produced geraniol is hydroxylated to 10-hydroxygeraniol under the catalysis of geraniol 10-hydroxy (G10H) [9,10,11], and then oxidized to 10-oxogeranial by 10-hydroxy geraniol oxidoreductase (10-HGO). Next, 10-oxogeraniol is converted to iridodial under the action of iridodial synthase, followed by conversion to iriotrial under the action of iridodial oxidoreductase (IO). Iriotrialis then converted to 7-deoxyloganetic acid under the action of IO. 7-deoxyloganetic acid is converted to 7-deoxyloganic acid by glucosyltransferase (7-DLGT) [6], and the product is then converted to loganic acid by 7-deoxyloganic acid hydroxylase (7-DLH) [6]. Lastly, secologanin, a precursor of CPT, is synthesized by secologanin synthase (SLS) [12, 13]. Another precursor tryptamine was produced from tryptophan under the catalyzation of tryptophan decarboxylase (TDC) [14]. Strictosidine is synthesized by condensation of tryptamine and secologanin catalyzed by strictosidine synthase (STR) [9, 15], and CPT is then formed via a series of catalytic reactions that have not yet been elucidated. A high-quality O. pumila genome has been assembled using next-generation sequencing, which led to a final genome assembly of 439.90 Mb, with contig and scaffold N50 values of 18.49 and 40.06 Mb, respectively. A total of 11 chromosomes were sequenced with sequential scaffolding strategy. Besides, more than 270 nitrogen-containing metabolites including different MIAs have been found [11]. Therefore, O. pumila has been regarded as a model plant for MIA biosynthesis [10].
To adapt to changes in the external environment and resist various biotic and abiotic stresses, plants have formed a series of complex and efficient regulatory networks causing changes in gene expression response to such stresses at multiple levels, and transcription factors (TFs) are critical regulators of these processes [16]. The GATA TF is one of the ubiquitous TF families in eukaryotes and is essential for many aspects of plant development, metabolism and signal conduction [17]. GATA proteins share a common feature of binding to the specific sequence (T/A) GATA (A/G) [18]. The DNA-binding domain of GATA contains a class IV zinc finger structure (C-X2-C-X17–20-C-X2-C), followed by a basal region [19]. Most GATA TFs in plants include a single C-X2-C-X18-C-X2-C motif and several contain C-X2-C-X20-C-X2-C. The first plant GATA TF was identified from tobacco and termed as NTL1 harboring C-X2-C-X18-C-X2-C motif [20]. GATA TFs have been identified and characterized in A. thaliana (29), and O. sativa (28) [17]. Based on phylogenetic relationships, DNA binding regions, and intron-exon structures, Arabidopsis and rice GATA family genes can be categorized into four families including I, II, III, and IV [21].
GATA TFs have been reported involved in plant metabolism. For example, A. thaliana GATA nitrate-inducible carbon-metabolism-involved (GNC) and cytokinin-responsive GATA1 (CGA1) regulated chlorophyll levels, chloroplast size, photosynthetic efficiency, and carbon and nitrogen metabolism [22]. Moreover, GNC and CGA1 show high expression in green tissues and are capable of mediating cytokinin to regulate plastid development [23]. GATA8 mediated the biomass accumulation and photosynthetic efficiency in O. sativa seedlings [24]. Transient overexpression of CrGATA1 in Catharanthus roseus seedlings increased vindoline production by activating D4H gene which contained GATA motifs in the promoter [25]. Under low nitrogen deposition, GATA44 and GATA58 genes exhibit low expression in soybean seedlings [26]. Additionally, in higher plants, the assimilation pathway of nitrate is tightly regulated. Nitrate is reduced from nitrate reductase to nitrite and then to ammonium nitrogen (NH4+) by nitrite reductase to participate in the synthesis of amino acids and proteins [27]. The promoter of nitrate reductase (NIA) in tomatoes covers the required cis-acting regulatory elements capable of specifically recognizing and binding to GATA protein and then regulating nitrogen metabolism [28]. Moreover, ammonia is required for alkaloid biosynthesis. Nitrogen is an important nutritional factor affecting plant alkaloid biosynthesis and accumulation [29], and CPT contains two nitrogen atoms in its molecular structure owing to its origin as an amino acid-derived alkaloid [11]. Thus, nitrogen metabolism may critically affect the regulation of CPT.
By analyzing the promoter sequence of CPT biosynthetic genes, multiple GATA motifs were deserved, suggesting exploring GATA TFs from O. pumila (OpGATAs) is of importance to study CPT biosynthesis. In this study, GATA family TFs distributed in O. pumila was systematically characterized. Based on phylogenetic relationship and expression pattern combined with co-expression analysis, candidate GATA genes regulating CPT biosynthesis were predicted. The results provided a comprehensive analysis of OpGATA family genes, which shed new lights on CPT biosynthesis in O. pumila.
