
Abstract
Background
Flavonoids are produced in all flowering plants in a wide range of tissues including in berry fruits. These compounds are of considerable interest for their biological activities, health benefits and potential pharmacological applications. However, transcriptomic and genomic resources for wild and cultivated berry fruit species are often limited, despite their value in underpinning the in-depth study of metabolic pathways, fruit ripening as well as in the identification of genotypes rich in bioactive compounds.
Results
To access the genetic diversity of wild and cultivated berry fruit species that accumulate high levels of phenolic compounds in their fleshy berry(-like) fruits, we selected 13 species from Europe, South America and Asia representing eight genera, seven families and seven orders within three clades of the kingdom Plantae. RNA from either ripe fruits (ten species) or three ripening stages (two species) as well as leaf RNA (one species) were used to construct, assemble and analyse de novo transcriptomes. The transcriptome sequences are deposited in the BacHBerryGEN database (http://jicbio.nbi.ac.uk/berries) and were used, as a proof of concept, via its BLAST portal (http://jicbio.nbi.ac.uk/berries/blast.html) to identify candidate genes involved in the biosynthesis of phenylpropanoid compounds. Genes encoding regulatory proteins of the anthocyanin biosynthetic pathway (MYB and basic helix-loop-helix (bHLH) transcription factors and WD40 repeat proteins) were isolated using the transcriptomic resources of wild blackberry (Rubus genevieri) and cultivated red raspberry (Rubus idaeus cv. Prestige) and were shown to activate anthocyanin synthesis in Nicotiana benthamiana. Expression patterns of candidate flavonoid gene transcripts were also studied across three fruit developmental stages via the BacHBerryEXP gene expression browser (http://www.bachberryexp.com) in R. genevieri and R. idaeus cv. Prestige.
Conclusions
We report a transcriptome resource that includes data for a wide range of berry(-like) fruit species that has been developed for gene identification and functional analysis to assist in berry fruit improvement. These resources will enable investigations of metabolic processes in berries beyond the phenylpropanoid biosynthetic pathway analysed in this study. The RNA-seq data will be useful for studies of berry fruit development and to select wild plant species useful for plant breeding purposes.
Similar content being viewed by others

Background
Berry fruit species span numerous plant families placing considerable demands on the genomics resources required to study fruit development, gene expression and biosynthesis of bioactive compounds. Over the past few years, genome sequences of woodland strawberry (Fragaria vesca) [1], highbush blueberry (Vaccinium corymbosum) [2, 3], cranberry (Vaccinium macrocarpon) [4], grapevine varieties (Vitis vinifera) [5, 6], black raspberry (Rubus occidentalis) [7] and more recently, wild blackberry (Rubus ulmifolius) [8] have been released. Fruit transcriptomes of red raspberry (Rubus idaeus cv. Nova) [9], Korean black raspberry (Rubus coreanus) [10], blue honeysuckle (Lonicera caerulea) [11], highbush blueberry varieties [12,13,14,15], cranberry [16], grapevine varieties [17,18,19,20,21,22], cultivated blackberry (Rubus sp. var. Lochness) [23], woodland [24] and cultivated strawberry (F. × ananassa) [25] are also available. A wealth of transcriptome information for organs and tissues of berry fruit species has also been reported. Here, we aimed to bridge some of the gaps currently existing in berry fruit RNA-seq resources by generating and analyzing the fruit transcriptomes of 12 species as well as the leaf transcriptome of an additional species as part of the the BacHBerry (BACterial Hosts for production of Bioactive phenolics from bERRY fruits) collaborative project [26].
Plant-based products like fruits and berries are essential parts of the human diet and are considered healthy and nutritious foods (reviewed in [27]). Many berries and fruits are valued for their high content of bioactive compounds, including specialised metabolites of the phenylpropanoid pathway such as flavonoids (flavonols, flavones, isoflavones, anthocyanins and proanthocyanidins). Berries and fruits also contain other beneficial compounds such as carotenoids, vitamins, minerals and terpenoids. Beneficial health effects have been studied in several species that were sequenced here including wild blackberries (Rubus vagabundus), blueberries (V. corymbosum), honeysuckle (L. caerulea), Maqui berry (Aristotelia chilensis), strawberry myrtle (Ugni molinae), raspberries (R. idaeus) [28,29,30,31,32,33,34] and crowberry (Corema album) [35, 36]. Health benefits have often been attributed to phenolic compounds, which have been shown to possess anti-inflammatory, anti-mutagenic, anti-microbial, anti-carcinogenic, anti-obesity, anti-allergic, antioxidant as well as neuro- and cardioprotective properties (for a review see [37] and references therein). Polyphenols also exhibit valuable functions in plants such as protecting against UV radiation and high light stress, acting as signaling molecules and helping to attract pollinators by means of floral pigments.
The plant species chosen in this study had been shown to contain a diverse profile of phenolic compounds, especially anthocyanins: A. chilensis [38,39,40,41], Berberis buxifolia (Calafate) [42], C. album [43], L. caerulea [44, 45], Rubus genevieri (blackberry) [26], R. idaeus [46], Ribes nigrum (blackcurrant) [47], R. vagabundus [33], U. molinae [48], V. corymbosum [48] and Vaccinium uliginosum (Bog bilberry) [49]. Some of these berries, such as Calafate, Maqui berry and strawberry myrtle, are often referred to as ‘superfruits’ because of their exceptionally high antioxidant capacities. These species were investigated for new bioactive compounds and new bioactivities together with the identification of their polyphenolic compounds such as anthocyanins [26, 50, 51].
The synthesis of phenylpropanoids, specifically anthocyanins and other flavonoids, has been studied in many plant species such as Arabidopsis thaliana (thale cress), Antirrhinum majus (snapdragon), Malus x domestica (apple), Petunia x hybrida (petunia), Solanum lycopersicum (tomato), V. vinifera and Zea mays (maize) (reviewed in [52]), although, phenylpropanoid biosynthesis has been less well investigated in berry fruit species. Anthocyanins are water-soluble plant pigments responsible for the red, purple or blue colouring of many plant tissues, especially flowers and fruits. Genes required for the formation of flavonoids are predominantly controlled at the transcriptional level. Members of several protein superfamilies mediate the transcriptional regulation of the flavonoid biosynthetic pathway, namely the MYB transcription factors (TFs), basic helix-loop-helix (bHLH) TFs and conserved WD40 repeat (WDR) proteins [53].
The MYB TFs that regulate flavonol, anthocyanin and proanthocyanidin (PA) biosynthesis harbor a highly conserved N-terminal MYB domain consisting of two imperfect tandem repeats (R2 and R3, R2R3-MYB) that function in DNA binding and protein-protein interactions (reviewed in [54]). Some MYB TFs can interact with bHLH transcriptional regulators and WDR proteins to form a dynamic transcriptional activation complex (MBW complex) that regulates the transcription of genes involved in anthocyanin and PA biosynthesis [55]. R2R3-type MYB TFs such as AtMYB12 from A. thaliana act independently of a bHLH cofactor and control the expression of genes encoding enzymes operating early in the flavonol biosynthetic pathway. MYB TFs are often specific for the genes and pathway/pathway branches they target, such as the flavonol-specific activators of the R2R3 MYB subgroup (SG) 7 (e.g., AtMYB12 [56]) whereas others are confined to regulating anthocyanin (MYB SG6, A. majus AmROSEA1 [57]) or PA biosynthesis (MYB SG5, A. thaliana AtTT2 [58]). Many R2R3-type MYB TFs, for instance MdMYB10 from M. domestica [59], activate flavonoid synthesis whereas some others can repress anthocyanin formation (P. hybrida MYB27 [60]). In contrast, bHLH proteins may have multiple regulatory targets [61] and can control transcription of several branches of the flavonoid pathway as shown, for instance, by AtTT8 from A. thaliana [58] and Noemi from Citrus medica [62] in the regulation of both anthocyanin and PA biosynthesis.
Among the large class of bHLH TFs, bHLH transcriptional regulators related to flavonoid synthesis (SG IIIf [63],) consist of a MYB-interacting region (MIR) at their N-terminus, a neighboring WD40/acidic domain (AD) necessary for interaction with WDR proteins and/or RNA polymerase II and a bHLH domain that has been shown to be involved in DNA binding [53]. Both the bHLH domain and the C-terminus of these proteins can mediate homo- or heterodimerization of bHLH proteins. Similar to the C-terminal part of MYB proteins, the N-terminal part of bHLH proteins is more variable.
The third component of the MBW complex, participating in flavonoid/anthocyanin biosynthesis, is the WDR protein. These proteins are generally characterized by WD40 motifs of about 40–60 amino acids that typically end with a WD dipeptide (reviewed by [64, 65]). WDR proteins may assist the formation of stable protein complexes, serve as docking platforms/rigid scaffolds for protein-protein interactions and are thought to have no DNA-binding activity. Similar to the bHLH proteins in the MBW complex, WDR proteins that regulate the flavonoid pathway can also coordinate other regulatory networks, such as Arabidopsis AtTTG1 that controls trichome and root hair formation as well as seed coat development [66].
Recent advances in sequencing and computational technologies have greatly facilitated the study of non-model, wild and emerging new crop plants and can play key roles in understanding the biosynthetic pathways for novel bioactive compounds. The genetic resources and tools we have developed are available via the web-based transcriptome sequence database BacHBerryGEN [67] and its BLAST portal [68]. Gene expression studies during fruit ripening can be investigated using the newly developed BacHBerryEXP expression browser [69] in two Rubus species. As proof of concept, we cloned and conducted the functional analysis of Myb, bHLH and WDR genes involved in regulating anthocyanin biosynthesis in a wild and a cultivated Rubus species, using the transcriptomic tools generated in this study. We also investigated transcript expression patterns of genes involved in flavonoid biosynthesis at three fruit developmental stages in wild blackberry (R. genevieri) and cultivated red raspberry (R. idaeus cv. Prestige).
Results and discussion
Transcriptome sequencing and de novo assembly
We conducted de novo assemblies of one leaf and 16 fruit transcriptomes from 13 wild and cultivated berry fruit species. These species belong to eight plant genera and seven families: Berberidaceae (B. buxifolia), Caprifoliaceae (L. caerulea), Elaeocarpaceae (A. chilensis), Ericaceae (C. album, V. corymbosum, V. uliginosum), Grossulariaceae (two cultivars of R. nigrum), Rosaceae (three species including two cultivars of R. idaeus, R. genevieri, R. vagabundus) and Myrtaceae (U. molinae) that are dispersed over seven orders and three clades in the plant kingdom; Eudicots (three species), Eudicots-Asterids (four species) and Eudicots-Rosids (six species) (Table 1, Additional file 1: Table S1 and Additional file 2: Figure S1). Ploidy levels varied from diploid (R. idaeus and V. corymbosum) to tetraploid for B. buxifolia, V. uliginosum, R. genevieri. Fruits and leaves utilised for transcriptome analysis were collected by members of the BacHBerry Consortium [26] in Chile, China, Portugal, Russia and the UK (Additional file 1: Table S1). The species that were used for RNA-seq were either woody deciduous shrubs (Asterids: L. caerulea, Vaccinium spp., Eudicots: Ribes spp. and Rosids: Rubus spp.), evergreen shrubs (Eudicots: B. buxifolia and Rosids: U. molinae), an evergreen dioecious tree (Rosids: A. chilensis) and a shrub (Asterids: C. album). Several berries and fruits such as blueberries, blackcurrants and raspberries are widely cultivated; whereas the distribution of the other species is mostly restricted to their native habitats, for example, A. chilensis and U. molinae grow in their native terrains, Chile and Argentina, as well as in New Zealand and Australia; R. genevieri grows only in its natural habitat, Portugal; V. uliginosum grows in cool temperate regions of the Northern Hemisphere and C. album grows on the Atlantic coast of France and the Iberian Peninsula.
The majority of the berry fruit species that were used for RNA sequencing and analysis (Table 1) lacked an available reference genome sequence, therefore, de novo assembly of the Illumina reads was carried out for each species using Trinity software. Ten transcriptomes were assembled from RNA-seq data derived from a single cDNA library corresponding to ripe/mature fruits for gene identification purposes. Furthermore, six transcriptomes were assembled from RNA sequences taken at three different stages during fruit development and ripening (green/unripe, immature/intermediate ripe and mature/ripe fruit) of two Rubus species, using three cDNA libraries per stage to enable quantitative analysis of gene expression levels. To allow comparisons to vegetative tissues and due to a predicted high content of polyphenols in leaves, a leaf transcriptome was also prepared for a single species (C. album) for qualitative analysis. The transcriptome datasets are presented in Table 2 and complementary information is provided in Additional file 3: Table S2 and Additional file 4: Table S3. The online BacHBerryGEN repository database [67] and its BLAST portal [68] were developed to allow mining of the transcriptomic data of the 13 wild and cultivated berry fruit species.
Phylogenetic analysis and estimation of species divergence time
We analysed the phylogenetic relationship of the twelve berry fruit transcriptomes and one leaf transcriptome together with the genome sequences of seven reference species. This included (i) four species classified among the Angiosperms/Eudicots/Rosids (A. thaliana, Populus trichocarpa, Glycine max and V. vinifera), (ii) a berry species that belongs to Angiosperms/Eudicots/Asterids (S. lycopersicum), (iii) an evergreen shrub that branches out at the base of the flowering plants (Amborella trichopoda) and (iv) a monocotyledonous species (Angiosperms/Monocots/Commelinids: Oryza sativa). In these 20 species, 56,232 gene families were identified using gene family clustering, of which 5387 were shared by all species and 205 of these shared families were single-copy gene families. The single-copy gene orthologues of the 20 species underwent homology searches to produce a super alignment matrix for the assembly of a phylogenetic tree (Fig. 1). The branching order displayed in the tree reflected the expected phylogenetic group classification for the clades, orders and families of the Angiosperms with members of the Rosids clade (A. chilensis, R. genevieri, R. idaeus, R. vagabundus, U. molinae, A. thaliana, P. trichocarpa, G. max and V. vinifera) and the clade of the Asterids (C. album, L. caerulea, V. corymbosum, V. uliginosum and S. lycopersicum) clustering together with an estimated time of divergence between the two clades of about 125 million years (My). Among the Rosids, U. molinae and A. chilensis separated from the Brassicales (A. thaliana) about 112–117 My ago, whereas the different Rubus spp. diverged about 66 My ago from the Fabales (G. max). R. nigrum spp. (Saxifragales) diverged about 117 My ago from the Vitales (V. vinifera), an order that represents an outgroup amongst Rosids. Among the Asterids, the Ericales separated from L. caerulea and S. lycopersicum approximately 117 My ago, while Vaccinium spp. (Ericales) diverged about 59 My ago from C. album. B. buxifolia (Ranunculales) split approximately 151 My ago from the other Eudicot orders. The monocot O. sativa is grouped outside the dicotyledonous species and diverged approximately 165 My ago. A. trichopoda represents a basal group of the Angiosperms that diverged about 129 My ago from the flowering plants.

