
Abstract
Background
Hypoxia mediated pulmonary hypertension (HPH) is a lethal disease and lacks effective therapy. CircRNAs play significant roles in physiological process. Recently, circRNAs are found to be m6A-modified. The abundance of circRNAs was influenced by m6A. Furthermore, the significance of m6A circRNAs has not been elucidated in HPH yet. Here we aim to investigate the transcriptome-wide map of m6A circRNAs in HPH.
Results
Differentially expressed m6A abundance was detected in lungs of HPH rats. M6A abundance in circRNAs was significantly reduced in hypoxia in vitro. M6A circRNAs were mainly from protein-coding genes spanned single exons in control and HPH groups. Moreover, m6A influenced the circRNA–miRNA–mRNA co-expression network in hypoxia. M6A circXpo6 and m6A circTmtc3 were firstly identified to be downregulated in HPH.
Conclusion
Our study firstly identified the transcriptome-wide map of m6A circRNAs in HPH.
M6A can influence circRNA–miRNA–mRNA network. Furthermore, we firstly identified two HPH-associated m6A circRNAs: circXpo6 and circTmtc3. However, the clinical significance of m6A circRNAs for HPH should be further validated.
Similar content being viewed by others


Background
Pulmonary hypertension (PH) is a lethal disease and defined as an increase in the mean pulmonary arterial pressure ≥ 25 mmHg at rest, as measured by right heart catheterization [1]. Hypoxia mediated pulmonary hypertension (HPH) belongs to group III PH according to the comprehensive clinical classification of PH, normally accompanied by severe chronic obstructive pulmonary disease (COPD) and interstitial lung diseases [2]. HPH is a progressive disease induced by chronic hypoxia [1]. Chronic hypoxia triggers over-proliferation of pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), and activation of quiescent fibroblasts, the hallmark of HPH [1, 3]. The pathological characteristics of HPH are pulmonary vascular remolding, pulmonary hypertension, and right ventricular hypertrophy (RVH) [4]. So far there is no effective therapy for HPH [2]. More effective therapeutic targets are needed to be discovered.
Circular RNAs (circRNAs) were firstly found abundant in eukaryotes using RNA-seq approach [5,6,7]. Pre-mRNA is spliced with the 5′ and 3′ ends, forming a ‘head-to-tail’ splice junction, then circRNAs are occurred [5]. According to the genome origin, circRNAs may be classified into four different subtypes: exonic circRNA, intronic circRNA, exon–intron circRNA and tRNA introns circRNA [5]. CircRNAs are reported to play crucial roles in miRNA binding, protein binding, regulation of transcription, and post-transcription [5, 8]. Recent reports indicated that circRNAs can translate to proteins [8, 9]. Moreover, circRNAs are widely expressed in human umbilical venous endothelial cells when stimulated by hypoxia [10, 11]. Up to date, only a few reports mentioned PH-associated circRNAs. CircRNAs expression profile is demonstrated in HPH and chronic thromboembolic pulmonary hypertension [12]. However, the post-transcript modification of circRNAs in HPH is still unknown.
N6-methyladenosine (m6A) is regarded as one part of “epitranscriptomics” and identified as the most universal modification on mRNAs and noncoding RNAs (ncRNAs) in eukaryotes [13, 14]. DRm6ACH (D denotes A, U or G; R denotes A, G; H denotes A, C, or U) is a consensus motif occurred in m6A modified RNAs [15,16,17]. M6A modification is mainly enriched around the stop codons, at 3’untranslated regions and within internal long exons [17,18,19]. Several catalyzed molecules act as “writers”, “readers”, and “erasers” to regulate the m6A modification status [14]. The methyltransferase complex is known as writers, including methyltransferase-like-3, − 14 and − 16 (METTL3/METTL14/METTL16), Wilms tumor 1-associated protein (WTAP), RNA binding motif protein 15 (RBM15), vir like m6A methyltransferase associated (KIAA1429) and zinc finger CCCH-type containing 13 (ZC3H13), appending m6A on DRACH [17, 20, 21]. METTL3 is regarded as the core catalytically active subunit, while METTL14 and WTAP play a structural role in METTL3’s catalytic activity [18, 22]. The “erasers”, fat mass and obesity related protein (FTO) and alkylation repair homolog 5 (ALKBH5), catalyze the N-alkylated nucleic acid bases oxidatively demethylated [22]. The “readers”, the YT521-B homology (YTH) domain-containing proteins family includes YTHDF (YTHDF1, YTHDF2, YTHDF3), YTHDC1, and YTHDC2, specifically recognizes m6A and regulates splicing, localization, degradation and translation of RNAs [14, 22, 23]. The YTHDF1 and YTHDF2 crystal structures forms an aromatic cage to recognize m6A sites in cytoplasm [24]. YTHDC1 is the nuclear reader and YTHDC2 binds m6A under specific circumstances or cell types [24]. Hypoxia may alter the balance of writers-erasers-readers and induce tumor growth, angiogenesis, and progression [25, 26].
Interestingly, circRNAs can be m6A-modified. M6A circRNAs displayed cell-type-specific methylation patterns in human embryonic stem cells and HeLa cells [14]. CircRNAs contained m6A modifications are likely to promote protein translation in a cap-independent pattern [9]. However, m6A circRNAs has not been elucidated in HPH yet. Here we are the first to identify the expression profiling of m6A circRNAs in HPH.