Results
Identification and phylogenetic analysis of GATA proteins
In this study, a total of 18 OpGATA genes were identified from the genome of O. pumila according to HMM search results, and renamed as OpGATA1-OpGATA18 according to their chromosome position. Fundamental characteristics of OpGATA1-OpGATA18 including coding sequence length, protein molecular weight, point isoelectric (pI) and subcellular location were analyzed (Table S1). The complete open reading frame (ORF) of OpGATAs varied from 160 bp (OpGATA4) to 543 bp (OpGATA15), and the molecular weights ranged from 17.73 kDa to 59.86 kDa. The pI values were predicted ranging from 5.05 (OpGATA4) to 10.11 (OpGATA6). All OpGATA proteins were predicted to localize in the nucleus (Table S1).
Phylogenetic analysis and classification of OpGATA
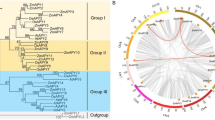
To determine the phylogenetic relationships of OpGATA proteins, a neighbor-joining tree was constructed by complying with the full-length GATA proteins from O. pumila, O. sativa, A. thaliana and C. roseus (Fig. 1; Fig. S1). OpGATA proteins were classified into four distinct subfamilies (I, II, III, and IV) (Fig. 2a). Seven OpGATAs (OpGATA1, OpGATA2, OpGATA7, OpGATA9, OpGATA11, OpGATA14, and OpGATA16) were classified as subfamily I; five OpGATAs (OpGATA3, OpGATA4, OpGATA8, OpGATA12, and OpGATA13) were assigned into subfamily II; five OpGATAs (OpGATA5, OpGATA6, OpGATA10, OpGATA17, and OpGATA18) were grouped into subfamily III and subfamily IV having only one OpGATA namely OpGATA15.

Neighbor-joining tree representing the relationship among GATA proteins of O. pumila, O. sativa and A. thaliana. Constructed with MEGA v7 using full-length amino acid sequences and the bootstrap test replicate was set as 1000 times

Schematic representation of phylogenetic relationships, conserved motifs and gene structures of the GATA genes in O. pumila. a A phylogenetic tree of 18 OpGATA proteins. b The motif composition of OpGATA proteins. The motifs are displayed in different colored boxes. c Exon/intron structures of OpGATA genes
Gene structure and motif composition of the OpGATA gene family
To gain insights into the characteristics of OpGATA proteins, the motifs of OpGATA proteins were analyzed by MEME. In total, 10 different conservative motifs were characterized (motifs 1–10) (Fig. 2b, Table S2). Motifs 1 and 5 were detected in all proteins except OpGATA3. Motifs 4, 6, 7, and 8 were mainly observed in subfamily I; motif 9 was mainly present in subfamily II; motif 2 and 3 remained in subfamily III; and motifs 1, 5, and 7 were mainly contained in subfamily IV. The exon and intron structures of OpGATA genes were obtained by comparing the corresponding genomic DNA sequences of O. pumila. Notably, subfamilies III and IV had more introns, whereas subfamilies I and II had only 1–3 introns (Fig. 2c). Overall, members within a single subfamily exhibited similar gene structures, and the results in this study showed similar gene structures and conserved motifs, strongly supporting the results of phylogenetic analysis of subfamily classification. Similar to previous studies of A. thaliana, rice, and other plants [17, 19, 21], OpGATAs classified into subfamilies I, II, and IV contain the conserved domain C-X2-C-X18-C-X2-C (except OpGATA3 and OpGATA15), while domain with the C-X2-C-X20-C-X2-C structure was existed in subfamily III (Fig. 3).

Alignments of GATA domain sequences of the GATA family members in O. pumila
Chromosomal distribution and synteny analysis of the OpGATA gene family
A physical location map of all OpGATA genes in the genome of O. pumila was drawn (Fig. 4). The distribution of OpGATA genes on chromosomes was not homogeneous. The maximum number of OpGATA genes was distributed on Opu_Chr02 (OpGATA2-OpGATA7), whereas Opu_chr05 and Opu_chr11 had no OpGATA genes and the other chromosomes harbored 1–2 OPGATA genes, such as OpGATA1 distributed on Opu_Chr01, OpGATA8 and OpGATA9 on Opu_Chr03, OpGATA10 and OpGATA11 on Opu_Chr04, OpGATA12 on Opu_Chr06, OpGATA13 on Opu_Chr07, OpGATA14 and OpGATA15 on Opu_Chr08, OpGATA16 on Opu_Chr09, OpGATA17 and OpGATA18 on Opu_Chr10.

The chromosomal distribution and synteny analysis of OpGATA genes in O. pumila. The locations of all the OpGATA genes are depicted in the chromosomes. Red-colored genes belong to subfamily I, blue-colored genes belong to subfamily II, green-colored genes belong to subfamily III, purple-colored genes belong to subfamily IV. Background gray lines indicate all O. pumila genome synteny blocks, and the Colored lines highlight the duplicated OpGATA gene pairs. ID of the chromosomes is indicated at the bottom
Moreover, replication events of OpGATAs were analyzed. The result showed that no tandem repeats were identified among the 18 genes, while four pairs of fragment repeats were detected between eight chromosomes, which were Opu_chr01 (OpGATA1)/Opu_chr09 (OpGATA16), Opu_chr02 (OpGATA4)/Opu_chr03 (OpGATA8), Opu_chr02 (OpGATA5)/Opu_chr10 (OpGATA18), and Opu_chr04 (OpGATA11)/Opu_chr08 (OpGATA14). Accordingly, some OpGATA genes may have been generated by gene replication, thereby critically affecting the amplification of OpGATA genes in O. pumila.