Phylogenetic analysis and estimation of species divergence time among 20 Angiosperm species. The twelve berry fruit transcriptomes and a berry leaf transcriptome were aligned together with the genome sequences of seven reference plant species (A. thaliana, A. trichopoda, G. max, O. sativa, P. trichocarpa, S. lycopersicum and V. vinifera) using single-copy gene orthologues (205). The estimated times of divergence are indicated at the tree nodes with the error values in parenthesis in million of years (My). The divergence time line is shown below the tree (in My)
Homology-based mining of candidate genes encoding enzymes involved in phenylpropanoid biosynthesis, particularly flavonoid biosynthesis
As a proof of concept, we used the transcriptome sequences developed in this study to identify candidate genes involved in phenylpropanoid biosynthesis, a pathway known to be very active in berry fruits. To identify transcripts encoding enzymes involved in the general phenylpropanoid pathway, its flavonoid branch as well as in the modification and decoration of its flavonoid products and to identify candidate regulatory genes, MassBlast [70] and the TBLASTN algorithm-based BacHBerryGEN BLAST server [68] were used with search parameters of ‘expect score cut-off’ of 1e-10, an open reading frame (ORF) length of a minimum of 100 amino acids (aa) and aa identity greater than 40% in the alignments.
Key plant enzymes involved in the general phenylpropanoid biosynthetic pathway and their corresponding sequences (60) including 23 experimentally validated genes from different plant species were used in a targeted search approach to mine the different transcriptomes for homologous transcripts encoding phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase (C4H), 4-coumarate CoA ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), flavonoid 3′,5′-hydroxylase (F3′5′H), flavonol synthase (FLS), dihydroflavonol 4-reductase (DFR), anthocyanidin synthase (ANS), anthocyanidin reductase (ANR), leucoanthocyanidin reductase (LAR), flavone synthase (FNS) and stilbene synthase (STS). These BLAST searches are detailed in Additional file 5: Table S4.
Published sequences from a total of 68 regulatory proteins (45 MYB TFs, 18 bHLH TFs and five WDRs) and 120 modifying and decorating enzymes (18 acyltransferases, 31 glucosyltransferases, 29 methyltransferases, 26 hydroxylases, nine reductases, two aurone synthases, two dehydrogenases, two dehydratases and one dirigent protein) from a range of plant species were also used in BLAST searches against the transcriptome sequences of the 13 species. Detailed BLAST search results are presented in Additional file 5: Table S4.
In total, 1248 sequences homologous to regulatory genes and 5150 sequences homologous to enzymes of the general phenylpropanoid pathway and its decoration and modification were identified from the different RNA-seq datasets (Table 3 and Additional file 5: Table S4). Multiple candidates encoding each type of decorating enzyme were identified in each transcriptome. Amongst putative modifying and decorating enzymes, 19 acyltransferases, 96 glucosyltransferases, 39 methyltransferases, 91 hydroxylases, 55 reductases, six aurone synthases, 16 dehydrogenases, 17 dehydratases and two dirigent protein candidate genes were identified on average per species. Generally, at least two to three homologues per decorating/modifying enzyme could be found in every species with glucosyltransferases and hydroxylases being the most abundant decorating enzymes. Different cultivars of R. idaeus (cv. Octavia and cv. Prestige) and R. nigrum (cv. Ben Hope and var. sibiricum cv. Biryusinka) exhibited similar patterns of homologue distribution amongst the transcripts encoding the different types of enzymes. R. genevieri, V. uliginosum, B. buxifolia and to a lesser extent L. caerulea and C. album exhibited a greater average number of homologues than the other species. This abundance of homologues is likely due to the higher ploidy levels of these accessions.
Comparison of BLAST search outputs of blackberry, blueberry, Maqui berry and strawberry myrtle also showed that transcripts encoding methyltransferases were the most conserved enzymes, with half to three-quarters of the sequences exhibiting high aa similarity levels, with the exception of blueberry (44.8% of genes). Reductases were also highly conserved between these species. In contrast, acyltransferases and glucosyltransferases were rarely detected with high levels of aa similarity. Approximately a third of the hydroxylases and glucosyltransferases were detected with high levels of aa similarity.
Amongst candidate regulatory genes controlling flavonol, anthocyanin or PA biosynthesis, on average, 85 Myb, five bHLH and four WDR candidate regulatory genes related to the phenylpropanoid pathway were detected per species.
In addition to the gene mining of the phenylpropanoid pathway, protein-coding sequences were predicted and functionally annotated in the transcriptomes of all the 13 species. The annotated ORFs for the transcriptomes of R. genevieri and R. idaeus cv. Prestige are shown in Additional file 6: Table S5.
Regulatory genes of the anthocyanin biosynthetic pathway isolated from R. genevieri and R. idaeus cv. Prestige
Using the transcriptomic data of R. genevieri (abbreviated as Rg) and R. idaeus cv. Prestige (abbreviated to Ri), several candidate regulatory genes of the anthocyanin biosynthetic pathway were identified in both species, cloned and characterised. The protein query sequences used for mining the fruit transcriptomic data were (1) M. domestica MdMYB10 as a representative member of the R2R3-type MYB gene subgroup 6 (SG6) family, responsible for the regulation of anthocyanin and PA biosynthesis [54, 71] which led to the isolation of RgMyb10 and RiMyb10; (2) A. thaliana AtMYB12 as a member of the R2R3-type MYB TFs of SG7 that control the activation of flavonol and flavone synthesis [54] which resulted in the isolation of RgMyb12 and RiMyb12; (3) bHLH TF homologues of P. hybrida ANTHOCYANIN1 (SG IIIf-1; PhAN1-type bHLHs) and A. majus DELILA (SG IIIf-2; AmDEL-type bHLHs) involved in the flavonoid/anthocyanin biosynthesis and epidermal cell fate [63] which generated the cloned RT-PCR products of RgAn1/RiAn1 and RgDel/RiDel respectively; as well as (4) M. domestica TRANSPARENT TESTA GLABRA 1 (MdTTG1) as a WD40 protein homologue which led to the cloning of RgTTG1 and RiTTG1 (Table 4, Additional file 7: Table S6 and Additional file 8: Table S7).
The cloned Myb genes were analysed for the presence of sequences encoding several known conserved aa motifs of R2R3-type MYB TFs (Additional file 9: Figure S2). The MYB domain consisting of the imperfect repeats R2 and R3 with regularly spaced tryptophan residues (R2 [−W-(x19)-W-(x19)-W-] … R3 [−F/I-(x18)-W-(x18)-W-] [54]) was highly conserved in the N-terminus of the four Rubus MYB TFs. Several regulators of the anthocyanin and PA pathways have been shown to contain an additional aa signature motif for bHLH interaction ([D/E]Lx2[R/K]x3Lx6Lx3R [61]) within the R3 repeat. The bHLH interaction motif and the anthocyanin-related SG6 MYB motif were present in the putative SG6 members RgMYB10 and RiMYB10 (Additional file 9: Figure S2) but were not present in the predicted SG7 homologues, RiMYB12 and RgMYB12. RgMYB10 and RiMYB10 also possessed domains present in other anthocyanin promoting MYB TFs such as the anthocyanin-related SG6 MYB motif of [R/K]Px[P/A/R]x2[F/Y] which lies downstream of the MYB domain as well as the small conserved ‘box A’ motif ([A/S/G]NDV) in the R3 repeat of the DNA binding domain [72]. In contrast, the SG7 homologues RiMYB12 and RgMYB12 contained a ‘box A’ motif ([D/E]N[E/D][I/V] [72]) characteristic of SG7 regulators in their R3 repeat. The conserved motif of flavonol synthesis-related SG7 R2R3-type MYBs (GRTxRSxMK [71] or [K/R][R/x][R/K]xGRT[S/x][R/G]x2[M/x]K [73]) was modified slightly in RiMYB12 (KxRx3GRTSRx2MK) and RgMYB12 (KRRx3GRNSRx2MK) (Additional file 9: Figure S2). This SG7 motif is also only partially conserved in the tomato SlMYB12 and grapevine VvMYB12 homologues [73]. The motif designated SG7-2 ([W/x][L/x]LS [73]) was fully conserved at the C-terminal ends of both RgMYB12 and RiMYB12 (Additional file 9: Figure S2). No motifs associated with MYBs that act as transcriptional repressors such as members of SG4 that contain an EAR (ethylene response factor-associated amphiphilic repression) motif (LxLxL or DLNxxP [74]) or the TLLLFR repression motif were found amongst the RgMYB and RiMYB SG6 TFs.
The Rubus Myb10 homologues (Table 4) were very similar with 92%/94% aa identity/similarity between RgMYB10 and RiMYB10. RiMYB10 was identical to a homologue characterized from another R. idaeus cultivar, cv. Latham (Accession no. EU155165) [72]. Another Myb10 homologue cloned from a Rubus hybrid cultivar (Accession no. JQ359611) has an aa identity/similarity of 89–91%/94% with Rg/RiMYB10. RuMYB1 from a cultivated blackberry (Rubus sp. var. Lochness) [75] shared 97% aa identity with RgMYB10 from wild blackberry and an aa identity/similarity of 93%/96% with the RiMYB10 from cultivated red raspberry. The Myb12 homologues of both Rubus species (Table 4) were also closely related (aa identity/similarity of 89%/91%). Phylogenetic analysis of the Rubus and several other R2R3-type MYB TFs showed clear separation of the flavonoid MYB regulators into two distinct clades (equivalent to SG6 and SG7 in A. thaliana [71]; Additional file 9: Figure S2).
Of the seven bHLH homologues cloned (Table 4), three encoded isoforms of RgAn1 (termed RgAn1-1, RgAn1-2, RgAn1-3 with 99% aa identity among the isoforms), RiAn1, RgDel and two isoforms of RiDel (named RiDel-1 and RiDel-2 that shared 99% identity at the aa level) had the general structure of flavonoid bHLH TFs (being about 600 aa in length, reviewed by [53]) (Additional file 10: Figure S3). The bHLH TFs each contained a N-terminal MYB-interacting region (MIR, aa 1 to approximately aa 200), a domain of interaction with WD40 and/or with the RNA polymerase II via the acidic domain (AD) (WD40/AD, extending from approximately aa 200 to aa 400) and a bHLH domain (approximately 60 aa, basic[~ 17 aa]-Helix 1[~ 16 aa]-Loop[~ 6–9 aa]-Helix 2[~ 15 aa]). The characteristic H-E-R aa motif (−H-(x3)-E-(x3)-R- [63]) within the basic part of the bHLH domain is preserved in all cloned bHLH TFs of the two Rubus species. The AmDEL homologues RgDEL and RiDEL-1/RiDEL-2 (SG IIIf-2) were closely related with a pairwise aa identity of 98% while the SG IIIf-1 PhAN1 homologues of R. genevieri (RgAN1-1 to RgAN1-3) and R. idaeus cv. Prestige (RiAN1) were slightly more diverged showing a 96–97% pairwise aa identity. Phylogenetic analysis showed clustering of the different Rubus bHLH TFs together with other plant bHLH homologues in two conserved clades of bHLH regulatory proteins (SGIIIf-1: PhAN1/AtTT8 clade and SGIIIf-2: AmDEL/PhJAF13 clade; Additional file 10: Figure S3).
When analysing WDR homologues, RgTTG1 (two isoforms named RgTTG1-1 and RgTTG1-2 that share a 99% identity at the aa level) and RiTTG1 were identified. These contained seven WD40 repeats (36–54 aa) as predicted using the WDSPdb database for WD40-repeat proteins [76, 77] (Additional file 11: Figure S4). Among these, four WD40 repeats corresponded to the domains previously identified in WDR proteins associated with anthocyanin biosynthesis [78]. The characteristic ‘WD’ dipeptide motif at the C-terminus of each WD40 repeat as well as the GH dipeptide delimiting the N terminus of several WD40 motifs were not fully conserved in many plant WDR homologues including those identified from Rubus (Additional file 11: Figure S4). Similarly, a D-H-[S/T]-W tetrad motif involved in the hydrogen bond network stabilising the propeller-like structure of certain WD40 proteins (reviewed by [79]) was conserved only partially between different WD40 proteins expressed in berry fruits. RiTTG1 was closely related to AtTTG1 (aa identity/similarity of 80%/88%) and MdTTG1 (aa identity/similarity of 92%/96%) whereas the two RgTTG1 isoforms were more distantly related (aa identity/similarity of 61%/78% with MdTTG1 and aa identity/similarity of 64%/79% with AtTTG1). The aa sequence of RiTTG1 was identical to that of another cultivar (R. idaeus cv. Moy TTG1, Accession no. HM579852). The phylogenetic analysis of RiTTG1 and RgTTG1 with other plant WDR homologues is shown in Additional file 11: Figure S4.
Candidate transcripts that are highly homologous to the Myb, bHLH and WDR regulatory genes cloned and functionally characterized in this study (Table 4) were identified in all the 13 berry fruit species and are listed in Additional file 12: Table S8.