Results
M6A level of circRNAs was reduced in HPH rats and most circRNAs contained one m6A peak
Three weeks treatment by hypoxia resulted in right ventricular systolic pressure (RVSP) elevating to 42.23 ± 1.96 mmHg compared with 27.73 ± 1.71 mmHg in the control (P < 0.001, Fig. 1a and b). The ratio of the right ventricle (RV), left ventricular plus ventricular septum (LV + S) [RV/ (LV + S)] was used as an index of RVH. RVH was indicated by the increase of RV/ (LV + S) compared with the control (0.25 ± 0.03 vs. 0.44 ± 0.04, P = 0.001, Fig. 1c). The medial wall of the pulmonary small arteries was also significantly thickened (19.28 ± 2.19% vs. 39.26 ± 5.83%, P < 0.001, Fig. 1d and e). Moreover, in the normoxia group, 53.82 ± 3.27% of the arterioles were non-muscularized (NM) vessels, and 25.13 ± 1.83% were fully muscularized (FM) vessels. In contrast, partially muscularized vessels (PM) and FM vessels showed a greater proportion (32.88 ± 3.15% and 41.41 ± 3.35%) in HPH rats, while NM vessels occupied a lower proportion (25.71 ± 2.55%) (Fig. 1f). Figure 1g displayed the heatmap of m6A circRNAs expression profiling in N and HPH. M6A abundance in 166 circRNAs was significantly upregulated. Meanwhile, m6A abundance in 191 circRNAs was significantly downregulated (Additional file 1: Data S1, filtered by fold change ≥4 and P ≤ 0.00001). Lungs of N and HPH rats were selected to measure m6A abundance in purified circRNAs. The m6A level in total circRNAs isolated from lungs of HPH rats was lower than that from controls (Fig. 1h). Moreover, over 50% circRNAs contained only one m6A peak either in lungs of N or HPH rats (Fig. 1i).

M6A level of circRNAs in HPH rats and the number of m6A peak in circRNAs Rats were maintained in a normobaric normoxic (N, FiO2 21%) or hypoxic (HPH, FiO2 10%) chamber for 3 weeks, then RVSP was detected (a, b). c The ratio of RV/ (LV + S). d H&E staining and immunohistochemical staining of α-SMA were performed in the lung sections. Representative images of pulmonary small arteries. Scale bar = 50 μm. Quantification of wall thickness (e) and vessel muscularization (f). g Heatmap depicting hierarchical clustering of altered m6A circRNAs in lungs of N and HPH rats. Red represents higher expression and yellow represents lower expression level. h Box-plot for m6A peaks enrichment in circRNAs in N and HPH. i Distribution of the number of circRNAs (y axis) was plotted based on the number of m6A peaks in circRNAs (x axis) in N and HPH. Values are presented as means ± SD (n = 6 in each group). Only vessels with diameter between 30 and 90 μm were analyzed. NM, nonmuscularized vessels; PM, partially muscularized vessels; FM, fully muscularized vessels. **0.001 ≤ P ≤ 0.009 (different from N); ***P < 0.001 (different from N)
M6A circRNAs were mainly from protein-coding genes spanned single exons in N and HPH groups
We analyzed the distribution of the parent genes of total circRNAs, m6A-circRNAs, and non-m6A circRNAs in N and HPH, respectively. N and HPH groups showed a similar genomic distribution of m6A circRNAs and non-m6A circRNAs (Fig. 2a and b). Moreover, about 80% of m6A circRNAs and non-m6A circRNAs were derived from protein-coding genes in both groups. A previous report indicated that most circRNAs originated from protein-coding genes spanned two or three exons [14]. While in our study, over 50 and 40% of total circRNAs from protein-coding genes spanned one exon in N and HPH groups, respectively (Fig. 2c and d). Similarly, m6A circRNAs and non-m6A circRNAs were mostly encoded by single exons. Therefore, it was indicated that m6A methylation was abundant in circRNAs originated from single exons in N and HPH groups.

The genomic origins of m6A circRNAs The distribution of genomic origins of total circRNAs (input, left), m6A circRNAs (eluate, center), and non-m6A circRNAs (supernatant, right) in N (a) and HPH (b). The percentage of circRNAs (y axis) was calculated according to the number of exons (x axis) spanned by each circRNA for the input circRNAs (left), m6A-circRNAs (red, right) and non-m6A circRNAs (blue, right) in N (c) and HPH (d). Up to seven exons are shown
The distribution and functional analysis for host genes of circRNAs with differentially expressed m6A peaks
The length of differentially-expressed m6A circRNAs was mostly enriched in 1–10,000 bps (Fig. 3a). The host genes of upregulated m6A circRNAs were located in chromosome 1, 2 and 10, while the downregulated parts were mostly located in chromosome 1, 2 and 14 (Fig. 3b).

The distribution and functional analysis for host genes of circRNAs with differentially expressed (DE) m6A peaks (a) Length of DE m6A circRNAs. b The chromosomes origins for host genes of DE m6A circRNAs. GO enrichment and KEGG signaling pathway analysis for host genes of upregulated (c) and downregulated (d) m6A circRNAs. GO enrichment analysis include biological process (BP) analysis, cellular component (CC) analysis, and molecular function (MF) analysis. P values are calculated by DAVID tool
Gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed to explore the host genes of circRNAs with differentially-expressed m6A peaks. In the GO analysis (Fig. 3c, left), the parent genes of circRNAs with upregulated m6A peaks were enriched in the protein modification by small protein conjugation or removal and macromolecule modification process in the biological process (BP). Organelle and membrane-bounded organelle were also the two largest parts in the cellular component (CC) analysis. Binding and ion binding were the two main molecular functions (MF) analysis. The top 10 pathways from KEGG pathway analysis were selected in the bubble chart (Fig. 3c, right). Among them, the oxytocin signaling pathway, protein processing in endoplasmic reticulum and cGMP-PKG signaling pathway were the top 3 pathways involved. In addition, vascular smooth muscle contraction pathway was the most associated pathway in PH progression [27].