To gain insights into the evolution of O. pumila GATA family, represented comparative system diagrams comparing O. pumila and five other dicotyledonous plants (A. thaliana, G. max, S. lycopersicum, V. vinifera and C. roseus) and one monocotyledonous plant (O. sativa) was analyzed (Fig. 5, Table S3). In total, OpGATA genes displayed different syntenic relationship with G. max (41), S. lycopersicum (35), V. vinifera (23), A. thaliana (26), C. roseus (23) and O. sativa (9), respectively, suggesting that GATA genes from O. pumila relatively had a more similar relationship with that in soybean. Furthermore, OpGATA4, OpGATA8, OpGATA12, and OpGATA14 exhibited syntenic relationship in the six plant species, demonstrating that these four proteins may critically affect evolution.

Synteny analysis of GATA genes between O. pumila and five representative plant species (A. thaliana, O. sativa, G. max, S. lycopersicum, V. vinifera and C. roseus). Gray lines in the background indicate the collinear blocks within O. pumila and other plant genomes, while red lines highlight syntenic GATA gene pairs
Expression profiles of OpGATA genes and key enzyme genes in different samples
Expression profiles of OpGATA and vital enzyme genes were evaluated in three distinct tissues and organs (roots, stems and leaves), together with cell suspension cultures and hairy roots. A heat map was built according to the QRT-PCR analysis. The results showed that most of the CPT-producing pathway genes (CMS, DXS, 7-DLGT, PMK, IO, 8-HGO, G10H, HMGR, 10-HGO, CMK, HDS, IPPI, HDR, HMGS, 7-DLH, GES, TSB, LAMT, SLS, IS, STR and TDC) expressed highly in roots or hairy roots (Fig. 6a). Several genes including MECS, CPR, MK, ASA, and GPPS showed higher expression in leaves. As indicated in Fig. 6b, most of OpGATA genes were highly expressed in stems, while expression level of OpGATA9 and OpGATA12 was higher in leaves, and OpGATA7, OpGATA14 as well as OpGATA15 expressed higher in roots. In addition, correlations between OpGATAs and pathway genes were analyzed (Fig. 7, Table S4). The results showed that OpGATA7 exhibited positive associations with key enzyme genes showing high expression in roots. Among this, HMGS and HDR showed pole-strength correlations (p < 0.05, r > 0.8) with OpGATA7, along with CMK, GPPS, HDS, HMGR IPPS showed strong correlations (p < 0.05, 0.6 < r < 0.8). OpGATA1, OpGATA4, OpGATA5, OpGATA6, OpGATA8, OpGATA9, OpGATA12, OpGATA16, OpGATA17, and OpGATA18 were found to be correlated with ASA, MK, CPR, or GPPS which were highly expressed in leaves. Only OpGATA1 and OpGATA18 exhibited a positive correlation with all the four genes, with Pearson correlation coefficients of greater than 0.6. To identify the cis-element of OpGATA, the 3000 bp promoter sequences of genes encoding vital enzymes in the CPT biosynthesis pathway were analyzed using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). As predicted, the GATA motif was present in the promoters of several key enzyme genes (TSB, SLS, MK, MDC, IPPI, HMGR, HDR, GES, G10H, DXS, AACT, and 8-HGO) (Table S5). Thus, these key biosynthetic genes may be regulated by GATA TFs.

The expression patterns of key enzyme genes and OpGATAs in leaves, roots, stems, cell suspension cultures and hairy roots examined by QRT-PCR. The color scale represents relative expression levels from high (red color) to low (blue color)

The correlation between the gene expression patterns of OpGATA and key enzyme genes. Red: positively correlated; blue: negatively correlated. Significant difference was calculated, *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
Camptothecin (CPT) is a widely known monoterpene indole alkaloid with excellent anticancer activity. CPT has been isolated from different plant species. CPT- producing weedy plant O. pumila has brought about widespread attention, and the whole genome of O. pumila has been sequenced [29]. Recently, metabolic engineering has been applied in O. pumila to elevate CPT content. For example, individual introduction of G10H or SLS, and co-expression of G10H and SLS significantly enhanced CPT content in transgenic O. pumila hairy roots [30]. Besides, transcription regulation of CPT biosynthesis has been studied. RNA interference of OpERF2 suppressed expression level of genes involved in MEP and secologanin-strictosidine pathways [31]. The transcription repressor OpMYB1 reduced CPT biosynthesis by downregulating expression level of TDC [32]. OpWRKY2 acted as a positive regulator of CPT biosynthesis by directly targeting TDC [10]. OpWRKY1 inhibited CPT biosynthesis by directly down-regulating CPR transcription in O. pumila [13]. Nevertheless, transcription regulation of CPT biosynthesis needs further study.