Functional characterisation of regulatory genes of the anthocyanin biosynthetic pathway isolated from R. genevieri and R. idaeus cv. Prestige
To characterise the MYB, bHLH and WDR proteins functionally (Table 4), transient and stable expression studies were carried out in two accessions of N. benthamiana, a laboratory isolate (JIC-LAB) and an ecotype from the Australian Northern Territory (NT) [80]. Agroinfiltrations were performed with the candidate regulatory genes from Rubus on their own and in combinations with putative partners (Additional file 13: Figure S5). The anthocyanin biosynthetic pathway is generally not active in leaves of N. benthamiana, although colourless flavonols are produced. Inoculated on their own, Rubus Myb10, Myb12, bHLH and WDR genes (Fig. 2 and Additional file 13: Figure S5) did not induce red-purple pigmentation observable visually in agroinfiltrated leaf patches of N. benthamiana. The lack of anthocyanin production in the infiltrated N. benthamiana leaves infiltrated with these genes was confirmed by analysing the methanol: water: HCl (80:20:1, v/v/v) extracts of leaf discs from infiltrated areas (Additional file 13: Figure S5).

Production of anthocyanins in leaves of N. benthamiana cv. NT following transient overexpression of Rubus Myb and bHLH regulatory genes in the presence or absence of a WDR component (TTG1). a Transient overexpression of flavonoid regulatory genes in N. benthamiana leaves at 3 days post infiltration (dpi) in comparison to the empty vector (ev) construct. The methanol extracts from each infiltration combination are presented below the infiltrated leaf used for extraction (i.e., 1.8-cm diameter leaf disc in 2 ml methanol: water: HCl (80:20:1, v/v/v). Bar = 1 cm. b Methanol extracts from N. benthamiana leaves (1.8-cm diameter leaf disc in 2 ml methanol: water: HCl (80:20:1, v/v/v) transiently expressing Rubus flavonoid regulatory genes with or without a WDR co-factor from 1 to 7 dpi. Extracts represent average absorbance values at 530 nm from eight leaf discs per time point. Leaf expression is shown at 7 dpi. Bar = 0.5 cm
However, when combined with most of the cloned Rubus bHLH TFs, inoculation of the Rubus Myb10 genes induced a strong red-purple colouration in infiltrated leaf patches to a level easily detectable by the naked eye (Fig. 2 and Additional file 13: Figure S5). For example, the three RgAn1-type bHLH isoforms from Rubus gave rise to similar pigmentation intensities when co-infiltrated with RgMyb10. RgAN1-2 was often the most effective bHLH partner among the RgAN1 isoforms. In contrast, the AmDEL-type RgDEL TF did not induce visual anthocyanin production in N. benthamiana leaves in combination with RgMYB10 or RiMYB10 (Additional file 13: Figure S5) suggesting that RgDEL might not be functional in activating anthocyanin biosynthesis or might have another regulatory role. Mixes of RgMyb10 and RgDel supplemented with either RgMyb12 and/or RgTTG1 also did not lead to visual pigmentation in leaves nor in methanol extracts of leaves (Additional file 13: Figure S5). In contrast, RiDEL was able to interact with RiMYB10 and RgMYB10 to induce anthocyanin biosynthesis and appeared to be as effective as RiAN1 in this partnership (Additional file 13: Figure S5). These results suggested that the DEL proteins from different species of Rubus that share a 98% aa identity (11 aa variations) have differential abilities to induce anthocyanin biosynthesis. Among the aa differences, only a few occur in (highly) conserved regions of plant bHLH TF (Additional file 10: Figure S3). For example, RgDEL that is unable to initiate anthocyanin production with Ri/RgMYB10 in contrast to RiDEL, contains an arginine at position 150 compared to lysine within the MIR domain and, in the WD40/AD domain, differences at aa positions 247, 248 and 251 (Additional file 10: Figure S3). These differences could be responsible for the lack of anthocyanin synthesis in RgDel and Ri/RgMyb10 co-infiltrated leaves.
RiMYB10 interacted with both the PhAN1-like RiAN1 and the two AmDEL-like bHLH homologues, RiDEL-1 and RiDEL-2, with the two RiDEL proteins producing similar pigmentation levels in combination with RiMYB10 (Additional file 13: Figure S5). However, there were noticeable differences in the intensity of pigmentation accumulating over time in these assays; anthocyanin production induced by RiMYB10 co-expressed with RiAN1 was weak early after infiltration and peaked 5 days post infiltration (dpi). In contrast, anthocyanin accumulation of RiMYB10 plus RiDEL peaked at 4 dpi at which time the leaf tissue often started to deteriorate in the highly anthocyanin-enriched areas. Similarly, RgMYB10 produced strong red pigmentation earlier with RiDEL-2 (at 3–4 dpi) than when co-infiltrated with RgAn1. This suggested that RiMYB10 and RgMYB10 might interact preferentially with the different bHLH homologues in a time/phase-dependent manner or that bHLH TFs possess different binding affinities towards their MYB partner leading to differences in the rate of forming the MBW complex. Alternatively, these phylogenetically distinct bHLH TFs might operate via a hierarchical mechanism, as has been suggested in regulating anthocyanin biosynthesis [60, 81]. For example, an AmDEL-type bHLH homologue (SG IIIf-2) might activate the expression of a PhAN1-type bHLH homologue (SG IIIf-1) for subsequent MBW complex formation, and analysis in N. benthamiana has provided experimental evidence to support this model [60, 81].
It has been suggested that anthocyanin promoting MYB TFs display selectivity in their interactions with different bHLH partners [82]. In several anthocyanin regulatory systems, it has been shown that a MYB10-like TF alone can stimulate anthocyanin production in N. tabacum and/or N. benthamiana [83,84,85], although always to a lesser extent than when co-expressed with a bHLH TF partner. However, R2R3-type MYB TFs from Rosaceous species, including a RiMYB10 homologue [72], three peach Myb10 genes [86] as well as a strawberry MYB10 homologue [87] could trigger pigmentation in N. tabacum and/or N. benthamiana leaves only in combination with an added bHLH partner. Overall, the most parsimonious explanation seems to be that where MYB SG6 proteins can stimulate anthocyanin production on their own in transient assays in N. tabacum and/or N. benthamiana, they use endogenous bHLH TFs and WD40 proteins expressed in N. tabacum or N. benthamiana leaves as partners in the MBW complex(es). Those SG6 TFs that require an added bHLH for anthocyanin induction likely require specific interacting bHLH partners for pigment formation, either in a hierarchical regulatory cascade or directly in the MBW complex that activates the expression of the genes encoding the enzymes of anthocyanin biosynthesis. Anthocyanin regulatory systems might vary between plant families/orders as they do for monocot and dicot species (reviewed by [88]) and might also involve selective binding to regulatory elements in the promoters of their target genes [89].
Agroinfiltration of Rubus Myb12 TF genes, RgMyb12 or RiMyb12, together with Rg/RiMyb10 and a bHLH gene (Rg/RiAn1 or RiDel) generally enhanced anthocyanin production in leaves of N. benthamiana (Additional file 13: Figure S5), as seen in earlier studies with AtMYB12, AmRos1 and AmDEL in tomato [90].
HPLC analysis of methanol: water: HCl extracts (80:20:1, v/v/v) from leaves of the N. benthamiana JIC-LAB isolate infiltrated with different combinations of Rubus Myb and bHLH homologues showed that the main anthocyanin compound produced corresponded to delphinidin-3-rutinoside, with maximum absorption at 530 nm. Flavonoids and other phenolics detected at about 350 nm included the flavonol myricetin-3-O-rutinoside (MyrRut; generally found in extracts from Rubus MYB12 co-expressing samples), myricetin (glucose)2 rhamnose (Myr(Glc)2Rha), kaempferol-3-O-rutinoside (KaeRut), kaempferol (glucose)2 rhamnose (Kae(Glc)2Rha), rutin (quercetin-3-O-rutinoside) and chlorogenic acids (CGA1 and CGA2). Delphinidin-3-rutinoside was also found to be the major product synthesized in N. benthamiana leaf tissues transiently overexpressing SG6 MYB (AmROS1) and bHLH (AmDEL) TFs [85].
To investigate the role of WDR proteins from Rubus in the MBW complex, transient assays in N. benthamiana leaves were carried out with the putative components of the R. idaeus MBW complex, RiMYB10, RiAN1 and RiTTG1. To score the amount of anthocyanins accumulated in infiltrated leaf patches in the presence or absence of a WDR co-factor over time, methanol: water: HCl extracts of leaf samples were analysed by absorbance at 530 nm. Anthocyanin accumulation increased approximately 4.3-fold between 4 and 7 dpi in the presence of a WDR component compared to an approximately 1.8-fold increase without a WDR co-factor and therefore, the addition of the WDR co-factor RiTTG1 almost doubled the anthocyanin content in N. benthamiana (JIC-LAB isolate) leaves infiltrated with RiMYB10 and RiAN1.
To examine the effect of a WDR co-factor in the infiltration mixes from very early stages post inoculation, anthocyanin accumulation was observed over 7 days, from 1 to 7 dpi in the response to agroinfiltration with RiMyb10 and RiAn1 in the NT accession of N. benthamiana (Fig. 2). At 1 dpi, anthocyanin formation was not visible by the naked eye nor detectable in methanol extracts of infiltrated leaves (eight leaf discs per treatment) and equalled to the one of the mock infiltrated leaves of avgA530nm of 0.06 (+/− SE of 0.00) that remained largely unchanged (avgA530nm of 0.05 to 0.07 (+/− SE of 0.00 to 0.01) over the time course. At 2 dpi, the effect of the addition of a WDR protein was already evident, as anthocyanin formation could be observed by the naked eye in RiMyb10, RiAn1 and RiTTG1-co-infiltrated leaves and pigment formation was estimated in methanol extracts of leaf discs as avgA530nm of 0.20 (+/− SE of 0.02). In contrast, samples lacking a WDR protein had similar anthocyanin levels to the control treatment (avgA530nm of 0.07 (+/− SE of 0.00) at 2 dpi and required 3–4 dpi to reach similar pigmentation levels as those leaves infiltrated with the WDR co-factor had exhibited at 2 dpi. At 3 dpi, the MBW co-expression mixes showed up to 3–5-fold stronger anthocyanin production that plateaued around 3–4 dpi and accurate scoring of anthocyanin accumulation after 7 dpi became very hard. Visual observation of pigment formation also suggested that incorporating the RgTTG1 isoforms in RgMyb10 and RgAn1-2 co-infiltration mixes increased (early) anthocyanin production. Overall, transient overexpression of a WDR protein enhanced the early accumulation of anthocyanins, leading to faster synthesis of pigments in vegetative tissues of N. benthamiana that normally do not synthesize coloured flavonoids (Fig. 2). The rapid formation of an ectopic MBW complex, when endogenous WDR homologues might not be accessible or are present at limiting levels might, therefore, boost the early induction of anthocyanin biosynthesis. Our data confirm a report on the induction of anthocyanin production in N. tabacum transient assays by the Myrica rubra MBW complex which was earlier and enhanced by the WDR component [91].
The wild accession of N. benthamiana from the Australian Northern Territories (NT) has been suggested to be particularly well-suited for anthocyanin-related studies [80]. Generally, anthocyanin pigmentation using berry fruit genes was visible in the NT accession well before (at 2 dpi) pigmentation could be observed by the naked eye in the JIC-LAB isolate (3–4 dpi). The yield of anthocyanins produced in the infiltrated leaves, as predicted by A530nm absorbance values of methanol: water: HCl (80:20:1, v/v/v) extracts from leaf discs, was also far higher (minimally 2–3-fold higher) in the NT isolate than in the JIC-LAB accession. Use of the NT accession for infiltrations confirmed all our observations using the JIC-LAB strain of N. benthamiana.
Functional analysis of candidate regulatory proteins in stable transformations of N. benthamiana
Stable (co)-transformations of N. benthamiana leaf and stem explants with RiMyb10 or RgMyb10 under the control of the constitutive CaMV 35S promoter led to anthocyanin induction with and without bHLH and/or WDR co-factors from the same species. Different levels of red pigmentation (varying from light red to dark red/purple) were initially observed in callus sectors of explants grown on selection media (Additional file 14: Figure S6). Anthocyanin pigmentation continued also into later stages of regeneration in leaves and stems of developing shoots. High levels of pigmentation were often associated with a severe delay in shoot development, shoot stunting and deformation as well as the absence of root formation. In the past, tomato plants overexpressing the grapevine R2R3 MYBs VvMYB5a and VvMYB5b showed also phenotypic alterations including dwarfism [92]. Similar to the transient expression studies, anthocyanin production was greatly enhanced in stably co-transformed tissue with Rubus Myb10, bHLH with or without WDR genes while a limited amount of red-purple pigmentation could be detected in tissues transformed with Rg/RiMyb10 alone. Our data indicated that transient assays do not always reflect the metabolic changes observed in stable transformations, the latter being more sensitive indicators (at specific stages during regeneration) of the ability of regulatory genes to ectopically induce anthocyanin production. This could reflect some inherent suppression mechanism of anthocyanin biosynthesis in maturing leaves of N. benthamiana. N. benthamiana leaf and stem explants transformed with RgMyb12 or RiMyb12 alone, developed calli and shoots with a wild-type appearance as observed in AtMYB12 ectopic overexpression studies [56].
Differential gene expression during fruit development and ripening in R. genevieri and R. idaeus cv. Prestige and expression patterns of genes related to anthocyanin biosynthesis
To study the gene expression levels during fruit ripening in the wild blackberry R. genevieri and the cultivated red raspberry R. idaeus cv. Prestige, the BacHBerryEXP expression browser [69] was developed using the RNA-seq data analysis and a visualization platform expVIP (expression Visualization and Integration Platform) [93]. The BacHBerryEXP browser uses the transcriptome data from three stages of fruit maturation for the two Rubus species (green fruit, intermediate/immature fruit and ripe fruit; Fig. 3a) and calculates gene expression levels by using the pseudoalignment tool Kallisto [94]. It displays either the expression units as raw counts or transcripts per million (tpm) and their log2 values that can be represented as heatmaps. BacHBerryEXP also contains a BLAST tool [95] to identify candidate transcript homologues for differential expression analysis of the two Rubus species.