In Fig. 3d left, the parent genes of circRNAs with downregulated m6A peaks were mainly enriched in the cellular protein modification process and protein modification process in BP. Organelle and membrane-bounded organelle made up the largest proportion in the CC classification. The MF analysis was focused on receptor signaling protein activity and protein binding. The parent genes of circRNAs with decreased m6A peaks were mainly involved in the tight junction and lysine degradation in the KEGG pathway analysis (Fig. 3d, right).
Hypoxia can influence the m6A level of circRNAs and circRNAs abundance
360 m6A circRNAs were shared in N and HPH groups. 49% of m6A circRNAs detected in N group were not detected in HPH group, and 54% of m6A circRNAs detected in HPH group were not detected in N group (Fig. 4a). To explore whether m6A methylation would influence circRNAs expression level, expression of the 360 common m6A circRNAs were identified. More circRNAs tended to decrease in HPH compared to N (Fig. 4b). Moreover, expression of m6A circRNAs was significantly downregulated compared with non-m6A circRNAs in hypoxia, suggesting that m6A may downregulate the expression of circRNAs in hypoxia (Fig. 4c, P = 0.0465).

The relationship of m6A level and circRNAs abundance in hypoxia (a) Venn diagram depicting the overlap of m6A circRNAs between N and HPH. b Two-dimensional histograms comparing the expression of m6A circRNAs in lungs of N and HPH rats. It showed that m6A circRNAs levels for all shared circRNAs in both groups. CircRNAs counts were indicated on the scale to the right. c Cumulative distribution of circRNAs expression between N and HPH for m6A circRNAs (red) and non-m6A circRNAs (blue). P value was calculated using two-sided Wilcoxon-Mann-Whiteney test
Construction of a circRNA–miRNA–mRNA co-expression network in HPH
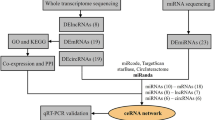
We found 76 upregulated circRNAs with increased m6A abundance, and 107 downregulated circRNAs with decreased m6A abundance (Fig. 5a, Additional file 2: Data S2, Additional files 3 and 4). As known, circRNAs were mostly regarded as a sponge for miRNAs and regulated the expression of corresponding target genes of miRNAs [28]. To explore whether circRNAs with differentially-expressed m6A abundance influence the availability of miRNAs to target genes, we selected differentially-expressed circRNAs with increased or decreased m6A abundance. GO enrichment analysis and KEGG pathway analysis were also performed to analyze target mRNAs. Target mRNAs displayed similar GO enrichment in the two groups (Fig. 5b and c). Two main functions were determined in BP analysis: positive regulation of biological process and localization. Intracellular and intracellular parts make up the largest proportion in CC part. Target mRNAs were mostly involved in protein binding and binding in MF part. In the KEGG pathway analysis, the top 10 most enriched pathways were selected (Fig. 5d and e). Wnt and FoxO signaling pathways were reported to be involved in PH progression [29,30,31]. Then, we analyzed the target genes involved in these two pathways. SMAD4 was associated with PH and involved in Wnt signaling pathways. MAPK3, SMAD4, TGFBR1, and CDKN1B were involved in FoxO signaling pathways. To explore the influence of circRNA-miRNA regulation on PH-associated genes expression, we constructed a circRNA-miRNA-mRNA network, integrating matched expression profiles of circRNAs, miRNAs and mRNAs (Fig. 5f and g). MicroRNAs sponged by the target genes of interest were analyzed. MiR-125a-3p, miR-23a-5p, miR-98-5p, let-7b-5p, let-7a-5p, let-7 g-5p, and miR-205 were analyzed because they were reported to be associated with PH [32, 33]. We filtered the key mRNAs and miRNAs, and founded that the two circRNAs were the most enriched, which were originated from chr1:204520403–204,533,534- (Xpo6) and chr7:40223440–40,237,400- (Tmtc3).

