
Abstract
Background
The whole repeat unit of 5S rDNA and the internal transcribed spacer (ITS) of four European Donax species were analysed. After amplifying, cloning and sequencing several 5S and ITS units, their basic features and their variation were described. The phylogenetic usefulness of 5S and ITS sequences in the inference of evolutionary relationships among these wedge clams was also investigated.
Results
The length of the 5S repeat presented little variation among species, except D. trunculus that differed from the rest of the Donax species in 170–210 bp. The deduced coding region covered 120 bp, and showed recognizable internal control regions (ICRs) involved in the transcription. The length of non-transcribed spacer region (NTS) ranged from 157 bp to 165 bp in Donax trunculus and from 335 bp to 367 bp in the other three species. The conservation degree of transcriptional regulatory regions was analysed revealing a conserved TATA-like box in the upstream region. Regarding ITS sequences, the four Donax species showed slight size differences among clones due to the variation occurring in the ITS1 and ITS2, except Donax variegatus did not display size differences in the ITS2. The total length of the ITS sequence ranged between 814 and 1014 bp. Resulting phylogenetic trees display that the two ribosomal DNA regions provide well-resolved phylogenies where the four European Donax species form a single clade receiving high support in nodes. The topology obtained with 5S sequences was in agreement with Donax evolutionary relationships inferred from several sequences of different nature in previous studies.
Conclusions
This is not only a basic research work, where new data and new knowledge is provided about Donax species, but also have allowed the authentication of these wedge clams and offers future applications to provide other genetic resources.
Similar content being viewed by others


Introduction
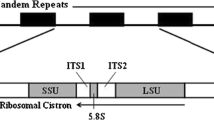
In higher eukaryotes, rDNA comprises two different multigene families [53], including the major 45S rDNA family encoding 18S, 5.8S, and 28S rRNA, and the minor 5S rDNA family encoding 5S rRNA, each composed of hundreds to thousands of copies, organized in tandem repeats, and consisting of coding regions and transcribed and non-transcribed spacers. The different evolutionary rates among different regions, the secondary structure of these genes and their organization in tandem repeats, make rDNA attractive candidate for species identification, population characterization, phylogenetic studies and evolutionary relationships and genomic structure [39, 47, 77].
The 5S rDNA consists of a highly conserved 120 bp coding sequence (5S rRNA gene) clustered in long direct tandem arrays and separated by variable non-transcribed flanking DNA sequences know as non-transcribed spacers or NTSs. Both together, the coding sequence and the NTS, form a repeat unit that can be found in hundreds to thousands of copies tandemly repeated in the genome. Even though the 5S rRNA gene is highly conserved, even among unrelated species, the NTS are variable both in length and sequence. These discrepancies have been used as molecular phylogenetic and species-specific markers in several bivalve mollusc species [24, 26, 55, 72], so that the 5S rDNA is a good candidate to identify molecular markers suitable for distinguish related species.
By the same token, the internal transcribed spacer (ITS) region of rDNA consists of one coding region (5.8S rRNA gene) and two non-coding regions (ITS1 and ITS2) located in the rDNA between 18S and 5.8S rRNA genes and between 5.8S and 28S rRNA genes, respectively. Due to ITS sequences show more variability than their flanking coding region [38], they have been also frequently used to infer phylogenetic relationships among bivalve species [6, 45, 100] and to differentiate related bivalve species [39, 54, 86]. For instance, ITS1 has been a widely chosen marker for assessing variation within species due to its high level of divergence, while ITS2 region has been proposed as an effective barcode similar to the cytochrome c oxidase subunit I (COI) for identifying species that are difficult to distinguish morphologically and allowing identify closely related species within different families and genera [105].
To date, numerous reports on the characterization of the 5S rDNA and the ITS region in several molluscan species, including bivalves, gastropods, and cephalopods have been published [44, 56, 101]. But bivalve molluscs stand out for being one of the most extensively studied group of organisms regarding 5S rDNA and ITS region, showing high levels of gene organization as well as a vast diversity of gene arrangements. Molecular organization of ITS region and 5S rDNA has been studied in cockles [26,27,28, 43], mussels [44], oysters [9, 10], scallops [41, 42, 45, 46, 55], razor clams [25, 99, 100] and Veneroida clams [3], but it have never been studied in the wedge clams of the genus Donax.