Transcriptome profiling of candidate genes encoding enzymes of the flavonoid core pathway, anthocyanin transporters, P-ATPases and flavonoid regulatory proteins during fruit maturation in R. genevieri (Rg) and R. idaeus cv. Prestige (Ri). a Gene expression was analysed in three developmental stages (unripe, immature and ripe fruits). b to d Candidate genes were identified via homology-based gene mining (BacHBerryGEN [68]) and expression patterns were visualized using the BacHBerryEXP expression browser (tpm and log2 values) [69]. b Candidate genes encoding enzymes of the phenylpropanoid core pathway and modifying proteins: Top panel - R. genevieri homologues: RgPAL-1 (TR124859|c0_g1_i1), RgPAL-2 (TR119394|c2_g1_i1), RgCHS (TR121228|c1_g2_i1), RgCHI-1 (TR87748|c0_g1_i2), RgCHI-2 (TR109085|c3_g1_i1), RgF3H-1 (TR65548|c1_g1_i2), RgF3H-2 (TR99162|c0_g1_i1), RgFNS (TR117515|c0_g1_i1), RgFLS-1 (TR82651|c1_g1_i1), RgFLS-2 (TR89606|c1_g1_i1), RgDFR (TR23878|c0_g1_i1), RgANS-1 (TR79533|c1_g1_i1), RgANS-2 (TR85881|c0_g1_i1), RgLAR-1 (TR97331|c0_g1_i1), RgLAR-2 (TR79474|c0_g3_i1), RgANR (TR77419|c0_g1_i1), RgUFGT (TR99106|c0_g1_i1); Lower panel - R. idaeus cv. Prestige homologues: RiPAL-1 (TR17637|c0_g1_i1), RiPAL-2 (TR60786|c0_g2_i1), RiCHS (TR38621|c0_g1_i3), RiCHI (TR60776|c0_g1_i1), RiF3H-1 (TR22747|c0_g2_i1), RiFNS-1 (TR17254|c0_g1_i1), RiFNS-2 (TR31274|c0_g1_i2), RiFLS (TR76353|c0_g1_i1), RiDFR-1 (TR26907|c0_g1_i1), RiDFR-2 (TR25484|c0_g1_i1), RiANS-1 (TR24906|c0_g1_i1), RiANS-2 (TR19248|c0_g2_i2), RiLAR-1 (TR24256|c0_g1_i1), RiLAR-2 (TR8288|c0_g1_i2), RiLAR-3 (TR58287|c1_g1_i1), RiANR (TR6460|c0_g1_i1), RiUFGT (TR3455|c0_g1_i1). c Candidate anthocyanin ABC and MATE transporters as well as P-ATPases. Top panel - R. genevieri homologues: RgABC-1 (TR71618|c2_g1_i1), RgABC-2 (TR72263|c2_g1_i5), RgABC-3 (TR114784|c2_g1_i2), RgABC-4 (TR73971|c3_g1_i3), RgMATE-1 (TR99523|c1_g1_i1), RgMATE-2 (TR81657|c3_g1_i1), RgMATE-3 (TR86341|c0_g1_i1), RgPH5-1 (TR72443|c2_g1_i1), RgPH5-2 (TR113411|c2_g1_i2), RgPH5-3 (TR72105|c0_g1_i1), RgPH1 (TR107023|c1_g2_i2); Lower panel - R. idaeus cv. Prestige homologues: RiABC-1 (TR41909|c1_g2_i1), RiABC-2 (TR66334|c1_g1_i2), RiABC-3 (TR27015|c1_g4_i3), RiMATE-1 (TR39949|c0_g1_i3), RiMATE-2 (TR11226|c0_g1_i1), RiMATE-3 (TR10226|c0_g1_i1), RiPH5-1 (TR570|c0_g1_i1), RiPH5-2 (TR41268|c0_g1_i1), RiPH1 (TR52475|c0_g1_i6). d Cloned and candidate Rubus regulatory proteins: Top panel - R. genevieri homologues: RgMYB10 (TR103098|c0_g1_i1), RgMYB12 (TR71550|c1_g1_i1), RgMYB6 (TR86812|c0_g1_i1), RgMYB5 (TR80732|c0_g11_i7), RgMYB2 (TR36560|c0_g1_i1), RgMYB1 (TR111295|c2_g2_i1), RgMYB4 (TR32557|c0_g1_i1), RgAN1 (TR110272|c1_g1_i1), RgDEL (TR110629|c1_g1_i1), RgTTG1 (TR29409|c0_g1_i1); Lower panel - R. idaeus cv. Prestige homologues: RiMYB10 (TR49283|c2_g2_i2), RiMYB12 (TR1036|c0_g1_i2), RiMYB6 (TR67691|c0_g1_i2), RiMYB5 (TR48317|c3_g1_i6), RiMYB2 (TR817|c0_g1_i2), RiMYB1 (TR75558|c0_g1_i1), RiMYB4 (TR16747|c0_g1_i1), RiAN1 (TR75681|c0_g1_i1), RiDEL (TR16024|c0_g1_i1) and RiTTG1 (TR7065|c0_g2_i1)
To illustrate the transcriptional expression levels of key structural and regulatory genes related to the general phenylpropanoid pathway and anthocyanin biosynthesis, we conducted a homology-based search of candidate genes using the BacHBerryGEN BLAST server [68] and a set of characterised plant protein homologues. We identified a range of candidate transcripts encoding proteins associated with phenylpropanoid metabolism as well as the modification, decoration and transport of its flavonoid products in the two Rubus species. For most candidate genes, up to five transcripts, with the highest homology scores (but not necessarily a full-length transcript), were selected and their expression profiles were analysed during fruit maturation using BacHBerryEXP [69] (Fig. 3 and Additional file 15: Table S9). For anthocyanin regulators such as the Rubus R2R3-type MYB, bHLH and WDR homologues cloned in this study, the expression levels of the cloned transcripts were assessed (Fig. 3 and Additional file 15: Table S9).
The formation of anthocyanin pigments in ripening fruits involves the coordinated expression of genes encoding a series of enzymes in the phenylpropanoid pathway. Heatmaps representing the transcriptomic profiling of key flavonoid pathway and anthocyanin biosynthetic enzymes (PAL, CHS, CHI, F3H, FLS, FNS, DFR, ANS, ANR and LAR), modifying enzymes (e.g., UFGT) and transcription (co-) factors (MYB, bHLH and WDR) obtained using BacHBerryEXP [69] is shown in Fig. 3 and transcript expression levels are presented in Additional file 15: Table S9. In some cases, gene transcript profiles exhibited a consensus trend, although for many genes, transcript-to-transcript profile variations were observed allowing us to group and select homologues according to their expression patterns. Expression of genes encoding enzymes involved in the phenylpropanoid pathway during fruit development has already been reported in Rubus sp. var. Lochness [75]. Transcription of genes encoding different isoforms of PAL either increased strongly from green to intermediate ripe fruits (RgPAL-1, RiPAL-1/- 2) or declined throughout fruit ripening stages (RgPAL-2) (Fig. 3b); presumably the isoforms induced during ripening are most closely associated with anthocyanin accumulation. Transcription of CHS homologues was mainly upregulated from green to the intermediate fruit stages (Fig. 3b). Transcripts encoding RgCHI-1 and RiCHI were upregulated until the intermediate ripening stage and then downregulated in ripe fruit, whereas RgCHI-2 was downregulated during fruit maturation (Fig. 3b) suggesting that it may not make a major contribution to anthocyanin biosynthesis. Coinciding with pigment accumulation in fruits, RgF3H-1 and RiF3H-1 were upregulated from green to immature fruits, with the highest levels at the ripe fruit stage (Fig. 3b) as previously observed for F3H in blackberry [75], while the transcript expression of RgF3H-2 and RiF3H-2 peaked at the intermediate ripe fruit stage and declined in ripe fruit (Fig. 3b and Additional file 15: Table S9). FLS homologues, RgFLS-1 and RiFLS, showed increased transcript levels from green to ripe fruits, whereas other FNS/FLS transcript levels, such as RgFNS and RiFNS-1, declined during fruit ripening confirming previous observations [75] or remained mainly unchanged (RiFNS-2 and RgFLS-2) during fruit ripening (Fig. 3b). The RiDFR-1 orthologue showed upregulation from green to intermediate ripe fruits, while transcript levels in the wild Rubus species (RgDFR) peaked at the immature ripening stage and declined in ripe fruits (Fig. 3b). Other DFR transcript isoforms (e.g., RiDFR-2, Fig. 3b) exhibited steady downregulation from green to ripe fruits. RiANS-1 and RgANS-1 encode candidate ANS isoforms involved in the synthesis of coloured anthocyanidins and were strongly expressed up to the immature fruit stage, whereas other ANS homologues (e.g., RgANS-2 and RiANS-2) were not induced during ripening (Fig. 3b).
Like other genes induced during ripening, transcripts encoding flavonol 3-O-glucosyltransferase (UFGT) in both Rubus species (Ri/RgUFGT, Fig. 3b) showed strong induction of transcript levels during ripening in line with the function of UFGT in stabilizing anthocyanidins by glucosylating them on the hydroxyl group of carbon 3, prior to transport to the vacuoles of the cells in coloured ripe fruits. Transcript levels for the PA biosynthesis gene LAR increased from green to the intermediate ripe fruits (RiLAR-1) or expression decreased steadily during fruit development (RgLAR-1, RgLAR-2, RiLAR-2 and RiLAR-3, Fig. 3b). For the PA-biosynthetic enzyme ANR, transcript levels decreased (RgANR) similarly to RuANR2 during fruit development [75] or expression levels remained similar during fruit development (RiANR) (Fig. 3b). Overall, it appeared that many flavonoid genes showed a coordinated expression pattern from the early stage to the production of anthocyanins in later stages of fruit development.
During fruit ripening, transport of anthocyanins and PAs is mediated mainly by two families of transporters, the ATP-binding cassette (ABC) transporter family and Multidrug and Toxic Compound Extrusion (MATE) transporters as well as glutathione S-transferases. Transcription of putative Rubus anthocyanin ABC transporters varied widely during fruit development (Fig. 3c and Additional file 15: Table S9). Candidates for anthocyanin and PA MATE homologues (e.g., RgMATE-1 and RiMATE-1) were found either to be downregulated in ripening fruits or transcript levels peaked in intermediate ripe fruits (e.g., RgMATE-2, RgMATE-3, RiMATE-2, RiMATE-3) (Fig. 3c). In blueberry, genes involved in vacuolar localization of PA exhibited developmental stage-specific expression patterns such as ANS, UFGT, LAR and ANR [2].
The transport of metabolites through endomembranes like the vacuolar tonoplast can be energized through proton pumps generated by P-ATPases such as PH5 whose gene expression is controlled directly by (pro)anthocyanin MBW complexes [96]. PH5 can act alone or in combination with another P-ATPase, PH1 that is absent in many plant species and boosts the activity of PH5 [97]. In R. genevieri and R. idaeus, some PH5 and PH1 homologues were downregulated (RgPH5-1, RiPH5-1), some upregulated (RgPH5-2) or their expression peaked at the immature fruit stage (RgPH5-3, RiPH5-2, RgPH1 and RiPH1) (Fig. 3c and Additional file 15: Table S9) suggesting roles in fruit hyperacidification like in pigmented grapevine where the expression of VvPH5 and VvPH1 peaked when berries changed colour at véraison [97].
Expression of genes encoding regulators of flavonoid biosynthesis during fruit development in Rubus species
Expression of the regulatory genes such as RiMyb10 and RgMyb10 was strongly upregulated during fruit ripening, especially from unripe to intermediate ripe fruits, whereas RiMyb12, RgMyb12, RiAn1 and RiDel were not differentially expressed at different stages of fruit ripening (Fig. 3d and Additional file 15: Table S9). A positive correlation between MYB10 transcript levels, anthocyanin synthesis and fruit colouration has also been reported in apple [59], blackberry [75], wild and cultivated strawberry as well as in sweet cherry [72]. RgAN1 was slightly upregulated from green to immature red fruits and moderately downregulated in ripe black fruits (Fig. 3d). In contrast, the expression of RubHLH1, a Ri/RgAN1 homologue, was consistently low in all stages of blackberry fruit ripening [75]. Interestingly, RgDEL expression was downregulated from green to ripe fruits (Fig. 3d), indicating that it is unlikely to be directly involved in anthocyanin formation in wild blackberry fruits but might activate the expression of RgAN1 as a part of the MBW complex involved in anthocyanin biosynthesis [60, 81] as also suggested by our expression assays where transient co-expression of RgDEL with RiMYB10 or RgMYB10 did not lead to leaf pigmentation in N. benthamiana while RiDEL and Ri/RgMYB10 did (Additional file 13: Figure S5). The transcript levels of Ri/RgTTG1 did not change during fruit development (Fig. 3d), whereas [75] reported that RuTTG1 showed slightly higher expression in green blackberries compared to later fruit ripening stages. Overall, Rg/RiDEL (tpm ≤ 1–2) and Rg/RiMYB12 (tpm ≤ 1) were both expressed at very low levels in fruit, whereas Ri/RgAN1 (up to tpm = 30–40), Rg/RiTTG1 (up to tpm = 10–20) and Rg/RiMYB10 (up to tpm = 35–120) were expressed at substantially higher levels in ripening fruits (Additional file 15: Table S9). This suggests that the highly abundant PhAN1-type bHLH TFs (RgAN1/RiAN1) are the dominant players and partners of the MYB10 TFs regulating anthocyanin production and fruit colouration during berry fruit ripening. The ability of RiDEL to activate anthocyanin biosynthesis in combination with Rg/RiMYB10 compared to the inability of RgDEL from the same genus to activate anthocyanin biosynthesis is noteworthy and suggests that the bHLH partners in the MBW complex may play slightly different roles even between different species in the same genus.