Construction of a circRNA–miRNA–mRNA co-expression network in HPH (a) Comparison of the relationship between m6A level and expression of circRNAs between N and HPH. The fold-change ≥2.0 was considered to be significant, which was the m6A abundance of HPH relative to N. Red dots represents circRNAs with upregulated m6A level and blue dots represents circRNAs with downregulated m6A level. IP/Input referred to the m6A abundance in circRNAs detected in MeRIP-Seq (IP) normalized to that detected in input. b and c GO enrichment analysis includes BP analysis, CC analysis, and MF analysis. P values are calculated by DAVID tool. d and e KEGG signaling pathway analysis for the downstream mRNAs which was predicted to be ceRNA of DE cirRNAs. Methy. down & exp. down represents downregulated cirRNAs with decreased m6A level. Methy. up & exp. up represents upregulated cirRNAs with increased m6A level. f and g CeRNA analysis for DE circRNAs. Network map of circRNA-miRNA-mRNA interactions. Green V type node: miRNA; yellow circular node: DE circRNAs; blue hexagon node: target genes of miRNAs; red hexagon node: PH-related genes
M6A circXpo6 and m6A circTmtc3 were downregulated in PASMCs and PAECs in hypoxia
M6A abundance was significantly reduced in PASMCs and PAECs when exposed to hypoxia (0.107% ± 0.007 vs. 0.054% ± 0.118, P = 0.023 in PASMCs; 0.114% ± 0.011 vs. 0.059% ± 0.008, P = 0.031 in PAECs, Fig. 6a). M6A abundance in circRNAs was lower than it in mRNAs (0.1–0.4%) [17, 18]. Next, we confirmed the back-splicing of circXpo6 and circTmtc3 by CIRI software. The sequence of linear Xpo6 and Tmtc3 mRNA was analyzed. Then we identified that circXpo6 was spliced form exon 7, 8, and 9 of Xpo6. CircTmtc3 was spliced form exon 8, 9, 10, and 11 (Fig. 6b). Using cDNA and genomic DNA (gDNA) from PASMCs and PAECs as templates, circXpo6 and circTmtc3 were only amplified by divergent primers in cDNA, while no product was detected in gDNA (Fig. 6c). To identify whether circXpo6 and circTmtc3 were modified by m6A, we performed M6A RNA Immunoprecipitation (MeRIP)-RT-PCR and MeRIP-quantitative RT-PCR (MeRIP-qRT-PCR) to detect the expression of circXpo6 and circTmtc3 (Fig. 6d and e). m6A circXpo6 and m6A circTmtc3 were significantly decreased in PASMCs and PAECs when exposed to hypoxia (P = 0.002, and P = 0.015 in PASMCs and P = 0.02, and P = 0.047 in PAECs).

The expression profiling of m6A circXpo6 and m6A circTmtc3 in pulmonary arterial smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs) in hypoxia. a M6A levels of total circRNAs were determined based on colorimetric method in vitro. PASMCs and PAECs were exposed to 21% O2 and 1% O2 for 48 h, respectively. Total RNA was extracted and treated by RNase R. M6A levels were determined as a percentage of total circRNAs. b Schematic representation of exons of the Xpo6 and Tmtc3 circularization forming circXpo6 and circTmtc3 (black arrow). c RT-PCR validation of circXpo6 and circTmtc3 in PASMCs and PAECs exposed to 21% O2. Divergent primers amplified circRNAs in cDNA, but not in genomic DNA (gDNA). The size of the DNA marker is indicated on the left of the gel. d and e RT-PCR and qRT-PCR was performed after m6A RIP in PASMCs and PAECs exposed to 21% (N) and 1% O2 (H) for 48 h, respectively. Input was used as a control (d). IgG was used as a negative control (d and e). Values are presented as means ± SD. *P ≤ 0.05 (different from 21% O2 or the N-anti-m6A); **0.001 ≤ P ≤ 0.009 (different from the N-anti-m6A), (n = 3 each)
Discussion
In this study, we identified the transcriptome-wide map of m6A circRNAs in hypoxia mediated pulmonary hypertension. On the whole, we found that m6A level in circRNAs was reduced in lungs when exposed to hypoxia. M6A circRNAs were mainly derived from single exons of protein-coding genes in N and HPH. M6A abundance in circRNAs was downregulated in hypoxia in vitro. M6A influenced the circRNA–miRNA–mRNA co-expression network in hypoxia. Moreover, circXpo6 and circTmtc3 were the novel identified circRNAs modified by m6A in hypoxia mediated pulmonary hypertension.
M6A plays important roles in various biological processes. M6A is associated with cancer progression, promoting the proliferation of cancer cells and contributing to the cancer stem cell self-renewal [18, 21]. Lipid accumulation was reduced in hepatic cells when m6A abundance in peroxisome proliferator-activator (PPaR) was decreased [34]. Enhanced m6A level of mRNA contributed to compensated cardiac hypertrophy [35]. Also, m6A modification of large intergenic noncoding RNA 1281 was necessary for mouse embryonic stem cells differentiation [36].
Although it has been reported that m6A mRNAs were influenced by hypoxia, there is no report about m6A circRNAs in HPH yet. Up to now, no consistent conclusion was reached about the link between m6A and hypoxia. Previous reports found that the m6A abundance in mRNA was increased under hypoxia stress in HEK293T cells and cardiomyocytes [37, 38]. The increased m6A level stabilized the mRNAs of Glucose Transporter 1 (Glut1), Myc proto-oncogene bHLH transcription factor (Myc), Dual Specificity Protein Phosphatase 1 (Dusp1), Hairy and Enhancer of Split 1 (Hes1), and Jun Proto-Oncogene AP-1 Transcription Factor Subunit (Jun) without influencing their protein level [37]. In contrast, another reported that m6A level of total mRNA was decreased when human breast cancer cell lines were exposed to 1% O2 [26]. Hypoxia increased demethylation by stimulating hypoxia-inducible factor (HIF)-1α- and HIF-2α–dependent over-expression of ALKBH5 [26]. In addition, transcription factor EB activates the transcription of ALKBH5 and downregulates the stability of METTL3 mRNA in hypoxia/reoxygenation-induced autophagy in ischemic diseases [38]. Our study found that m6A abundance in total circRNAs was decreased in hypoxia exposure. Moreover, our study indicated that circXpo6 and circTmtc3 were the novel identified circRNAs modified by m6A in HPH. M6A abundance in circXpo6 and circTmtc3 was decreased in hypoxia. It is probably because of HIF-dependent and ALKBH5-mediated m6A demethylation [26].