Four Donax species, Donax semistriatus, Donax trunculus, Donax variegatus and Donax vittatus, are common along the European littoral and live sympatrically in some areas [1, 20, 21, 31, 84]. These marine bivalves play an important socioeconomic role in some European coastal regions. For instance, the wedge clam D. trunculus is an exploited and economically important traditional seafood in several European countries, including France [95], Italy [106], Portugal [4], Spain [61] and Turkey [69], which could emerge if managed properly on the local scale. However, natural beds of this species in Galicia (north – west coast of Spain) have been intensively exploited, and they have suffered a severe decrease. In fact, the amount harvested of this wedge clam has declined within the last 16 years from ~ 17 t (2001) to 171.10 kg (2017) (Consellería do Mar, Xunta de Galicia) and at present, only the fishermen’s association of Arousa (108.05 kg) and Cedeira (365.39 kg) commercialise this bivalve mollusc (data from [7]). On a much larger scale, only in the Iberian Peninsula, the recorded captures has suffered a sharp decline, with a maximum production of 1042 t in 2005, but reaching only 195 t in 2016 (FAO-FIGIS, 2018). In point of fact, currently some D. trunculus localities seem to be at high long-term risk of extinction [57]. Furthermore, D. trunculus may account for most of the recorded catches of FAO in these countries for genus Donax. Nevertheless, FAO statistics [18] do not distinguish between species. Therefore, it is likely to find in the fish market other Donax species with lower economic value being sold as D. trunculus. However, despite the economic importance that genus Donax has for the European seafood sector and of being an overexploited species, until a few years ago basic genetic studies in this organism were neglected. To date, with respect to the Donax genus, the complete female mitochondrial genomes have been sequenced and characterized [20] and three methods for the molecular identification of European Donax species have been developed [22, 65, 72]. Concerning D. vittatus, a work that deals with the description and study of the karyotype of D. vittatus and compares it with the karyotype of D. trunculus has recently been published [30] and its genetic diversity and population structure have been evaluated with mitochondrial and nuclear markers [21]. Regarding D. trunculus, most of the works have focused on studying the karyotype of the species [8, 34, 58], as well as on the analysis of mobile elements and satellite DNA [74, 75, 78,79,80, 87]. In addition, Theologidis et al. [96] have studied the mode of inheritance of mitochondrial DNA. Recently, microsatellite markers have also been developed [64, 82] and population genetic analyses based on this type of molecular markers [57, 63, 82] and mitochondrial markers [23] have been carried out.
In this work, the 5S rDNA and the ITS region of four wedge clams, D. semistriatus, D. trunculus, D. variegatus and D. vittatus, of the bivalve family Donacidae present in Europe were analysed. The aim of this analysis was to amplify, clone, and sequence the 5S rDNA and ITS repeats units to i) provide their basic characteristics, ii) assess their variability, iii) estimate their divergence and iv) report their utility in evolutionary relationships.
Material and methods
Sampling and DNA extraction
Twelve Donax trunculus specimens were collected from natural beds in Vilarrube (northwestern Spain) while twelve D. semistriatus, eight D. variegatus and twenty D. vittatus samples came from Portuguese coast (Table 1). Field work was conducted in accordance with local legislation and with regulations and guidelines established by the University of A Coruña. No endangered or protected species were involved. Specimens were taxonomically identified using a species-specific PCR-RFLP analysis of COI capable discriminating among the four Donax species [65]. Total genomic DNA was extracted from ethanol-preserved foot using a Chelex-100 (Sigma-Aldrich, USA) protocol based on Walsh et al. [102].
PCR amplification, cloning and sequencing
For 5S rDNA, amplification reactions were carried out using a set of primers designed by Fernández-Tajes and Méndez [24] annealing to the coding region in opposite orientations. They were carried out in 25 μl containing 150 ng of genomic DNA, 0.6 μM of each primer, 0.25 μM of each dNTP, 2 mM of MgCl2, 0.6 U of Taq polymerase (Roche Applied Science) and the buffer recommended by the polymerase suppliers. Cycling conditions were 2 min denaturing at 95 °C; (30 s at 95 °C, 30 s at 55 °C, and 1 min at 72 °C) × 35; and a final extension step at 72 °C for 5 min.