The expression of RgMYB6 (Fig. 3d), the closest homologue of the activators of flavonol biosynthesis in G. max (GmMYB12B2) and blackberry (RuMYB6), increased in ripe fruits like RuMYB6 [75]. On the other hand, RiMYB6 decreased with fruit ripening (Fig. 3d). For the RuMYB5 homologues, RiMYB5 and RgMYB5, expression peaked at the intermediate ripe fruit stage (Fig. 3d) which may relate to increases in PA synthesis in developing fruits in both species. This was confirmed by the identification of the PAs catechin and epicatechin in intermediate to ripe fruits (approximately 1/10 of the anthocyanin content) [51]. Fluctuations in MYB5 homologue expression have been reported previously. RuMYB5 from cultivated blackberry, has been predicted to interact with RuTTG1 and RubHLH1 in PA synthesis, and showed decreasing transcript levels during ripening [75]. In strawberry, FaMYB5 transcripts accumulate steadily during fruit development. It has been suggested that FaMYB5 may play a role in fine-tuning both PA biosynthesis during early fruit development and anthocyanin biosynthesis during fruit ripening [98]. Variations in expression patterns could also relate to the fact that another MYB5 homologue, AtMYB5 has been considered to be a general flavonoid pathway activator [73].
Homologues of RuMYB2, a putative PA synthesis activator by analogy to AtTT2, were strongly downregulated (RgMYB2) similarly to RuMYB2 or almost exclusively expressed in green fruits (RiMYB2) (Fig. 3d).
Transcript levels of RgMyb1 and RiMyb1, encoding homologues of FaMYB1 (a transcriptional repressor of anthocyanin/PA biosynthesis in strawberry) were upregulated from green to intermediate ripe fruits and were transcribed abundantly in R. genevieri or at low level in R. idaeus (Fig. 3d). The AtMYB4 phenylpropanoid repressor homologues in both Rubus species (RgMYB4 and RiMYB4, Fig. 3d) were downregulated during ripening as was RuMYB4 in blackberry [75].
Conclusions
We report transcriptome sequences and analytical tools for gene identification, cloning and functional analysis from 13 berry fruit species coming from Europe, South America and Asia, spanning eight plant genera and seven families. Tools and resources are accessible and searchable online via the BacHBerryGEN database [67, 68] and the BacHBerryEXP gene expression browser [69]. These resources will assist gene expression and functional genomic studies in berry fruit species as well as contributing to the understanding of the synthesis of polyphenols, the molecular mechanisms underlying phenylpropanoid, and particularly flavonoid, synthesis and the regulatory processes controlling phenylpropanoid metabolism during fruit ripening. These tools have already contributed to identifying the genes involved in the synthesis of novel biologically active compounds in berry fruits [26]. Ultimately, studies of metabolic pathways should facilitate breeding programmes for fleshy fruits by providing markers for shortening the long process of breeding and by identifying valuable and better varieties, resulting in benefits to both consumers and farmers.
The usefulness of these transcriptomic resources has been demonstrated by the cloning and characterisation of regulators of the anthocyanin pathway from these berry fruit species, namely R2R3-type MYBs, bHLH and WDR homologues, which regulate anthocyanin and PA biosynthesis in two Rubus species. Functional validation of Rubus homologues of MdMYB10, AtMYB12, PhAN1/AmDEL and AtTTG1/MdTTG1 was undertaken in N. benthamiana leaves. The regulators Rg/RiMYB10, Rg/RiAN1, RiDEL and Ri/RgTTG1, are likely to be part of red raspberry (R. idaeus) and wild blackberry (R. genevieri) MBW complexes, respectively regulating the expression of flavonoid genes late in anthocyanin biosynthesis. However, the DEL homologue from wild blackberry was unable to induce pigment formation with Ri/RgMYB10 perhaps as a result of the few aa differences found between RgDEL and RiDEL.
There is a growing interest in the exploitation of wild fruits and berries as part of the rising demand for novel health promoting foods. In berries, high antioxidant activity is most often associated with berries from wild species, together with a broad variety of anthocyanins and high total polyphenol contents compared to cultivated varieties. In commercial cultivars, the flavonoid content has often been altered and reduced during domestication with an accompanying increase in susceptibility towards pests/insects [99]. In this study, we investigated a wide range of wild and cultivated berry fruit species representing diverse plant families (Berberidaceae, Caprifoliaceae, Elaeocarpaceae, Ericaceae, Grossulariaceae, Rosaceae and Myrtaceae) to provide a broad platform for classification of genes and their products and to establish fruit-specific expression patterns to gain new insights into the complex regulation of metabolic pathways during berry fruit development.
The demand for varied, nutritious and healthy food has been growing in both the developed and developing world. Diets that include berries and fruits are rich in polyphenols including monolignols, flavonoids (anthocyanins, PAs, flavonols, flavones, flavanones, isoflavonoids and phlobaphenes), various phenolic acids and stilbenes. All these polyphenols have been shown to play important roles in plant growth and development, biotic and abiotic defence mechanisms as well as conferring benefits for human health.
Methods
Plant materials and isolation of total RNA
Plant tissues from 13 berry fruit species were collected by partners of the BacHBerry Consortium [26] in Chile, China, United Kingdom, Portugal and Russia (Table 1 and Additional file 1: Table S1). Plants were grown either in their natural habitat or under cultivated conditions (Additional file 1: Table S1). Fruits were harvested at different developmental ripening stages (unripe, intermediate and/or ripe fruits) and leaf material was collected from fully developed leaves between January to August 2014 and July to August 2015 (Additional file 1: Table S1).
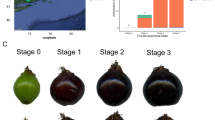
RNA was extracted from 13 berry fruit species using (i) ripe fruits of ten species, i.e., A. chilensis, B. buxifolia, L. caerulea, R. nigrum cv. Ben Hope, R. nigrum var. sibiricum cv. Biryusinka (also described as R. nigrum subsp. sibiricum cv. Biryusinka), R. idaeus cv. Octavia, R. vagabundus, U. molinae, V. corymbosum and V. uliginosum; (ii) unripe (green), intermediate (pale red) and ripe (dark red) fruits of R. idaeus cv. Prestige; (iii) unripe (green), intermediate (red) and ripe (black) fruits of R. genevieri and (iv) leaf material of C. album (Table 1 and Additional file 1: Table S1). Plant material was frozen in liquid nitrogen immediately after harvest, stored at -80 °C and transported in dry ice prior to RNA extraction. Leaves as well as deseeded whole berries and fruits were ground to a fine powder with liquid nitrogen. Total RNA of A. chilensis, B. buxifolia, R. idaeus cv. Octavia, R. vagabundus, V. corymbosum, V. uliginosum and U. molinae was isolated from 200 mg of frozen fruit tissue based on a protocol for plant tissues rich in polyphenols and polysaccharides [100] and included an additional DNase I treatment (RQ1 RNase-Free DNase, Promega) before phenol: chloroform extraction within step III of the protocol. For C. album, L. caerulea, R. nigrum cv. Ben Hope, R. nigrum var. sibiricum cv. Biryusinka, R. genevieri and R. idaeus cv. Prestige, total RNA was extracted from 200 mg of frozen fruit tissue and leaves, respectively by using the Spectrum Plant Total RNA kit (Sigma) following the manufacturer’s guidelines and protocol A (using 750 μl binding solution). The optional step of on-column DNase digestion was performed and total RNA was eluted with 60 μl elution buffer. For each sample, the total RNA of 10–15 fruits was pooled. The RNA concentration and quality were evaluated via spectrophotometric analysis and the RNA integrity was also analysed by gel electrophoresis. Overall, 17 total RNA samples were produced for the 13 species: ten species (ripe fruit), two species (three fruit ripening stages) and one species (leaf).
Synthesis of cDNA and RNA sequencing
cDNA library preparation and sequencing were carried out by the Earlham Institute, formerly The Genome Analysis Centre (TGAC) Norwich, UK. The libraries were constructed on the Sciclone NGS Workstation (PerkinElmer) following the Illumina TruSeq RNA sample preparation v2 guide and using the TruSeq RNA Library Preparation Kit v2 (Illumina). The library preparation involved several quality control analysis steps, including the use of the Quant-iT™ RNA Assay Kit (Life Technologies) for RNA quantification, the Quant-iT™ dsDNA Assay Kit (Life Technologies) for double-stranded DNA quantification as well as the LabChip GX Automated Electrophoresis System (PerkinElmer) and High Sensitivity DNA kit (Agilent) for RNA/dsDNA quantification and verification of the cDNA library insert size. Shortly, the RNA-seq workflow included (1) purification and fragmentation of mRNA from 1 μg of total RNA with a poly(A)-pull down using oligo-dT attached magnetic beads; (2) first strand cDNA synthesis with random hexamer primers and SuperScript II reverse transcriptase (Invitrogen); (3) second strand cDNA synthesis using DNA polymerase I and RNase H; (4) cDNA end repair/blunting; (5) cDNA fragment 3′ end adenylation; (6) ligation of multiple indexing adapters to cDNA fragments and purification of ligated products via bead-based size selection using AMPure XP beads (Beckman Coulter); (7) PCR enrichment of adapter-ligated cDNA fragments with a PCR primer cocktail that anneals to the adapter ends; (8) quantitative and qualitative validation of cDNA library; (9) normalisation and equimolar pooling of indexed DNA libraries; (10) dilution of library pool to a final concentration of 10 pM and spiking of each library pool with 1% PhiX Control v3 (Illumina); (11) flow cell clustering using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina); and (12) sequencing of flow cell using the Illumina HiSeq™ 2000 platform with the TruSeq SBS Kit v3-HS (Illumina) and HiSeq Control Software 2.2.58 and RTA 1.18.64. Reads in bcl format were de-multiplexed based on the 6 bp Illumina index by the CASAVA 1.8 package allowing for a one base-pair mismatch per library and converted to FASTQ format by bcl2fastq.
De novo assembly of transcriptomes
Illumina data from total RNA samples of the 13 berry fruit species were assembled by the Earlham Institute (Norwich, UK) into individual de novo transcriptomes using Trinity [101] and these assemblies were then used as a reference for mapping, quantification of expression and functional annotation. The alignment of RNA-seq reads to a transcriptome reference was performed using TopHat2 [102] with a minimum anchor length of 12 and a maximum of 20 multihits. Adapter/primer sequences were clipped, and low-quality reads were removed. Quality control of the raw data was performed using FastQC [103] and the contamination screening and filtering tool Kontaminant [104]. Gene/isoform expression was quantified using Cufflinks [105]. Transdecoder [106] was used to extract ORFs from the de novo transcriptome assemblies. Peptides of these ORFs were annotated via an in-house pipeline (AnnotF [107]) that compares the results of Blast2GO and InterProSCAN (Additional file 6: Table S5). Transcriptome sequences of R. genevieri and R. idaeus cv. Prestige fruits at three different stages of maturation (with three biological replicates per ripening stage) were either kept separate to undertake subsequent differential expression studies or pooled together to generate a consensus fruit sequence.
Mining of fruit transcriptomes, BLAST searches, expression profiling and phylogenetic analysis
The BacHBerryGEN database [67] was created to deposit the transcriptomic data of the 13 berry fruit species. A BLAST search engine [68] was developed to conduct homology-based searches of candidate genes. The workflow MassBlast [70, 108] was also used to identify homologues and orthologues of enzymes responsible for the synthesis, decoration and regulation of phenylpropanoid compounds.
The BacHBerryEXP expression browser [69] was established based on [93] to facilitate the differential expression analysis of candidate genes of two Rubus species, R. genevieri and R. idaeus cv. Prestige, during three developmental stages of fruit ripening (green/immature/ripe). Six transcriptome sequences of R. genevieri and R. idaeus cv. Prestige (two species x three fruit ripening stages) were uploaded in the BacHBerryEXP expression browser [69] and used to analyse differential transcription of genes involved in the anthocyanin biosynthetic pathway. The transcript identification ID can be retrieved either in the BacHBerryGEN database [68] or by using the built-in BLAST search module of the BacHBerryEXP expression browser [69].
The phylogenetic analysis of the transcriptomes of the 13 berry fruit species together with the genome sequences of seven reference species (A. thaliana [109], P. trichocarpa [110], G. max [111], V. vinifera [5], S. lycopersicum [112], O. sativa [113] and A. trichopoda [114]) was carried out following the comparative genomic analysis detailed in [115]. EvidentialGene [116] was used to translate and validate the ORFs. The longest translated ORFs from each gene with at least 100 aa were aligned with BLAST 2.26 [117] against each other and orthologue groups were identified using OrthoMCL 2.0.9 [118]. A multiple alignment for each of the 214 single-copy gene families was produced using MUSCLE v3.8.1551 [119]. The longest contiguous block of each aa sequence, defined as more than 20 aa with less than five contiguous positions with a gap in any sequence, were concatenated to produce a super alignment matrix containing 205 gene families (with nine gene families filtered out due to the lack of a single contiguous block) using a BioRuby script [120] (Additional file 16). ProtTest 3.4.2 [121] was used to find the best fitting model to produce the phylogenetic tree which was JTT + I + G + F. The phylogenetic tree was assembled using RAxML 8.2.12 [122] with the option PROTGAMMAJTT and declaring O. sativa and A. trichopoda as outgroups. The divergence time among the 20 species was estimated using MCMCtree 4.8a from the PAML software [123] (Additional file 16). The species divergence time was predicted using the calibration points of (i) A. thaliana and P. trichocarpa (107–109 million years (My) ago), (ii) A. thaliana and G. max (107–109 My ago), (iii) S. lycopersicum and P. trichocarpa (107–125 My ago), (iv) O. sativa and A. thaliana (140–200 My ago) and (v) V. vinifera and A. thaliana (113–114 My ago) [124]. The Additional file 16 details the programme execution.
Identification and cloning of regulatory genes from R. genevieri and R. idaeus cv. Prestige
Orthologues of (i) R2R3-type MYB transcription factors (TFs): MdMYB10 (M. domestica cv. Maypole MYB10, Accession no. AB744002.1) and AtMYB12 (A. thaliana MYB domain protein 12, Accession no. NM_130314.4) TFs; (ii) bHLH TFs: PhAN1 (P. hybrida ANTHOCYANIN 1, Accession no. AF260919) and AmDEL (A. majus DELILA, Accession no. M84913.1) TFs and (iii) a WD40-repeat gene: MdTTG1 (M. domestica TRANSPARENT TESTA GLABRA1; Accession no. GU173814.1) were identified in R. genevieri and R. idaeus cv. Prestige by mining the fruit transcriptomic data deposited in the BacHBerryGEN database [67] using the TBLASTN programme of the BacHBerryGEN BLAST server [68] and a protein sequence query (Additional file 7: Table S6).