The GATA TF family involved in many aspects of physiology-related processes has been broadly explored in a range of plants including Arabidopsis, rice [17], grapes [33], Moso bamboo [34], and Gossypium sp. [21]. The present study reported GATA TFs in O. pumila. Totally, 18 GATA TFs were identified and named OpGATA1-OpGATA18 according to their physical location on the chromosome. The whole OpGATA family in O. pumila could be classified into four groups, similar to those in A. thaliana. In subfamily III, the GATA domain harbored 20 residues in the zinc finger domain, making up a C-X2C-X20-C-X2-C structure, and the other three subfamilies showed that C-X2C-X18-C-X2-C structure, containing 18 residues. The CCT and TIFY domains were specifically identified within subfamily III. The CCT domain was initially found in Arabidopsis Constans protein, which facilitates root and hypocotyl development within A. thaliana and mediates flowering [35]. Previously, the family with a completely conserved TIFY domain was termed TIFY [36]. However, in recent studies, the TIFY domain has been shown to exist extensively in jasmonate ZIM domain protein family and PEAPOD proteins, which are associated with the jasmonic acid pathway [37].
Motif analysis showed that all OpGATAs contained motif 1 and 5 except OpGATA3, and specific motifs were detected in other groups. For example, motif 4 was only observed in subfamily I, motif 9 was only detected in subfamily II, and motifs 2 and 3 were only detected in subfamily III, suggesting that although some motifs of GATA family genes are highly conserved, new evolutionary motifs may have distinct functions in some plants, and the functions of these new evolutionary motifs need to be further verified. The homology of GATA genes from O. pumila with those from Arabidopsis, rice, soybeans, tomatoes, and grapes was also explored. Notably, the Arabidopsis GATA TFs AtGATA1 (AT3G24050), AtGATA2 (AT3G60530), and AtGATA4 (AT2G45050) have been reported to facilitate light-dependent regulation of gene expression and photomorphogenesis [38]. Accordingly, the homologous genes OpGATA9 (Opuchr03_g0010130–1.1) and OpGATA2 (Opuchr03_g0010130–1.1) may also affect light-dependent regulation of genes [38]. AtGATA22 (AT4G26150), which is homologous with OpGATA12 (Opuchr06_g0009000–1.1), affects the response to cytokinins and hinders root growth in A. thaliana [39]. Additionally, GNC (AT5G56860), which is homologous to OpGATA12, adversely affects seed germination, flowering, and leaf elongation, and overexpression of GNC inhibits the germination, leaf expansion, and flowering of A. thaliana [40]. AtGATA12 (AT5G25830), which is homologous to OpGATA14, is involved in primary dormancy in A. thaliana [41].
Expression level of most genes encoding the key enzymes in the CPT biosynthesis pathway (CMS, DXS, 7-DLGT, PMK, IO, 8-HGO, G10H, HMGR, 10-HGO, CMK, HDS, IPPI, HDR, HMGS, 7-DLH, GES, TSB, LAMT, SLS, IS, STR and TDC) were significantly higher in roots or hairy roots compared with those in other tissues, whereas genes involved in the MVA and MEP pathways (ASA, GPPS, MK, and MECS) were mostly expressed in leaves. OpGATA7 exhibited positive associations with CPT biosynthesis pathway genes, which showed significant expression in roots. Plant terpenoids are synthesized mainly through the MVA and MEP pathways [42]. Genes that are highly expressed in the leaves are typically involved in the MEP and MVA pathways [43], demonstrating that OpGATA1, OpGATA4, OpGATA5, OpGATA6, OpGATA8, OpGATA9, OpGATA12, OpGATA16, OpGATA17, and OpGATA18 may regulate CPT biosynthesis by participating in the upstream pathway. Among the genes mentioned above, GATA motifs were found in the promoters of key enzyme genes (i.e., SLS, MDC, IPPI, HMGR, HDR, GES, G10H, AACT, and 8-HGO) highly expressed in roots or hairy roots and key enzyme genes (i.e., MK, DXS) highly expressed in leaves, demonstrating that these key enzyme genes may be directly regulated by OpGATA genes and then affect the biosynthesis of CPT. Of which, the genes highly expressed in roots or hairy roots which is pole-strength correlations with OpGATA7 is a higher possibility of being directly regulated. It is interesting that OpGATA3 expressed ubiquitously in the stem, roots, leaves, cell suspension cultures and hairy roots, we analyzed and found that the promoter of OpGATA3 has phytohormone responsive elements and biotic and abiotic stress (Fig. S2), which is in close relation to the regulation of plant growth.
Overall, this comprehensive analysis of GATA family genes from O. pumila provided insights into the characteristics of OpGATA genes and may improve our understanding of the mechanisms regulating CPT biosynthesis in O. pumila.
Conclusion
In this study, OpGATA TF family in O. pumila were characterized and identified. Overall, a total of 18 OpGATA genes showing different chromosomal distribution were classified into four subfamilies. Synteny analysis of GATA genes were conducted within several plant species including O. pumila, Arabidopsis, grapes, tomatoes, soybeans and C. roseus, and the functions of some homologous genes were predicted. OpGATA genes showed different expression patterns within a range of samples (leaves, stems, roots, cell suspension cultures and hairy roots) in correlation to key pathway genes, highlighting the potential roles of some OpGATA genes in the regulation of CPT biosynthesis in O. pumila. This study provides novel OpGATA TFs involved in regulating CPT biosynthesis.