Previous reports indicated that m6A methylation close to 3’UTR and stop codon of mRNA is inversely correlated with gene expression [14, 39]. Low m6A level is negatively associated with circRNAs expression, while high m6A level is not linked to circRNAs expression in human embryonic stem cells and HeLa cells [14]. Consistent with the previous reports [14, 39], our study found that m6A reduced the total circRNAs abundance in hypoxia. The association between m6A level and specific gene abundance is remained as an open question. Some previous reports indicated that m6A level was positively associated with long non-coding RNA (lncRNA) or mRNA expression [40, 41]. M6A was positively associated with RP11–138 J23.1 (RP11) expression when ALKBH5 was overexpressed in colorectal cancer [40]. mRNAs were downregulated after METTL14 deletion in β-cells [41]. On the contrary, another reports insisted that m6A level was negatively associated with RNA expression [42,43,44]. the mRNA lifetime of Family with Sequence Similarity 134, Member B (FAM134B) was prolonged when the m6A site was mutant [42]. The decreased m6A level resulted in the increased expression of N-methyl-D-aspartate receptor 1 (NMDAR1) in Parkinson’s disease [43]. Forkhead Box protein M1 (FOXM1) abundance was increased when ALKBH5 was upregulated in glioblastoma [44]. Our study indicated that the expression of circXpo6 and circTmtc3 was decreased with the downregulated m6A level. The association between m6A level and circRNAs abundance was not determined yet. We suspected that m6A may influence the expression of circXpo6 and circTmtc3 through similar manners as before [40, 41]. But it needs further validation.
Competing endogenous RNA (CeRNA) mechanism was proposed that mRNAs, pseudogenes, lncRNAs and circRNAs interact with each other by competitive binding to miRNA response elements (MREs) [45, 46]. M6A acts as a post-transcript regulation of circRNAs and influences circRNAs expression, thus we suggested that m6A could also regulate the circRNA–miRNA–mRNA co-expression network. When the circRNAs were classified, we found that these downstream targets regulated by circRNA–miRNA of interest were mostly enriched in PH-associated Wnt and FoxO signaling pathways [30, 31]. The Wnt/β-catenin (bC) pathway and Wnt/ planar cell polarity (PCP) pathway are the two most critical Wnt signaling pathways in PH [30]. As known, the two important cells associated with HPH are PASMCs and PAECs [1, 3]. The growth of PASMCs was increased when Wnt/bC and Wnt/PCP pathways were activated by platelet derived growth factor beta polypeptide b (PDGF-BB) [30, 47]. In addition, the proliferation of PAECs was enhanced when Wnt/bC and Wnt/PCP pathways were activated by bone morphogenetic protein 2 (BMP2). Furthermore, the FoxO signaling pathway is associated with the apoptosis-resistant and hyper-proliferative phenotype of PASMCs [31]. Reactive oxygen species is increased by hypoxia and activates AMPK-dependent regulation of FoxO1 expression, resulting in increased expression of catalase in PASMCs [48]. Our study firstly uncovered that m6A influenced the stability of circRNAs, thus affecting the binding of circRNAs and miRNA, resulting in the activation of Wnt and FoxO signaling pathways.
Conclusion
In conclusion, our study firstly identified the transcriptome-wide map of m6A circRNAs in HPH. M6A level in circRNAs was decreased in lungs of HPH and in PASMCs and PAECs exposed to hypoxia. M6A level influenced circRNA–miRNA–mRNA co-expression network in HPH. Moreover, we firstly identified two downregulated m6A circRNAs in HPH: circXpo6 and circTmtc3. CircRNAs may be used as biomarkers because it is differentially enriched in specific cell types or tissues and not easily degraded [6]. Also, the aberrant m6A methylation may contribute to tumor formation and m6A RNAs may be a potential therapy target for tumor [17].
Limitations still exist in the study. First, we did not analyze the m6A level between circRNAs and the host genes. Second, the exact mechanism of hypoxia influences m6A was not demonstrated. Thirdly, the function of circXpo6 and circTmtc3 in HPH was not elaborated. Lastly, besides hypoxia mediated pulmonary hypertension, many other significant PH models should also be noted, such as monocrotaline mediated PH, monocrotaline + pneumonectomy mediated PH, and so on. It is insufficient that we explored the expression profiling of m6A circRNAs only in hypoxia mediated pulmonary hypertension. We plan to explore the expression profiling of m6A circRNAs in monocrotaline-induced PH and other PH models. Moreover, the clinical significance of m6A circRNAs for HPH should be further validated.
Methods
Hypoxia mediated PH rat model and measurement of RVSP and RVH
Sprague-Dawley rats (SPF, male, 180–200 g, 4 weeks) were obtained from the Animal Experimental Center of Zhejiang University, China. Rats were maintained in a normobaric normoxia (FiO2 21%, n = 6) or hypoxic chamber (FiO2 10%, n = 6) for 3 weeks [3, 49]. Rats were then anesthetized by intraperitoneal injection of 1% sodium pentobarbital (130 mg/kg) [50]. Then, rats were fixed in supine position on the board. All of the operations were performed after rats were anesthetized and became unconscious. RVSP was measured as below. Right ventricle catheterization was performed through the right jugular using a pressure-volume loop catheter (Millar) as the previous reports [49, 51]. After measurement of RVSP, all rats were put into a confined and transparent euthanasia device (to observe whether the rats were sacrificed), then 100% CO2 was released into the device continuously until all the rats sacrificed. The criteria for sacrifice were that rats did not have spontaneous breath for 2–3 min and blink reflex. Then, heart tissues were removed and segregated. The ratio of [RV/ (LV + S)] was used as an index of RVH. Lung were removed and immediately frozen at liquid nitrogen or fixed in 4% buffered paraformaldehyde solution. All experimental procedures were conducted in line with the principles approved by the Institutional Animal Care and Use Committee of Zhejiang University.