Total RNA (1 μg) extracted from ripe fruits of both Rubus species was used for first strand cDNA synthesis with oligo (dT)18 primers (Sigma) using SuperScript III reverse transcriptase (Invitrogen) and RNaseOUT (Recombinant Ribonuclease Inhibitor, Invitrogen) following the manufacturer’s instructions. First strand cDNAs were amplified using primers specific to the 5′ and 3′ ends of each gene (Additional file 8: Table S7) and PfuUltra® II Fusion HS DNA Polymerase (Agilent Genomics). The PCR amplification was carried out with an initial denaturation of 2 min at 94 °C followed by 40 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min (for MdMYB10 homologues), for 1.5 min (for AtMYB12 and MdTTG1 homologues) or for 2 min (for bHLH homologues) and a final elongation of 3 min at 72 °C.
To facilitate the cloning of the various TF RT-PCR products, the CaMV 35S promoter (pro) and soybean poly(A) (SPA) terminator (ter) sequences [125] were cloned into three basic vectors (i) pGreenII0029 (containing a NOS-pro::nptII::NOS-ter plant selectable marker gene) [126], (ii) pGreenII00179 (harbouring a CaMV35S-pro::hpt::CaMV35S-ter plant selectable marker gene) [126] and (iii) pGreenII00229 (possessing a NOS-pro::bar::NOS-ter plant selectable marker gene) [126]. The RT-PCR fragments of the Rubus regulatory genes were inserted between the CaMV 35S-pro and SPA-ter sequences as blunt-end fragments or BamHI/XbaI-PstI/SmaI/XhoI/NsiI-digested fragments (Additional file 7: Table S6 and Additional file 8: Table S7). The TF RT-PCR fragments and vector combinations are indicated in Additional file 7: Table S6.
Agrobacterium-mediated transient expression and stable transformation of regulatory genes in N. benthamiana
The different pGreen-based vectors were introduced together with pSoup [126] or a pSoup derivative containing the viral suppressor of gene silencing P38 from Turnip Crinkle Virus (pBOOST-S; CaMV35S-pro::P38::SPA-ter cassette in vector pCLEAN-S161 [125]) into the Agrobacterium tumefaciens strain AGL1 via a freeze-thaw method [127]. The presence of the different regulatory genes in the Agrobacterium strains was confirmed by PCR amplification of the full-length genes from Agrobacterium plasmid preps and sequencing of the PCR products. Transient assays in N. benthamiana were carried out as described in [125] in two accessions of N. benthamiana, in the Australian ecotype Northern Territory (NT; seeds kindly provided by Prof. Peter Waterhouse, Queensland University of Technology, Brisbane, Australia) [80] and the John Innes Centre laboratory isolate (JIC-LAB; predicted to be of the same origin as the LAB isolate of [80]). For co-infiltration assays using Agrobacterium strains harbouring Myb, bHLH or WDR homologues of R. genevieri and R. idaeus cv. Prestige, Agrobacterium strains (OD600 = 1.0) were mixed equally. A so-called empty vector strain without a gene of interest (pGreenII00179 + pBOOST-S in AGL1) was used as a negative control or as a component of the co-infiltration mixes to complement for co-factors (e.g., WDR). Four- to five-week old N. benthamiana plants were used for patch infiltration to monitor the production of polyphenolic compounds (mainly anthocyanins and flavonols). The abaxial side of three to four leaves per plant were infiltrated, the leaves were observed from 1 to 14 days post infiltration (dpi) and samples were harvested from 1 to 7 dpi.
Regulatory genes were stably transformed into N. benthamiana (JIC-LAB isolate) using either leaf or stem explants following the Agrobacterium-mediated transformation protocol of Moricandia arvensis [128]. Single selection based on kanamycin (100 mg/l) was used for the transformation with Myb genes. Dual selection based on kanamycin (30 mg/l) and hygromycin (10 mg/l) was applied for the co-transformation with Myb and bHLH genes. Triple selection based on kanamycin (30 mg/l), hygromycin (10 mg/l) and DL-phosphinothricin (PPT, 3 mg/l) was used for the co-transformation with Myb, bHLH and WDR genes.
Detection and analysis of polyphenolic compounds
Leaf discs were cut from patch-infiltrated N. benthamiana (JIC-LAB isolate and NT accession) leaves using a standard cork borer (diameter of 1.8 cm). The top two to three infiltrated leaves were sampled avoiding main veins. In time course experiments eight leaf discs were collected from two different plants (i.e., four to six infiltrated leaves from two plants) from 1 dpi to up to 7 dpi per time point. Each infiltration mix was tested several times in both N. benthamiana isolates. The leaf discs with a fresh weight of 36.89 ± 0.92 mg (JIC-LAB isolate) or 51.29 ± 1.47 mg (NT) were cut into quarters and immediately merged into 2 ml extraction solution (methanol: water: HCl, 80:20:1, v/v/v), quickly vortexed and incubated with gentle shaking overnight (16 h) at 4 °C in the dark. Following 3 h of moderate rocking at room temperature, extracts were centrifuged at 13,000 rpm for 15 min and the supernatants were analysed using a spectrophotometer at A530nm for their anthocyanin content. The absorption at 530 nm was averaged for the eight leaf discs (avgA530) and the standard error was calculated for each treatment and time point.
Leaf disc samples from Agrobacterium-infiltrated leaf patches of N. benthamiana (JIC-LAB isolate) were taken 5 dpi and analysed by HPLC/photodiode array detector (PDA) and mass spectrometry (MS) in comparison to chlorogenic acid and rutin standards (serial dilutions in 20% methanol) using the Shimadzu IT-ToF and a Kinete× 2.6 μm EVOC18, 100 Å pore size LC column (100-× 2.1-mm) according to [90]. Polyphenolic compounds (mainly anthocyanins and flavonols) were identified based on their mass and mass of their fragments, respectively.
Availability of data and materials
The transcriptome sequences generated during this current study are available from the BacHBerryGEN repository (http://jicbio.nbi.ac.uk/berries). BLAST and expression analyses can be performed using the BacHBerryGEN database (http://jicbio.nbi.ac.uk/berries/blast.html) and BacHBerryEXP gene expression browser (http://www.bachberryexp.com), respectively. Nucleotide sequences of Rubus flavonoid regulatory genes cloned in this study are deposited in GenBank. Additional data generated and/or analysed during this study are included either in this published article as supplementary information or can be requested from the authors (e.g., transcriptome ORF/peptide annotations and Trinotate annotations of secondary metabolite biosynthesis /phenylpropanoid pathway for each transcriptome). Metabolomics datasets of the 13 berry fruit species studied in this manuscript are available at https://ics.hutton.ac.uk/germinate-berrybase/# (Dr. Alexandre Foito and Prof. Derek Stewart; The James Hutton Institute, Invergowrie, UK). Berry fruit material can be requested from the institutions listed in Table 1 and Additional file 1: Table S1.
Abbreviations
- 4CL:
-
4-coumarate CoA ligase
- aa:
-
amino acid
- ABC transporter:
-
ATP-binding cassette transporter
- Am:
-
Antirrhinum majus
- ANR:
-
Anthocyanidin reductase
- ANS:
-
Anthocyanidin synthase
- At:
-
Arabidopsis thaliana
- avg:
-
average
- bar :
-
phosphinothricin acetyl transferase
- bHLH:
-
basic helix-loop-helix protein
- C4H:
-
Cinnamate 4-hydroxylase
- CaMV35S:
-
Cauliflower Mosaic Virus 35S
- CHI:
-
Chalcone isomerase
- CHS:
-
Chalcone synthase
- DFR:
-
Dihydroflavonol 4-reductase
- dpi:
-
days post infiltration
- F3’5’H:
-
Flavonoid 3’,5’-hydroxylase
- F3’H:
-
Flavonoid 3’-hydroxylase
- F3H:
-
Flavanone 3-hydroxylase
- Fa:
-
Fragaria × ananassa
- FLS:
-
Flavonol synthase
- FNS:
-
Flavone synthase
- Gm:
-
Glycine max
- hpt :
-
hygromycin phosphotransferase
- LAR:
-
Leucoanthocyanidin reductase
- MATE transporter:
-
Multidrug and Toxic Compound Extrusion transporter
- MBW:
-
MYB, bHLH and WDR protein complex
- Md:
-
Malus x domestica
- My:
-
Million years
- MYB:
-
MYB-domain containing protein
- NOS:
-
Nopaline synthase
- nptII :
-
neomycin phosphotransferase II
- nt:
-
nucleotide
- ORF:
-
Open reading frame
- PA:
-
Proanthocyanidin
- PAL:
-
Phenylalanine ammonia lyase
- Ph:
-
Petunia x hybrida
- Rg:
-
Rubus genevieri
- Ri:
-
Rubus idaeus cv. Prestige
- Ru:
-
Rubus sp. var. Lochness
- SG:
-
subgroup
- Sl:
-
Solanum lycopersicum
- SPA:
-
Soybean poly(A) signal
- STS:
-
Stilbene synthase
- TF:
-
Transcription factor
- tpm:
-
transcripts per million
- UFGT:
-
UDP glucose: flavonol 3-O-glucosyltransferase
- Vv:
-
Vitis vinifera
- WDR:
-
WD40 motif containing protein
References
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2011;43:109–16.
Colle M, Leisner CP, Wai CM, Ou S, Bird KA, Wang J, Wisecaver JH, Yocca AE, Alger EI, Tang H, Xiong Z, Callow P, Ben-Zvi G, Brodt A, Baruch K, Swale T, Shiue L, Song GQ, Childs KL, Schilmiller A, Vorsa N, Buell CR, VanBuren R, Jiang N, Edge PP. Haplotype-phased genome and evolution of phytonutrient pathways of tetraploid blueberry. GigaScience. 2019;8:1–15.
Gupta V, Estrada AD, Blakley I, Reid R, Patel K, Meyer MD, Andersen SU, Brown AF, Lila MA, Loraine AE. RNA-Seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening, biosynthesis of bioactive compounds, and stage-specific alternative splicing. GigaScience. 2015;4(5):1–22.
Polashock J, Zelzion E, Fajardo D, Zalapa J, Georgi L, Bhattacharya D, Vorsa N. The American cranberry: first insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014;14(1):165.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quetier F, Wincker P. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–5.
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, LM FG, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Demattè L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Van de Peer Y, Salamini F, Viola R. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE. 2007;2:e1326.
VanBuren R, Bryant D, Bushakra JM, Vining KJ, Edger PP, Rowley ER, Priest HD, Michael TP, Lyons E, Filichkin SA, Dossett M, Finn CE, Bassil NV, Mockler TC. The genome of black raspberry (Rubus occidentalis). Plant J. 2016;87:535–47.
Wellcome Sanger Institute. 2018. 25 Genomes for 25 Years project. https://www.sanger.ac.uk/science/collaboration/25-genomes-25-years.
Hyun TK, Lee S, Kumar D, Rim Y, Kumar R, Lee SY, Lee CH, Kim J-Y. RNA-seq analysis of Rubus idaeus cv. Nova: transcriptome sequencing and de novo assembly for subsequent functional genomics approaches. Plant Cell Rep. 2014b;33(10):1617–28.
Hyun TK, Lee S, Rim Y, Kumar R, Han X, Lee SY, Lee CH, Kim J-Y. De-novo RNA sequencing and metabolite profiling to identify genes involved in anthocyanin biosynthesis in Korean black raspberry (Rubus coreanus Miquel). PLoS ONE. 2014a;9(2):e88292.
Huo J-W, Liu P, Wang Y, Qin D, Zhao LJ. De novo transcriptome sequencing of blue honeysuckle fruit (Lonicera caerulea L.) and analysis of major genes involved in anthocyanin biosynthesis. Acta Physiol Plant. 2016;38:180.
Li X, Sun H, Pei J, Dong Y, Wang F, Chen H, Sun Y, Wang N, Li H, Li Y. De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants. Gene. 2012;511(1):54–61.
Li L, Zhang H, Liu Z, Cui X, Zhang T, Li Y, Zhang L. Comparative transcriptome sequencing and de novo analysis of Vaccinium corymbosum during fruit and color development. BMC Plant Biol. 2016;16(1):223.
Rowland LJ, Alkharouf N, Darwish O, Ogden EL, Polashock JJ, Bassil NV, Main D. Generation and analysis of blueberry transcriptome sequences from leaves, developing fruit, and flower buds from cold acclimation through deacclimation. BMC Plant Biol. 2012;12(1):46–64.
Song Y, Liu HD, Zhou Q, Zhang HJ, Zhang ZD, Li YD, Wang HB, Liu FZ. High-throughput sequencing of highbush blueberry transcriptome and analysis of basic helix-loop-helix transcription factors. J Integr Agr. 2017;16(3):591–604.
Sun H, Liu Y, Gai Y, Geng J, Chen L, Liu H, Kang L, Tian Y, Li Y. De novo sequencing and analysis of the cranberry fruit transcriptome to identify putative genes involved in flavonoid biosynthesis, transport and regulation. BMC Genomics. 2015;16(652):1–17.
Dal Santo S, Tornielli G, Zenoni S, Fasoli M, Farina L, Anesi A, Guzzo F, Delledonne M, Pezzotti M. The plasticity of the grapevine berry transcriptome. Genome Biol. 2013;14:R54.
Guo D-L, Xi F-F, Yu Y-H, Zhang X-Y, Zhang G-H, Zhong G-Y. Comparative RNA-Seq profiling of berry development between table grape ‘Kyoho’ and its early-ripening mutant ’Fengzao’. BMC Genomics. 2016;17:795.