Materials and methods
Identification of OpGATAs
The hidden Markov model (PF00320) of the GATA domain originating from the Pfam database (http://pfam.xfam.org) was used to identify the OpGATA family. To avoid probable GATA members were missing, a BLASTP-algorithm based search using GATA amino acid sequences from Arabidopsis as queries was conducted: e-value ≤1e− 3 [44]. The Pfam database (http://pfam.xfam.org/search/sequence), NCBI CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) were employed to verify the integrity of the GATA domain, with an e-value cutoff of 0.01 [45]. The ProSite ExPASy server (http://web.expasy.org/protparam/) was adopted to predict the physical and chemical properties of OpGATA proteins. Subcellular localization of GATA proteins was predicted using CELLO (http://cello.life.nctu.edu.tw/).
Multiple sequence alignment and phylogenetic analysis
The GATA proteins from A. thaliana Information Resources (www.arabidopsis.org/index.jsp) and the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/cgi-bin/ORF_infopage.cgi) were downloaded [19]. MAFFT software was employed for multiple sequence alignment of GATA proteins [46]. The neighbor-joining tree of GATA TF families from A. thaliana, O. sativa, O. pumila and C. roseus were built by MEGA v7 [47], with the parameters of Poisson model, pairwise deletion, and 1000 bootstrap tests.
Motifs and gene structures
The MEME was employed (http://meme.sdsc.edu/meme/itro.html) to identify the conserved motif of GATA protein in O. pumila, with the following parameters were adopted: 0 or 1 occurrence per sequence; maximum number of motifs = 10; and optimum motif length = 6–50 residues. Exon-intron structure of the GATA members was investigated by analyzing the O. pumila genome, and gene structure was visualized with TBtools [48].
Chromosomal distribution and gene duplication of GATA genes
The method for mapping GATA genes on the chromosome of O. pumila was identical to that of FtAP2/ERF genes [49]. Gene replication events were investigated using the multiple collinear scanning toolkit (MCScanX) and BLASTP method. TBtools software (https://github.com/CJ-Chen/TBtools) was adopted to build syntenic analysis maps for determining the syntenic relationships between OpGATA proteins and GATA proteins from A. thaliana, O. sativa, G. max, S. lycopersicum, V. vinifera and C. roseus.
Expression analysis by quantitative real-time PCR (QRT-PCR)
Total RNA was extracted using a Plant RNAprep Pure Kit (TIANGEN, China). Corresponding sequences of OpGATA genes and key enzyme genes were acquired from the O. pumila genome sequence database (http://pumila.kazusa.or.jp/). Primers used for QRT-PCR analysis were designed using Primer 5 software (Table S6). Relative expression levels were calculated using the 2−ΔΔCt method, with housekeeping gene OpActin from O. pumila as the internal control [10]. All QRT-PCR analyses were performed with three biological replicates. The heatmap was constructed by TBtools software base on QRT-PCR analysis. Pearson’s correlation coefficient was analyzed using the OmicStudio tools at https://www.omicstudio.cn, significant difference was tested at significance levels of 0.05, 0.01, and 0.001.
Availability of data and materials
The genomic information of Ophiorrhiza pumila was downloaded from Ophiorrhiza pumila Genome DateBase (http://pumila.kazusa.or.jp/). All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- O. pumila :
-
Ophiorrhiza pumila
- A. thaliana :
-
Arabidopsis thaliana
- O. sativa :
-
Oryza sativa
- G. max :
-
Glycine max
- S. lycopersicum :
-
Solanum lycopersicum
- V. vinifera :
-
Vitis vinifera
- CPT:
-
Camptothecin
- pI:
-
Point isoelectric
- TFs:
-
Transcription factors
- MIAs:
-
Monoterpene indole alkaloid
- MEP:
-
2-C-methyl-d-erythritol 4-phosphate
- MVA:
-
Mevalonate
- G10H:
-
Geraniol 10-hydroxy
- 10-HGO:
-
10-hydroxy geraniol oxidoreductase
- IO:
-
Iridodial oxidoreductase
- 7-DLGT:
-
7-deoxyloganic acid by glucosyltransferase
- 7-DLH:
-
7-deoxyloganic acid hydroxylase
- SLS:
-
Secologanin synthase
- TDC:
-
Tryptophan decarboxylase
- STR:
-
Strictosidine synthase
- ASA:
-
Anthranilate synthase
- CPR:
-
Cytochrome P450 reductases
- DXR:
-
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- DXS:
-
1-deoxy-D-xylulose-5-phosphate synthase
- HDR:
-
1-hydroxy-2-methyl-2(E)-butenyl-4-diphosphate reductase
- TSB:
-
The beta-subunit of tryptophan synthase
- QRT-PCR:
-
Quantitative real-time PCR
- 8-HGO:
-
8-hydroxy-geraniol oxidoreductase
- AACT:
-
Acetyl-CoA C-acetyltransferase
- CMK:
-
4-(cytidine 5-diphospho)-2-C-methylerythritolkinase
- CMS:
-
4-(cytidine 5-diphospho)-2-C-methylerythritol synthase
- GES:
-
Geraniol synthase
- GPPS:
-
Geranyl diphosphate synthase/geranyl pyrophosphate synthase
- HDS:
-
Hydroxymethylbutenyl 4-diphosphate synthase
- HMGR:
-
3-hydroxy-3-methylglutaryl-CoA reductase
- HMGS:
-
3-hydroxy-3-methylglutaryl-CoA synthase
- IPPI:
-
Isopentenyl diphosphate isomerase
- IS:
-
Iridoid synthase
- LAMT:
-
Loganic acid O-methyltransferase
- MDC:
-
Mevalonate 5-diphosphate decarboxylase/mevalonate (diphospho)-decarboxylase
- MECS:
-
2-C-methylerythritol-2,4-cyclodiphosphate synthase
- MK:
-
Mevalonate kinase
- PMK:
-
Phosphomevalonate kinase
- CSC:
-
Cell suspension cultures
- HR:
-
Hairy roots
References
Lorence A, Nessler CL. Camptothecin, over four decades of surprising findings. Phytochemistry. 2004;65:2735–49. https://doi.org/10.1016/j.phytochem.2004.09.001.