Histological analysis
Lung tissues were embedded in paraffin, sectioned at 4 μm and stained with hematoxylin and eosin (H&E) and α-smooth muscle actin (α-SMA, 1:100, ab124964, Abcam, USA). The ratio of pulmonary small artery wall thickness and muscularization were calculated [3].
Isolation and hypoxia-treatment of PASMCs and PAECs
PASMCs and PAECs were isolated using the methods according to previous reports [32, 50, 52]. PASMCs and PAECs were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 20% FBS for 48 h, respectively [32, 53]. The cells were incubated in a 37 °C, 21% O2 or 1% O2–5% CO2 humidified incubator. PASMCs at 70–80% confluence in 4 to 7 passages were used in experiments. PAECs at 80–90% confluence in 4 to 5 passages were used in experiments [54].
RNA isolation and RNA-seq analysis of circRNAs
Total RNA (10 mg) was obtained using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) from lungs (1 g) of control and HPH rats. The extracted RNAs were purified with Rnase R (RNR07250, Epicentre) digestion to remove linear transcripts. Paired-end reads were harvested from Illumina Hiseq Sequence after quality filtering. The reads were aligned to the reference genome (UCSC RN5) with STAR software. CircRNAs were detected and annotated with CIRI software [55]. Raw junction reads were normalized to per million number of reads mapped to the genome with log2 scaled.
MeRIP and library preparation
Total RNA was extracted as the methods described above. Then, rRNA was depleted following DNase I treatment. RNase R treatment (5 units/mg) was performed in duplicate with 5 mg of rRNA-depleted RNA input. High-throughput m6A and circRNAs sequencing were performed by Cloudseq Biotech Inc. (Shanghai, China). Fragmented RNA was incubated with anti-m6A polyclonal antibody (Synaptic Systems, 202,003) in IPP buffer for 2 h at 4 °C. The mixture was then incubated with protein A/G magnetic beads (88,802, Thermo Fisher) at 4 °C for an additional 2 h. Then, bound RNA was eluted from the beads with N6-methyladenosine (PR3732, BERRY & ASSOCIATES) in IPP buffer and extracted with Trizol reagent (15,596,026, Thermo Fisher). NEBNext® Ultra™ RNA Library Prep Kit (E7530L, NEB) was used to construct RNA-seq library from immunoprecipitated RNA and input RNA. The m6A-IP and input samples were subjected to 150 bp paired-end sequencing on Illumina HiSeq sequencer. Methylated sites on circRNAs were identified by MetPeak software.
Construction of circRNA–miRNA–mRNA co-expression network
The circRNA–miRNA–mRNA co-expression network was based on the ceRNA theory that circRNA and mRNA shared the same MREs [45, 46]. Cytoscape was used to visualize the circRNA–miRNA–mRNA interactions based on the RNA-seq data. The circRNA-miRNA interaction and miRNA–mRNA interaction of interest were predicted by TargetScan and miRanda.
Measurement of Total m6A, MeRIP-RT-PCR and MeRIP-qRT-PCR
Total m6A content was measured in 200 ng aliquots of total RNA extracted from PASMCs and PAECs exposed to 21% O2 and 1% O2 for 48 h using an m6A RNA methylation quantification kit (P-9005, Epigentek). MeRIP (17–701, Millipore) was performed according to the manufacturer’s instruction. A 1.5 g aliquot of anti-m6A antibody (ABE572, Millipore) or anti-IgG (PP64B, Millipore) was conjugated to protein A/G magnetic beads overnight at 4 °C. A 100 ng aliquot of total RNA was then incubated with the antibody in IP buffer supplemented with RNase inhibitor and protease inhibitor. The RNA complexes were isolated through phenol-chloroform extraction (P1025, Solarbio) and analyzed via RT-PCR or qRT-PCR assays. Primers sequences are listed in Table 1.
Data analysis
3′ adaptor-trimming and low quality reads were removed by cutadapt software (v1.9.3). Differentially methylated sites were identified by the R MeTDiff package. The read alignments on genome could be visualized using the tool IGV. Differentially expressed circRNAs were identified by Student’s t-test. GO and KEGG pathway enrichment analysis were performed for the corresponding parental mRNAs of the DE circRNAs. GO enrichment analysis was performed using the R topGO package. KEGG pathway enrichment analysis was performed according to a previous report [56]. GO analysis included BP analysis, CC analysis, and MF analysis. MicroRNAs sponged by the target genes were predicted by TargetScan and microRNA websites. P values are calculated by DAVID tool for GO and KEGG pathway analysis. The rest statistical analyses were performed with SPSS 19.0 (Chicago, IL, USA) and GraphPad Prism 5 software (La Jolla, CA). N refers to number of samples in figure legends. The statistical significance was determined by Student’s t-test (two-tailed) or two-sided Wilcoxon-Mann-Whiteney test. P < 0.05 was considered statistically significant. All experiments were independently repeated at least three times.
Availability of data and materials
The raw high-throughput m6A and circRNAs sequencing generated during the current study has been uploaded into the repository named Zenodo, the persistent web links are https://doi.org/10.5281/zenodo.3596039, https://doi.org/10.5281/zenodo.3596178, https://doi.org/10.5281/zenodo.3596016, and https://doi.org/10.5281/zenodo.3596276.