Sun L, Fan X, Zhang Y, Jiang J, Sun H, Liu C. Transcriptome analysis of genes involved in anthocyanins biosynthesis and transport in berries of black and white spine grapes (Vitis davidii). Hereditas. 2016;153:17.
Sun R, He F, Lan Y, Xing R, Liu R, Pan Q, Wang J, Duan C. Transcriptome comparison of Cabernet Sauvignon grape berries from two regions with distinct climate. J Plant Physiol. 2015;178:43–54.
Sweetman C, Wong DCJ, Ford CM, Drew DP. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics. 2012;13:691.
Li W, Liu F, Zeng S, Xiao G, Wang G, Wang Y, Peng M, Huang H. Gene expression profiling of development and anthocyanin accumulation in kiwifruit (Actinidia chinensis) based on transcriptome sequencing. PLoS ONE. 2015;10:e0136439.
Garcia-Seco D, Zhang Y, Gutierrez-Mañero FJ, Martin C, Ramos-Solano B. RNA-Seq analysis and transcriptome assembly for blackberry (Rubus sp. var. Lochness) fruit. BMC Genomics. 2015;16:5.
Zhang Y, Li W, Dou Y, Zhang J, Jiang G, Miao L, Han G, Liu Y, Li H, Zhang Z. Transcript quantification by RNA-seq reveals differentially expressed genes in the red and yellow fruits of Fragaria vesca. PLoS ONE. 2015;10(12):e0144356.
Sánchez-Sevilla J, Vallarino JG, Osorio S, Bombarely A, Posé D, Merchante C, Botella MA, Iraida Amaya I, Valpuesta V. Gene expression atlas of fruit ripening and transcriptome assembly from RNA-seq data in octoploid strawberry (Fragaria × ananassa). Sci Rep. 2017;7:13737.
BacHBerry Consortium, Dudnik A, Almeida AF, Andrade R, Avila B, Bañados P, Barbay D, Bassard J-E, Benkoulouche M, Bott M, Braga A, Breitel D, Brennan R, Bulteau L, Chanforan C, Costa I, Costa RS, Doostmohammadi M, Faria N, Feng C, Fernandes A, Ferreira P, Ferro R, Foito A, Freitag S, Garcia G, Gaspar P, Godinho-Pereira J, Hamberger B, Hartmann A, Heider H, Jardim C, Julien-Laferriere A, Kallscheuer N, Kerbe W, Kuipers OP, Li S, Love N, Marchetti-Spaccamela A, Marienhagen J, Martin C, Mary A, Mazurek V, Meinhart C, Sevillano DM, Menezes R, Naesby M, MHH N, Okkels FT, Oliveira J, Ottens M, Parrot D, Pei L, Rocha I, Rosado-Ramos R, Rousseau C, Sagot M-F, dos Santos CN, Schmidt M, Shelenga T, Shepherd L, Silva AR, da Silva MH, Simon O, Stahlhut SG, Solopova A, Sorokin A, Stewart D, Stougie L, Su S, Thole V, Tikhonova O, Trick M, Vain P, Veríssimo A, Vila-Santa A, Vinga S, Vogt M, Wang L, Wang L, Wei W, Youssef S, Neves AR, Forster J. BacHBerry: BACterial Hosts for production of Bioactive phenolics from bERRY fruits. Phytochem Rev. 2018;17:291–326.
Martin C, Li J. Medicine is not health care, food is health care: plant metabolic engineering, diet and human health. New Phytol. 2017;216:699–719.
Jofré I, Pezoa C, Cuevas M, Scheuermann E, Freires IA, Rosalen PL, de Alencar SM, Matias S, Romero F. Antioxidant and vasodilator activity of Ugni molinae Turcz. (Murtilla) and its modulatory mechanism in hypotensive response. Oxid Med Cell Longev. 2016;2016:1-11. https://doi.org/10.1155/2016/6513416.
Overall J, Bonney S, Wilson M, Beermann A, Grace MH, Esposito D, Lila MA, Komarnytsky S. Metabolic effects of berries with structurally diverse anthocyanins. Int J Mol Sci. 2017;18:422.
Rao AV, Snyder DM. Raspberries and human health: a review. J Agric Food Chem. 2010;58(7):3871–83.
Rojo LE, Ribnicky D, Logendra S, Poulev A, Rojas-Silva P, Kuhn P, Dorn R, Grace MH, Lila MA, Raskin I. In vitro and in vivo anti-diabetic effects of anthocyanins from Maqui berry (Aristotelia chilensis). Food Chem. 2012;131:387–96.
Tsuda T. Dietary anthocyanin-rich plants: Biochemical basis and recent progress in health benefits studies. Mol Nutr Food Res. 2012;56:159–70.
Tavares L, Figueira I, McDougall GJ, Vieira HLA, Stewart D, Alves PM, Ferreira RB, Santos CN. Neuroprotective effects of digested polyphenols from wild blackberry species. Eur J Nutr. 2013;52:225–36.
Wang YH, Li B, Lin Y, Ma Y, Zhang Q. Meng XJ Effects of Lonicera caerulea berry extract on lipopolysaccharide-induced toxicity in rat liver cells: Antioxidant, anti-inflammatory, and anti-apoptotic activities. J Funct Foods. 2017;33:217–26.
León-González AJ, López-Lázaro M, Espartero JL, Martín-Cordero C. Cytotoxic activity of dihydrochalcones isolated from Corema album leaves against HT-29 colon cancer cells. Nat Prod Commun. 2013;8(9):1255–6.
Macedo D, Tavares L, McDougall GJ, Miranda HV, Stewart D, Ferreira RB, Tenreiro S, Outeiro TF, Santos CN. (Poly)phenols protect from α-synuclein toxicity by reducing oxidative stress and promoting autophagy. Hum Mol Genet. 2015;24(6):1717–32.
Costa C, Tsatsakis A, Mamoulakis C, Teodoro M, Briguglio G, Caruso E, Tsoukalas D, Marginae D, Dardiotis E, Kouretasg D, Fenga C. Current evidence on the effect of dietary polyphenols intake on chronic diseases. Food Chem Toxicol. 2017;110:286–99.
Brauch JE, Buchweitz M, Schweiggert RM, Carle R. Detailed analyses of fresh and dried Maqui (Aristotelia chilensis (Mol.) Stuntz) berries and juice. Food Chem. 2016;190:308–16.
Escribano-Bailón MT, Alcalde-Eon C, Muñoz O, Rivas-Gonzalo JC, Santos-Buelga C. Anthocyanins in berries of Maqui (Aristotelia chilensis (Mol.) Stuntz). Phytochem Anal. 2006;17(1):8–14.
González B, Vogel H, Razmilic I, Wolfram E. Polyphenol, anthocyanin and antioxidant content in different parts of Maqui fruits (Aristotelia chilensis) during ripening and conservation treatments after harvest. Ind Crops Prod. 2015;76:158–65.
Fredes C, Yousef GG, Robert P, Grace MH, Lila MA, Gómez M, Gebauer M, Montenegro G. Anthocyanin profiling of wild Maqui berries (Aristotelia chilensis [Mol.] Stuntz) from different geographical regions in Chile. J Sci Food Agric. 2014;94(13):2639–48.
Arena ME, Zuleta A, Dyner L, Constenlac D, Ceci L, Curvetto N. Berberis buxifolia fruit growth and ripening: Evolution in carbohydrate and organic acid contents. Sci Hortic. 2013;158:52–8.
León-González AJ, Truchado P, Tomás-Barberán FA, López-Lázaro M, Barradas MCD, Martín-Cordero C. Phenolic acids, flavonols and anthocyanins in Corema album (L.) D. Don berries. J Food Compos Anal. 2013;29:58–63.
Chaovanalikit A, Thompson MM, Wrolstad RE. Characterization and quantification of anthocyanins and polyphenolics in blue honeysuckle (Lonicera caerulea L.). J Agric Food Chem. 2004;52:848–52.
Wang Y, Zhu J, Meng X, Liu S, Mu J, Ning C. Comparison of polyphenol, anthocyanin and antioxidant capacity in four varieties of Lonicera caerulea berry extracts. Food Chem. 2016;197:522–9.
Beekwilder J, Jonker H, Meesters P, Hall RD, van der Meer IM, de Vos CHR. Antioxidants in raspberry: On-line analysis links antioxidant activity to a diversity of individual metabolites. J Agric Food Chem. 2005;53:3313–20.
Slimestad R, Solheim H. Anthocyanins from black currants (Ribes nigrum L.). J Agric Food Chem. 2002;50(11):3228–31.
Brito A, Areche C, Sepúlveda B, Kennelly EJ, Simirgiotis MJ. Anthocyanin characterization, total phenolic quantification and antioxidant features of some Chilean edible berry extracts. Molecules. 2014;19:10936–55.
Rui L, Ping W, Qing-qi G, Zhen-yu W. Anthocyanin composition and content of the Vaccinium uliginosum berry. Food Chem. 2011;125:116–20.
Kallscheuer N, Menezes R, Foito A, Henriques da Silva MD, Braga A, Dekker W, Sevillano DM, Rosado-Ramos R, Jardim C, Oliveira J, Ferreira P, Rocha I, Silva AR, Sousa M, Allwood JW, Bott M, Faria N, Stewart D, Ottens M, Naesby M, Nunes dos Santos C, Marienhagen J. Identification and microbial production of the raspberry phenol salidroside that is active against Huntington's disease. Plant Physiol. 2019;179(3):969–85.
Foito A, Steward D. Berry Metabolomics Database. https://ics.hutton.ac.uk/germinate-berrybase/##. Accessed 2015-2018.
Tohge T, de Souza LP, Fernie AR. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J Exp Bot. 2017;68(15):4013–28.
Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot. 2011;62(8):2465–83.
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15(10):573–81.
Ramsay NA, Glover BJ. MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci. 2005;10(2):63–70.
Mehrtens F, Kranz H, Bednarek P, Weisshaar B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005;138:1083–96.
Schwinn K, Venail J, Shang Y, Mackay S, Alm V, Butelli E, Oyama R, Bailey P, Davies K, Martin C. A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell. 2006;18:831–51.
Nesi N, Jond C, Debeaujon I, Caboche M, Lepiniec L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell. 2001;13:2099–114.
Espley RV, Hellens RP, Putterill J, Stevenson DE, Kutty-Amma S, Allan AC. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007;49(3):414–27.
Albert NW, Davies KM, Lewis DH, Zhang H, Montefiori M, Brendolise C, Boase MR, Ngo H, Jameson PE, Schwinn KE. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell. 2014;26:962–80.
Zimmermann IM, Heim MA, Weishaar B, Uhrig JF. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like bHLH proteins. Plant J. 2004;40:22–34.
Butelli E, Licciardello C, Ramadugu C, Durand-Hulak M, Celant A, Reforgiato Recupero G, Froelicher Y, Martin C. Noemi controls production of flavonoid pigments and fruit acidity and illustrates the domestication routes of modern citrus varieties. Curr Biol. 2019;29(1):158–64.
Heim MA, Jacoby M, Werber M, Martin C, Weisshaar B, Bailey PC. The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol Biol Evol. 2003;20(5):735–47.
Miller JC, Chezem WR, Clay NK. Ternary WD40 repeat-containing protein complexes: Evolution, composition and roles in plant immunity. Front Plant Sci. 2016;7(6):1108.
Zhang B, Schrader A. TRANSPARENT TESTA GLABRA 1-dependent regulation of flavonoid biosynthesis. Plants. 2017;6(4):65.
Walker AR, Davison PA, Bolognesi-Winfield AC, James CM, Srinivasan N, Blundell TL, Esch JJ, Marks MD, Gray JC. The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell. 1999;11:1337–49.
Trick M, Thole V, Martin C. 2018. BacHBerryGEN Database. http://jicbio.nbi.ac.uk/berries.
Trick M, Thole V, Martin C. 2018. BacHBerryGEN Database BLAST portal. http://jicbio.nbi.ac.uk/berries/blast.html.
Ramírez-González R, Ghasemi Afshar B, Thole V, Martin C. 2019. BacHBerryEXP gene expression browser. http://www.bachberryexp.com.
Veríssimo A, Bassard J-E, Julien-Laferrière A, Sagot M-F, Vinga S. MassBlast: A workflow to accelerate RNA-seq and DNA database analysis. bioRxiv. 2017. https://doi.org/10.1101/131953.
Stracke R, Werber M, Weisshaar B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol. 2001;4:447–56.
Lin-Wang K, Bolitho K, Grafton K, Kortstee A, Karunairetnam S, McGhie TK, Espley RV, Hellens RP, Allan ACA. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 2010;10:50.
Czemmel S, Stracke R, Weisshaar B, Cordon N, Harris NN, Walker AR, Robinson SP, Bogs J. The grapevine R2R3-MYB transcription factor VvMYBF1 regulates flavonol synthesis in developing grape berries. Plant Physiol. 2009;151(3):1513–30.
Albert NW. Subspecialization of R2R3-MYB repressors for anthocyanin and proanthocyanidin regulation in forage legumes. Front Plant Sci. 2015;6:1165.
Garcia-Seco D, Zhang Y, Gutierrez-Mañero FJ, Martin C, Ramos-Solano B. Application of Pseudomonas fluorescens to blackberry under field conditions improves fruit quality by modifying flavonoid metabolism. PLoS ONE. 2015;10(11):e0142639.
Wang Y, Hu, XJ, Zou XD, Wu XH, Ye ZQ, Wu YD. 2015. WDSPdb Database. http://wu.scbb.pkusz.edu.cn/wdsp/. Accessed Oct 2018.
Wang Y, Hu XJ, Zou XD, Wu XH, Ye ZQ, Wu YD. WDSPdb: a database for WD40-repeat proteins. Nucleic Acids Res. 2015;43:D339–44.
Brueggemann J, Weisshaar B, Sagasser M. A WD40-repeat gene from Malus x domestica is a functional homologue of Arabidopsis thaliana TRANSPARENT TESTA GLABRA1. Plant Cell Rep. 2010;29(3):285–94.