Rather GA, Sharma A, Jeelani SM, Misra P, Kaul V, Lattoo SK. Metabolic and transcriptional analyses in response to potent inhibitors establish MEP pathway as major route for camptothecin biosynthesis in Nothapodytes nimmoniana (Graham) Mabb. BMC Plant Biol. 2019;19:301. https://doi.org/10.1186/s12870-019-1912-x.
Pu X, Zhang CR, Zhu L, Li QL, Huang QM, Zhang L, et al. Possible clues for camptothecin biosynthesis from the metabolites in camptothecin-producing plants. Fitoterapia. 2019;134:113–28. https://doi.org/10.1016/j.fitote.2019.02.014.
Raveendran VV, Vijayan FP, Padikkala J. Antitumor activities of an anthraquinone fraction isolated from in vitro cultures of Ophiorrhiza rugosa var decumbens. Integr Cancer Ther. 2012;11:120–8. https://doi.org/10.1177/1534735411403478.
Zhang L, Ma D, Zhang Y, He W, Yang J, Li C, et al. Characterization of DNA topoisomerase-1 in Spodoptera exigua for toxicity evaluation of camptothecin and hydoxy-camptothecin. PLoS One. 2013;8:e56458. https://doi.org/10.1371/journal.pone.0056458.
Kai G, Wu C, Gen L, Zhang L, Cui L, Ni X. Biosynthesis and biotechnological production of anti-cancer drug camptothecin. Phytochem Rev. 2015;4:525–39. https://doi.org/10.1007/s11101-015-9405-5.
Coleman RL. Emerging role of topotecan in front-line treatment of carcinoma of the ovary. Oncologist. 2002;7:46–55. https://doi.org/10.1634/theoncologist.7-1-46.
Sirikantaramas S, Yamazaki M, Saito K. Camptothecin: biosynthesis, biotechnological production and resistance mechanism(s). Advances in Botanical Research. 2013;68:139–61.
Cui L, Ni X, Ji Q, Teng X, Yang Y, Wu C, et al. Co-overexpression of geraniol-10-hydroxylase and strictosidine synthase improves anti-cancer drug camptothecin accumulation in Ophiorrhiza pumila. Sci Rep. 2015;5:8227 https://doi.org/10.1038/srep08227.
Hao X, Xie C, Ruan Q, Zhang X, Wu C, Han B, et al. The transcription factor OpWRKY2 positively regulates the biosynthesis of the anticancer drug camptothecin in Ophiorrhiza pumila. Hortic Res. 2021;8:7. https://doi.org/10.1038/s41438-020-00437-3.
Rai A, Hirakawa H, Nakabayashi R, Kikuchi S, Hayashi K, Rai M, et al. Chromosome-level genome assembly of Ophiorrhiza pumila reveals the evolution of camptothecin biosynthesis. Nat Commun. 2021;12:405. https://doi.org/10.1038/s41467-020-20508-2.
Wang C, Wu C, Wang Y, Xie C, Shi M, Nile S, et al. Transcription factor OpWRKY3 is involved in the development and biosynthesis of camptothecin and its precursors in Ophiorrhiza pumila hairy roots. Int J Mol Sci. 2019;20:3996. https://doi.org/10.3390/ijms20163996.
Xu M, Wu C, Zhao L, Wang Y, Wang C, Zhou W, et al. WRKY transcription factor OpWRKY1 acts as a negative regulator of camptothecin biosynthesis in Ophiorrhiza pumila hairy roots. Plant Cell Tissue Organ Cult. 2020;142:69–78. https://doi.org/10.1007/s11240-020-01833-2.
You D, Feng Y, Wang C, Sun C, Wang Y, Zhao D, et al. Cloning, characterization, and enzymatic identification of a new tryptophan decarboxylase from Ophiorrhiza pumila. Biotechnol Appl Biochem. 2020. https://doi.org/10.1002/bab.1935.