Abbreviations
- ALKBH5:
-
Alkylation repair homolog 5
- BMP2:
-
Bone morphogenetic protein 2
- BP:
-
Biological process analysis
- CC:
-
Cellular component analysis
- CeRNA:
-
Competing endogenous RNA
- circRNAs:
-
Circular RNAs
- COPD:
-
Chronic obstructive pulmonary disease
- Dusp1:
-
Dual Specificity Protein Phosphatase 1
- FAM134B:
-
Family with Sequence Similarity 134, Member B
- FOXM1:
-
Forkhead Box protein M1
- FTO:
-
Fat mass and obesity related protein
- Glut1:
-
Glucose Transporter 1
- GO:
-
Gene ontology analysis
- Hes1:
-
Hairy and Enhancer of Split 1
- HIF-1α, HIF-2α:
-
Hypoxia-inducible factor-1α,-2α
- HPH:
-
Hypoxia mediated pulmonary hypertension
- Jun:
-
Jun Proto-Oncogene AP-1 Transcription Factor Subunit
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KIAA1429:
-
Vir like m6A methyltransferase associated
- LncRNA:
-
Long non-coding RNA
- m6A:
-
N6-methyladenosine
- MeRIP:
-
M6A RNA Immunoprecipitation
- MeRIP-qRT-PCR:
-
MeRIP-quantitative RT-PCR
- METTL3, METTL14, METTL16:
-
Methyltransferase-like-3, − 14 and − 16
- MF:
-
Molecular functions analysis
- MREs:
-
MiRNA response elements
- Myc:
-
Myc proto-oncogene bHLH transcription factor
- ncRNAs:
-
Noncoding RNAs
- NMDAR1:
-
N-methyl-D-aspartate receptor 1
- PAECs:
-
Pulmonary artery endothelial cells
- PASMCs:
-
Pulmonary artery smooth muscle cells
- PDGF-BB:
-
Platelet derived growth factor beta polypeptide b
- PH:
-
Pulmonary hypertension
- PPαR:
-
Peroxisome proliferator-activator
- RBM15:
-
RNA binding motif protein 15
- RP11:
-
RP11–138 J23.1
- RVH:
-
Right ventricular hypertrophy
- RVSP:
-
Right ventricular systolic pressure
- WTAP:
-
Wilms tumor 1-associated protein
- YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2:
-
YT521-B homology domain-containing proteins family
- ZC3H13:
-
Zinc finger CCCH-type containing 13
References
Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol. 2015;308:L229–52.
Galiè N, Humbert M, Luc Vachiery J, Gibbs S, Lang I, Torbicki A, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2015;2015(46):879–82.
Zhang RF, Shi LH, Zhou L, Zhang GS, Wu XH, Shao FC, et al. Transgelin as a therapeutic target to prevent hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;306:L574–83.
Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. 2014;189(3):314–24.
Meng X, Li X, Zhang P, Wang J, Zhou Y, Chen M. Circular RNA: an emerging key player in RNA world. Brief Bioinform. 2017;18(4):547–57.
Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32(5):309–16.
Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–8.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of CircRNAs. Mol Cell. 2017;66(1):1–13.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–41.
Boeckel JN, Jae N, Heumuller AW, Chen W, Boon RA, Stellos K, et al. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res. 2015;117(10):884–90.
Dang RY, Liu FL, Li Y. Circular RNA hsa_circ_0010729 regulates vascular endothelial cell proliferation and apoptosis by targeting the miR-186/HIF-1alpha axis. Biochem Biophys Res Commun. 2017;490(2):104–10.
Wang J, Zhu MC, Kalionis B, Wu JZ, Wang LL, Ge HY, et al. Characteristics of circular RNA expression in lung tissues from mice with hypoxiainduced pulmonary hypertension. Int J Mol Med. 2018;42(3):1353–66.
Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, et al. High-resolution N(6) -methyladenosine (m(6) a) map using photo-crosslinking-assisted m(6) a sequencing. Angew Chem. 2015;54(5):1587–90.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20(9):2262–76.
Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y, et al. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2018;46(D1):D139–45.
Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46(D1):D327–34.
Wang S, Chai P, Jia R, Jia R. Novel insights on m(6)a RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. 2018;17(1):101.
Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9(2):124.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Wang X, Huang J, Zou T, Yin P. Human m(6)a writers: two subunits, 2 roles. RNA Biol. 2017;14(3):300–4.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–17.
Wu B, Li L, Huang Y, Ma J, Min J. Readers, writers and erasers of N(6)-methylated adenosine modification. Curr Opin Struct Biol. 2017;47:67–76.
Liao S, Sun H, Xu C. YTH domain: a family of N(6)-methyladenosine (m(6)a) readers. Genomics Proteomics Bioinformatics. 2018;16(2):99–107.
Patil DP, Pickering BF, Jaffrey SR. Reading m(6)a in the Transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28(2):113–27.
Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, et al. Cross-talk among writers, readers, and erasers of m6A regulates cancer growth and progression. Sci Adv. 2018;4:eaar8263.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci. 2016;113(14):E2047–56.
Rowan SC, Keane MP, Gaine S, McLoughlin P. Hypoxic pulmonary hypertension in chronic lung diseases: novel vasoconstrictor pathways. Lancet Respir Med. 2016;4(3):225–36.
Chan JJ, Tay Y. Noncoding RNA:RNA regulatory networks in Cancer. Int J Mol Sci. 2018;19(5):1310.
Baarsma HA, Konigshoff M. 'WNT-er is coming': WNT signalling in chronic lung diseases. Thorax. 2017;72(8):746–59.
de Jesus PV, Yuan K, Alastalo TP, Spiekerkoetter E, Rabinovitch M. Targeting the Wnt signaling pathways in pulmonary arterial hypertension. Drug Discov Today. 2014;19(8):1270–6.
Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20(11):1289–300.
Su H, Xu X, Yan C, Shi Y, Hu Y, Dong L, et al. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir Res. 2018;19(1):254.
Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30(4):716–23.
Zhong X, Yu J, Frazier K, Weng X, Li Y, Cham CM, et al. Circadian clock regulation of hepatic lipid metabolism by modulation of m(6)a mRNA methylation. Cell Rep. 2018;25(7):1816–28.
Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. The N(6)-Methyladenosine mRNA Methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139(4):533–45.
Yang DD, Qiao J, Wang GY, Lan YY, Li GP, Guo XD, et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential, vol. 46; 2018.
Nate J. Fry BAL, Olga R. Ilkayeva, Christopher L. Holley, and Kyle D. Mansfield: N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. Rna 2017, 23(9):1444–1455.
Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, et al. METTL3 and ALKBH5 oppositely regulate m(6)a modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019:1–19.
Luo GZ, MacQueen A, Zheng G, Duan H, Dore LC, Lu Z, et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun. 2014;5:5630.
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18(1):87.
Yao Y, Bi Z, Wu R, Zhao Y, Liu Y, Liu Q, et al. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPbeta pathway via an m(6)A-YTHDF2-dependent manner. FASEB J. 2019; fj201802644R.
Cai M, Liu Q, Jiang Q, Wu R, Wang X, Wang Y. Loss of m(6) a on FAM134B promotes adipogenesis in porcine adipocytes through m(6) A-YTHDF2-dependent way. IUBMB Life. 2019;71(5):580–6.
Chen X, Yu C, Guo M, Zheng X, Ali S, Huang H, et al. Down-regulation of m6A mRNA methylation is involved in dopaminergic neuronal death. ACS Chem Neurosci. 2019.
Fitzsimmons CM, Batista PJ. It's complicated... m(6)A-dependent regulation of gene expression in cancer. Biochim Biophys Acta Gene Regul Mech. 2019;1862(3):382–93.
Chen L, Zhang S, Wu J, Cui J, Zhong L, Zeng L, Ge S. circRNA_100290 plays a role in oral cancer by functioning as a sponge of the miR-29 family. Oncogene. 2017;36(32):4551–61.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–8.
JD JW, Austin ED, Gaskill C, Baskir R, Bilousova G, Jean JC, et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2014;307:C415–30.
Awad H, Nolette N, Hinton M, Dakshinamurti S. AMPK and FoxO1 regulate catalase expression in hypoxic pulmonary arterial smooth muscle. Pediatr Pulmonol. 2014;49(9):885–97.
Cowburn AS, Crosby A, Macias D, Branco C, Colaco RD, Southwood M, et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci U S A. 2016;113(31):8801–6.
Yun X, Jiang HY, Lai N, Wang J, Shimoda LA. Aquaporin 1-mediated changes in pulmonary arterial smooth muscle cell migration and proliferation involve β-catenin. Am J Physiol Lung Cell Mol Physiol. 2017;313:L889–98.
Yamazato Y, Ferreira AJ, Hong KH, Sriramula S, Francis J, Yamazato M, et al. Prevention of pulmonary hypertension by angiotensin-converting enzyme 2 gene transfer. Hypertension. 2009;54(2):365–71.
Xiao R, Su Y, Feng T, Sun M, Liu B, Zhang J, et al. Monocrotaline induces endothelial injury and pulmonary hypertension by targeting the extracellular calcium-sensing receptor. J Am Heart Assoc. 2017;6(4):e004865.
Zhang HY, Liu Y, Yan LX, Du W, Zhang XD, Zhang M, et al. Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J Cell Physiol. 2018;233(5):4077–90.
Omura J, Satoh K, Kikuchi N, Satoh T, Kurosawa R, Nogi M, et al. Protective roles of endothelial AMP-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ Res. 2016;119(2):197–209.
Thomas B, Hansen MTV, Christian K. Damgaard and Jørgen Kjems: comparison of circular RNA prediction tools. Nucleic Acids Res. 2015;44(6):e58.
Tian L, Greenberg SA, Kong SW, Altschuler J, Kohane IS, Park PJ. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci U S A. 2005;102(38):13544–9.
Acknowledgements
We thanked all subjects who participated in this study. We thank Cloud-Seq Biotech Ltd. Co. (Shanghai, China) for the MeRIP-Seq service and the subsequent bioinformatics analysis.
Funding
This study was supported by the National Natural Science Foundation of China (81570043), the National Natural Science Foundation of China (81270107), the Zhejiang Provincial Natural Science Foundation of China (LY19H010003). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HS performed the experiments, analyzed the data, and made the figures; GWW, LFW and XQM analyzed the results; KJY and RFZ designed the study, analyzed the data, explained the findings and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol of this research was submitted to and approved by the Institutional Animal Care and Use Committee of Zhejiang University, China (permit number: SYXK 2017–0006).
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Data S1.
Differentially expressed m6A abundance in circRNAs.
Additional file 2: Data S2.
Differentially expressed m6A abundance linked with differentially expressed circRNAs abundance.
Additional file 3.
Differentially expressed m6A circRNAs in the lungs of HPH rat model.
Additional file 4.
Differentially expressed circRNAs in the lungs of HPH rat model.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Su, H., Wang, G., Wu, L. et al. Transcriptome-wide map of m6A circRNAs identified in a rat model of hypoxia mediated pulmonary hypertension. BMC Genomics 21, 39 (2020). https://doi.org/10.1186/s12864-020-6462-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6462-y