Hu X-J, Li T, Wang Y, Xiong Y, Wu X-H, Zhang D-L, Zhi-Qiang Ye Z-Q, Wu Y-D. Prokaryotic and highly-repetitive WD40 proteins: A systematic study. Sci Rep. 2017;7:10585.
Bally J, Nakasugi K, Jia F, Jung H, Ho SYW, Wong M, Paul CM, Naim F, Wood CC, Crowhurst RN, Hellens RP, Dale JL, Waterhouse PM. The extremophile Nicotiana benthamiana has traded viral defence for early vigour. Nat Plants. 2015;1:15165.
Montefiori M, Brendolise C, Dare AP, Lin-Wang K, Davies KM, Hellens RP, Allan AC. In the Solanaceae, a hierarchy of bHLHs confer distinct target specificity to the anthocyanin regulatory complex. J Exp Bot. 2015;66(5):1427–36.
Butelli E, Licciardello C, Zhang Y, Liu J, Mackay S, Bailey P, Reforgiato-Recupero G, Martin C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012;24:1242–55.
Appelhagen I, Wulff-Vester AK, Wendell M, Hvoslef-Eide A-K, Russell J, Oertel A, Martens S, Mock H-P, Martin C, Matros A. Colour bio-factories: Towards scale-up production of anthocyanins in plant cell cultures. Metab Eng. 2018;48:218–32.
Lim SH, Kim DH, Kim JK, Lee JY, Ha SH. A radish basic helix-loop-helix transcription factor, RsTT8 acts a positive regulator for anthocyanin biosynthesis. Front Plant Sci. 2017;8:1917.
Outchkourov NS, Carollo CA, Gomez-Roldan V, de Vos RCH, Bosch D, Hall RD, Beekwilder J. Control of anthocyanin and non-flavonoid compounds by anthocyanin-regulating MYB and bHLH transcription factors in Nicotiana benthamiana leaves. Front Plant Sci. 2014;5:519.
Rahim MA, Busatto N, Trainotti L. Regulation of anthocyanin biosynthesis in peach fruits. Planta. 2014;240(5):913–29.
Lin-Wang K, McGhie TK, Wang M, Liu Y, Warren B, Storey R, Espley RV, Allan ACA. Engineering the anthocyanin regulatory complex of strawberry (Fragaria vesca). Front Plant Sci. 2014;5:651.
Petroni K, Tonelli C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011;181:219–29.
Espley RV, Brendolise C, Chagné D, Kutty-Amma S, Green S, Volz R, Putterill J, Schouten HJ, Gardiner SE, Hellens RP, Allan AC. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell. 2009;21:168–83.
Zhang Y, Butelli E, Alseekh S, Tohge T, Rallapalli G, Luo J, Kawar PG, Hill L, Santino A, Fernie AR, Martin C. Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nat Commun. 2015;26(6):8635.
Liu X, Feng C, Zhang M, Yin X, Xu C, Chen K. The MrWD40-1 gene of Chinese Bayberry (Myrica rubra) interacts with MYB and bHLH to enhance anthocyanin accumulation. Plant Mol Biol Rep. 2013;31:1474–84.
Mahjoub A, Hernould M, Joubes J, Decendit A, Mars M, Barrieu F, Hamdi S, Delrot S. Overexpression of a grapevine R2R3-MYB factor in tomato affects vegetative development, flower morphology and flavonoid and terpenoid metabolism. Plant Physiol Biochem. 2009;47:551–61.
Borrill P, Ramírez-González R, Uauy C. expVIP: a customisable RNA-seq data analysis and visualisation platform. Plant Physiol. 2016;170:2172–86.
Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
Priyam A, Woodcroft BJ, Rai V, Munagala A, Moghul I, Ter F, Gibbins MA, Moon H, Leonard G, Rumpf W, Wurm Y. Sequenceserver: a modern graphical user interface for custom BLAST databases. biorxiv. 2015. https://doi.org/10.1101/033142.
Schumacher K. pH in the plant endomembrane system - an import and export business. Curr Opin Plant Biol. 2014;22:71–6.
Li Y, Provenzano S, Bliek M, Spelt C, Appelhagen I, Machado de Faria L, Verweij W, Schubert A, Sagasser M, Seidel T, Weisshaar B, Koes R, Quattrocchio F. Evolution of tonoplast P‐ATPase transporters involved in vacuolar acidification. New Phytol. 2016;211:1092–107.
Schaart JG, Dubos C, Romero De La Fuente I, van Houwelingen AM, de Vos RC, Jonker HH, Xu W, Routaboul JM, Lepiniec L, Bovy AG. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria × ananassa) fruits. New Phytol. 2013;197(2):454–67.
Chacon-Fuentes M, Parra L, Lizama M, Seguel I, Urzua A, Quiroz A. Plant flavonoid content modified by domestication. Environ Entomol. 2017;46(5):1080–9.
Vasanthaiah HKN, Katam R, Sheikh MB. Efficient protocol for isolation of functional RNA from different grape tissue rich in polyphenols and polysaccharides for gene expression studies. Electron J Biotechnol. 2008;11(no. 3):1–8.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat Biotechnol. 2011;29(7):644–52.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
FastQC Database. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed Nov 2014 to Feb 2016.
Leggett RM, Ramirez-Gonzalez RH, Clavijo BJ, Waite D, Davey RP. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front Genet. 2013;4:288.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
AnnotF Annotation Pipeline. https://github.com/EI-CoreBioinformatics/AnnotF. Accessed 2014 to 2016.
Veríssimo A, Bassard J-E, Julien-Laferrière A, Sagot M-F, Vinga S. 2017. MassBlast Database. https://averissimo.github.io/mass-blast/. Accessed 2016-2017.
Cheng C-Y, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017;89:789–804.
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Dejardin A, Depamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, Henrissat B, Holligan D, Holt R, Huang W, Islam-Faridi N, Jones S, Jones-Rhoades M, Jorgensen R, Joshi C, Kangasjarvi J, Karlsson J, Kelleher C, Kirkpatrick R, Kirst M, Kohler A, Kalluri U, Larimer F, Leebens-Mack J, Leple JC, Locascio P, Lou Y, Lucas S, Martin F, Montanini B, Napoli C, Nelson DR, Nelson C, Nieminen K, Nilsson O, Pereda V, Peter G, Philippe R, Pilate G, Poliakov A, Razumovskaya J, Richardson P, Rinaldi C, Ritland K, Rouze P, Ryaboy D, Schmutz J, Schrader J, Segerman B, Shin H, Siddiqui A, Sterky F, Terry A, Tsai CJ, Uberbacher E, Unneberg P, Vahala J, Wall K, Wessler S, Yang G, Yin T, Douglas C, Marra M, Sandberg G, Van de Peer Y, Rokhsar D. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006;313:1596–604.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–83.
The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41.
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, Childs KL, Davidson RM, Lin H, Quesada-Ocampo L, Vaillancourt B, Sakai H, Lee SS, Kim J, Numa H, Itoh T, Buell CR, Matsumoto T. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:1–10.
Pissis S, Project AG. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1467.
Zhao Q, Yang J, Cui M-Y, Liu J, Fang Y, Yan M, Qiu W, Shang H, Xu Z, Yidiresi R, Weng J-K, Pluskal T, Vigouroux M, Steuernagel B, Wei Y, Yang L, Hu Y, Chen X-Y, Martin C. The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol Plant. 2019;12:935–50.
Visser EA, Wegrzyn JL, Myburg AA, Naidoo S. Defence transcriptome assembly and pathogenesis related gene family analysis in Pinus tecunumanii (low elevation). BMC Genomics. 2018;19:632.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T. BioRuby: bioinformatics software for the Ruby programming language. Bioinformatics. 2010;26:2617–9.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Yang Z. PAML: Phylogenetic Analysis by Maximum Likelihood Programme Package. 2017. McMcTree for Bayesian estimation of species divergence time. http://abacus.gene.ucl.ac.uk/software/paml.html. Accessed July-August 2019.
Hedges SB, Julie M, Michael S, Madeline P, Sudhir K. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. 2015;32:835–45.
Thole V, Worland B, Snape J, Vain P. The pCLEAN dual binary vector system for Agrobacterium-mediated plant transformation. Plant Physiol. 2007;145:1211–9.
Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM. pGreen: a versatile and flexible binary vector for Agrobacterium-mediated transformation. Plant Mol Biol. 2000;42:819–32.
An G, Ebert PR, Mitra A, Ha SB. Binary vectors. In SB Gelvin, RA Schilperoort, eds, Plant Molecular Biology Manual. Kluwer Academic Publishers, Dordrecht. The Netherlands. 1988;A3:1–19.
Thole V, Rawsthorne S. Development of a strategy for transgenic studies and monitoring of transgene expression in two closely related Moricandia species possessing a C3 or C3-C4 intermediate photosynthetic phenotype. Physiol Plant. 2003;119:155–64.
Acknowledgements
We would like to thank the coordinators of the BacHBerry project (BACterial Hosts for production of Bioactive phenolics from bERRY fruits, http://www.bachberry.eu/) Dr. Jochen Förster (Carlsberg Research Laboratory, Denmark) and Dr. Alexey Dudnik (The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark). We would like to specially thank Dr. Nicola Love (John Innes Centre, Norwich, UK), Bárbara Ávila (Pontificia Universidad Católica de Chile, Macul, Chile) and Pedro Oliveira (Instituto Nacional de Investigacao Agraria, Oeiras, Portugal) for their help. The authors thank the Earlham Institute (Norwich, UK), particularly Dr. Purnima Pachori, Dr. Helen Chapman and Heather Musk, for RNA-seq analyses. We are grateful to Prof. Peter Waterhouse (Queensland University of Technology, Brisbane, Australia) for providing the seeds of N. benthamiana cv. Northern Territory. Andrew Davis is thanked for photographic images and the John Innes Centre plant husbandry team for plant care.
Funding
This research was funded by the European Union Framework Program 7, Project BacHBerry [FP7–613793]. The authors also acknowledge support from the Institute Strategic Programmes ‘Designing Future Wheat’ (BB/P016855/1), ‘Understanding and Exploiting Plant and Microbial Secondary Metabolism’ (BB/J004596/1) and ‘Molecules from Nature’ (BB/P012523/1) from the UK Biotechnology and Biological Sciences Research Council to the John Innes Centre and the European funded COST ACTION FA1106 QualityFruit. VT, PV and CM have also received funding from the European Union’s Horizon 2020 research and innovation programme through the TomGEM project under grant agreement No. 679796. The funding bodies had no role in the design of the study, collection, analysis and interpretation of data nor in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CM, PV and VT designed and coordinated the experiments; AF, LS, SF, DS, CNS, RM, PB, LW, AS, OT and TS provided plant material; DB carried out RNA extractions; VT, J-EB, PV, MN and DB analysed transcriptome datasets; MT developed the BacHBerryGEN database and RR-G and BGA the BacHBerryEXP gene expression browser; RR-G performed the phylogenetic analysis of the transcriptome sequences; VT cloned and characterized Rubus spp. flavonoid regulatory genes; LH carried out HPLC analyses; VT, PV and CM interpreted the results and co-wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has not directly involved humans or animals. The collection of plant material complies with international guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Classification and description of berry fruit species.
Additional file 2: Figure S1.
Schematic representation of the phylogenetic relationship among the 13 berry fruit species studied.
Additional file 3: Table S2.
RNA-seq descriptors for 13 berry fruit species.
Additional file 4: Table S3.
RNA-seq descriptors for two Rubus species at three fruit ripening stages: green, immature and ripe.
Additional file 5: Table S4.
BLAST search output summaries for transcript candidates involved in the phenylpropanoid pathway for 13 berry fruit species: Enzymes of the core pathway and its decorating, modifying and regulatory proteins.
Additional file 6: Table S5.
Peptide annotation for the transcriptomes of R. genevieri and R. idaeus cv. Prestige.
Additional file 7: Table S6.
Identification and cloning of regulatory genes of the phenylpropanoid pathway from R. genevieri (A) and R. idaeus cv. Prestige (B).
Additional file 8: Table S7.
Primers used for the cloning of regulatory genes of the phenylpropanoid pathway from R. genevieri (A) and R. idaeus cv. Prestige (B).
Additional file 9: Figure S2.
Phylogenetic relationship and protein sequence alignment of a subset of R2R3-type MYB transcription factor homologues.
Additional file 10: Figure S3.
Phylogenetic relationship and protein sequence alignment of a subset of bHLH transcription factor homologues.
Additional file 11: Figure S4.
Phylogenetic relationship, prediction of WD40 motifs and protein sequence alignment of a subset of WDR homologues.
Additional file 12: Table S8.
Homologues of the regulatory genes cloned in this study in the collection of the 13 berry fruit species.
Additional file 13: Figure S5.
Production of anthocyanins in leaves of two accessions of N. benthamiana, JIC-LAB strain and cv. NT, following transient overexpression of regulatory genes from R. genevieri and R. idaeus cv. Prestige at various time points after infiltration (4 dpi to 14 dpi) alone or in combination.
Additional file 14: Figure S6.
Examples of anthocyanin formation in kanamycin, hygromycin and/or PPT-resistant N. benthamiana calli and shoots transformed with Rubus Myb, bHLH and WDR regulatory genes.
Additional file 15: Table S9.
Differential expression of candidate transcripts homologous to enzymes involved in the phenylpropanoid pathway of R. genevieri and R. idaeus cv. Prestige during three fruit ripening stages: Examples of candidate enzymes of the general phenylpropanoid pathway, flavonoid regulatory enzymes, transporters, decorating and modifying enzymes.
Additional file 16:
Step-by-step guide of the phylogenetic analysis of the transcriptomes of the 13 berry fruit species together with the reference genomes of A. thaliana, P. trichocarpa, G. max, V. vinifera, S. lycopersicum, O. sativa and A. trichopoda.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Thole, V., Bassard, JE., Ramírez-González, R. et al. RNA-seq, de novo transcriptome assembly and flavonoid gene analysis in 13 wild and cultivated berry fruit species with high content of phenolics. BMC Genomics 20, 995 (2019). https://doi.org/10.1186/s12864-019-6183-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6183-2