Lu Y, Wang H, Wang W, Qian Z, Li L, Wang J, et al. Molecular characterization and expression analysis of a new cDNA encoding strictosidine synthase from Ophiorrhiza japonica. Mol Biol Rep. 2009;36:1845–52. https://doi.org/10.1007/s11033-008-9389-y.
Liu M, Wen Y, Sun W, Ma Z, Huang L, Wu Q, et al. Genome-wide identification, phylogeny, evolutionary expansion and expression analyses of bZIP transcription factor family in tartaty buckwheat. BMC Genomics. 2019;20:483. https://doi.org/10.1186/s12864-019-5882-z.
Reyes JC, Muro-Pastor MI, Florencio FJ. The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol. 2004;134:1718–32. https://doi.org/10.1104/pp.103.037788.
Zhu W, Guo Y, Chen Y, Wu D, Jiang L. Genome-wide identification, phylogenetic and expression pattern analysis of GATA family genes in Brassica napus. BMC Plant Biol. 2020;20:543. https://doi.org/10.1186/s12870-020-02752-20018-009-0066-7.
Chen H, Shao H, Li K, Zhang D, Fan S, Li Y, et al. Genome-wide identification, evolution, and expression analysis of GATA transcription factors in apple (Malus×domestica Borkh.). Gene. 2017;627:460–72. https://doi.org/10.1016/j.gene.2017.06.049.
Daniel-Vedele F, Caboche M. A tobacco cDNA clone encoding a GATA-1 zinc finger protein homologous to regulators of nitrogen metabolism in fungi. Mol Gen Genet. 1993;240:365–73. https://doi.org/10.1007/BF00280388.
Zhang Z, Zou X, Huang Z, Fan S, Qun G, Liu A, et al. Genome-wide identification and analysis of the evolution and expression patterns of the GATA transcription factors in three species of Gossypium genus. Gene. 2019;680:72–83. https://doi.org/10.1016/j.gene.2018.09.039.
Chiang YH, Zubo YO, Tapken W, Kim HJ, Lavanway AM, Howard L, et al. Functional characterization of the GATA transcription factors GNC and CGA1 reveals their key role in chloroplast development, growth, and division in Arabidopsis. Plant Physiol. 2012;160:332–48. https://doi.org/10.1104/pp.112.198705.
Naito T, Kiba T, Koizumi N, Yamashino T, Mizuno T. Characterization of a unique GATA family gene that responds to both light and cytokinin in Arabidopsis thaliana. Biosci Biotechnol Biochem. 2007;71:1557–60. https://doi.org/10.1271/bbb.6069.
Nutan KK, Singla-Pareek SL, Pareek A. The Saltol QTL-localized transcription factor OsGATA8 plays an important role in stress tolerance and seed development in Arabidopsis and rice. J Exp Bot. 2020;71:684–98. https://doi.org/10.1093/jxb/erz368.
Liu Y, Patra B, Pattanaik S, Wang Y, Yuan L. GATA and phytochrome interacting factor transcription factors regulate light-induced vindoline biosynthesis in Catharanthus roseus. Plant Physiol. 2019;180:1336–50. https://doi.org/10.1104/pp.19.00489.
Zhang C, Hou Y, Hao Q, Chen H, Chen L, Yuan S, et al. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS One. 2015;10:e0125174. https://doi.org/10.1371/journal.pone.0125174.
Crawford NM, Glass ADM. Molecular and physiological aspects of nitrate uptake in plants. Trends Plant Sci. 1998;3:389–95. https://doi.org/10.1016/S1360-1385(98)01311-9.
Dorbe MF, Caboche M, Daniel-Vedele F. The tomato nia gene complements a Nicotiana plumbaginifolia nitrate reductase-deficient mutant and is properly regulated. Plant Mol Biol. 1992;18:363–75. https://doi.org/10.1007/BF00034963.
Bensaddek L, Gillet F, Saucedo JE, Fliniaux MA. The effect of nitrate and ammonium concentrations on growth and alkaloid accumulation of Atropa belladonna hairy roots. J Biotechnol. 2001;85:35–40. https://doi.org/10.1016/s0168-1656(00)00372-2.
Shi M, Gong H, Cui L, Wang Q, Wang C, Wang Y, et al. Targeted metabolic engineering of committed steps improves anti-cancer drug camptothecin production in Ophiorrhiza pumila hairy roots. Ind Crop Prod. 2020;148:112277. https://doi.org/10.1016/j.indcrop.2020.112277.
Udomsom N, Rai A, Suzuki H, Okuyama J, Imai R, Mori T, et al. Function of AP2/ERF transcription factors involved in the regulation of specialized metabolism in Ophiorrhiza pumila revealed by transcriptomics and metabolomics. Front Plant Sci. 2016;7:1861. https://doi.org/10.3389/fpls.2016.01861.
Rohani ER, Chiba M, Kawaharada M, Asano T, Oshima Y, Mitsuda N, et al. An MYB transcription factor regulating specialized metabolisms in Ophiorrhiza pumila. Plant Biotechnol. 2016;33:1–9. https://doi.org/10.5511/plantbiotechnology.15.1117a.
Zhang Z, Ren C, Zou L, Wang Y, Li S, Liang Z. Characterization of the GATA gene family in Vitis vinifera: genome-wide analysis, expression profiles, and involvement in light and phytohormone response. Genome. 2018;61:713–23. https://doi.org/10.1139/gen-2018-0042.
Wang T, Yang Y, Lou S, Wei W, Zhao Z, Ren Y, et al. Genome-wide characterization and gene expression analyses of GATA transcription factors in Moso bamboo (Phyllostachys edulis). Int J Mol Sci. 2019;21:14. https://doi.org/10.3390/ijms21010014.
Suárez-López P, Wheatley K, Robson F, Onouchi H, Valverde F, Coupland G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature. 2001;410:1116–20. https://doi.org/10.1038/35074138.
Vanholme B, Grunewald W, Bateman A, Kohchi T, Gheysen G. The tify family previously known as ZIM. Trends Plant Sci. 2007;12:239–44. https://doi.org/10.1016/j.tplants.2007.04.004.
Bai Y, Meng Y, Huang D, Qi Y, Chen M. Origin and evolutionary analysis of the plant-specific TIFY transcription factor family. Genomics. 2011;98:128–36. https://doi.org/10.1016/j.ygeno.2011.05.002.
Luo XM, Lin WH, Zhu S, Zhu JY, Sun Y, Fan XY, et al. Integration of light- and brassinosteroid-signaling pathways by a GATA transcription factor in Arabidopsis. Dev Cell. 2010;19:872–83. https://doi.org/10.1016/j.devcel.2010.10.023.
Köllmer I, Werner T, Schmülling T. Ectopic expression of different cytokinin-regulated transcription factor genes of Arabidopsis thaliana alters plant growth and development. J Plant Physiol. 2011;168:1320–7. https://doi.org/10.1016/j.jplph.2011.02.006.
Richter R, Behringer C, Müller IK, Schwechheimer C. The GATA-type transcription factors GNC and GNL/CGA1 repress gibberellin signaling downstream from DELLA proteins and phytochrome-interacting factors. Genes Dev. 2010;24:2093–104. https://doi.org/10.1101/gad.594910.
Ravindran P, Verma V, Stamm P, Kumar PP. A novel RGL2-DOF6 complex contributes to primary seed dormancy in Arabidopsis thaliana by regulating a GATA transcription factor. Mol Plant. 2017;10:1307–20. https://doi.org/10.1016/j.molp.2017.09.004.
Yu F, Utsumi R. Diversity, regulation, and genetic manipulation of plant mono- and sesquiterpenoid biosynthesis. Cell Mol Life Sci. 2009;66:3043–52. https://doi.org/10.1007/s00018-009-0066-7.
Vranová E, Coman D, Gruissem W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol. 2013;64:665–700. https://doi.org/10.1146/annurev-arplant-050312-120116.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. https://doi.org/10.1093/nar/gkr367.
Chanwala J, Satpati S, Dixit A, Parida A, Giri MK, Dey N. Genome-wide identification and expression analysis of WRKY transcription factors in pearl millet (Pennisetum glaucum) under dehydration and salinity stress. BMC Genomics. 2020;21:231. https://doi.org/10.1186/s12864-020-6622-0.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. https://doi.org/10.1093/molbev/mst010.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. https://doi.org/10.1093/molbev/msy096.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Liu M, Sun W, Ma Z, Zheng T, Huang L, Wu Q, et al. Genome-wide investigation of the AP2/ERF gene family in tartary buckwheat (Fagopyum Tataricum). BMC Plant Biol. 2019;19:84. https://doi.org/10.1186/s12870-019-1681-6.
Acknowledgements
Not applicable.
Funding
This work was supported by Zhejiang Provincial Ten Thousand Program for Leading Talents of Science and Technology Innovation [2018R52050]; National Natural Science Fund of China [82073963, 31571735, 81522049]; The Major Science and Technology Projects of Breeding New Varieties of Agriculture in Zhejiang Province [2021C02074]; Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents; Zhejiang Chinese Medical University Research Foundation [2021JKZDZC06]; The Opening Project of Zhejiang Provincial Preponderant and Characteristic Subject of Key University (Traditional Chinese Pharmacology), Zhejiang Chinese Medical University [ZYAOX2018009].
Author information
Authors and Affiliations
Contributions
M.S. and Q.H. performed bioinformatics analysis, sample collection, experiments and wrote the manuscript. Y.W., C.W., R.Z., and S.Z. helped analyze the data. G.K. designed the experiments and conceived the project, provided overall supervision of the study and revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ophiorrhiza pumila materials used in the study were kept as sterile in the greenhouse, which are maintained in accordance with the institutional guidelines of School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, China. This article did not contain any studies with human participants or animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, M., Huang, Q., Wang, Y. et al. Genome-wide survey of the GATA gene family in camptothecin-producing plant Ophiorrhiza pumila. BMC Genomics 23, 256 (2022). https://doi.org/10.1186/s12864-022-08484-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08484-x