For ITS1 and ITS2, PCR reactions were performed with a pair of primers that anneal at the 3’end of the 18S ribosomal gene and the 5’end of the 28S ribosomal gene [37] (ITSF: 5′-GTTTCCGTAGGTGAACCTG-3′ and ITSR: 5´-CTCGTCTGATCTGAGGTCG-3′). They were performed in 25 μl containing 100 ng of genomic DNA, 0.25 μM of each dNTP, 1.5 mM of MgCl2, 1 μM of each primer, 0.625 U of Taq polymerase (Roche Applied Science) and the buffer recommended by the polymerase suppliers. Cycling conditions were 3 min denaturing at 94 °C; (20 s at 94 °C, 20 s at 55 °C, and 45 s at 72 °C) × 30; and a final extension step at 72 °C for 5 min. PCR products were migrated on a 2.0% agarose gel electrophoresis. Gels were stained by immersion in 0.5 μg/ml ethidium bromide solution for 30 min, visualized and recorded on a transilluminator Gel Doc XR Systems (Bio-Rad, Barcelona, Spain).
PCR products were migrated on 2% agarose gel electrophoresis. Gels were stained by immersion in 0.5 μg/ml ethidium bromide solution for 30 min, visualized and recorded on a transilluminator Gel Doc XR Systems (Bio-Rad, Barcelona, Spain).
For three or four individuals of each species, the product obtained was ligated into the T&A™ cloning vector and transformed into Escherichia coli ECOS™ JM109 strain competent cells using T&A™ Cloning Vector Kit (Yeastern Biotech Co., Ltd). Recombinant colonies were screened by PCR amplifying with M13 forward and reverse primers to assess the size of the insert. PCR reaction mixture contained 5 μl of recombinant cells, 1x PCR buffer, 1.5 mM MgCl2, 0.2 μM of each dNTP, 0.6 μM of each primer, and 0.3 U of Taq polymerase (Roche Applied Science) in a final volume of 12.5 μl. The thermal cycle profile consisted of an initial denaturation of 10 min at 94 °C, 30 cycles of 1 min at 94 °C, 1 min at 55 °C and 1 min at 72 °C; and a final extension of 10 min at 72 °C. Several recombinant colonies (3–10 per individual) were selected at random and grown in LB medium and, in order to purify the plasmids, a QIAprep Spin Miniprep Kit (QIAGEN) was used. Plasmids were sequenced using M13 primers (forward and reverse) on an ABI PRISM 3120xl (Applied Biosystems, Foster City, CA, USA) at the Molecular Biology Unit of the University of A Coruña (Spain). The corresponding nucleotide sequences have been deposited in the GenBank database under accession numbers MG041608 – MG041761 (Table 1).
Sequence analysis
The identity of sequences obtained was corroborated using BLASTn searches of the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Sequence data were aligned via MAFFT [50] using the L-INS-i algorithm (recommended for < 200 sequences with one conserved domain and long gaps) and manually checked using the BioEdit v.7.2.5 sequence editor [35]. The number of variable sites, nucleotide diversity and sequence divergence were estimated using DnaSP v5.10.01 [52]. Differences between sequence pairs and Donax consensus sequences were calculated using Geneious Pro v.4.8.5 [15]. For the phylogenetic analyses, sequence data were aligned in MAFFT [50] using the L-INS-i algorithm. 5S and ITS alignments, consisting of 652 and 1110 pb and including 27 sequences from D. semistiatus, 20 from D. trunculus, 21 from D. variegatus, 15 from D. vittatus, and 22 sequences from D. semistiatus, 23 from D. trunculus, 13 from D. variegatus, 12 from D. vittatus, respectively; and Cerastoderma edule (GB accession numbers: AJ132199.1 for 5S and AM229683.1 for ITS) and Cerastoderma glaucum (GB accession numbers: AJ842010.1 for 5S and AM229691.1 for ITS) like outgroups were then analysed using Bayesian Inference (BI). Akaike Information Criterion was selected using jModelTest v.2.1.8 [12] for each gene partition, including codon positions of coding genes. The optimal chosen method for 5S rDNA and ITS, were HKY + I + G and GTR + G, respectively. BI analyses were run using MrBayes v.3.2.6 [83]. Two independent Markov chain Monte Carlo (MCMC) runs, each comprising four linked chains (one cold and three heated; as default settings), were performed for 5,000,000 generations, sampling every 1000 generations to allow adequate time for convergence. The convergence of the two runs was assessed by stopping the analysis when the average standard deviation was below 0.01 (stoprule = yes and stopval = 0.01 in the mcmc command). 1,311,000 and 3189,000generations were enough to reach adequate average standard deviation (< 0.01) in 5S rDNA and ITS, respectively. The first 25% trees were discarded as burn-in.
Results
5S rDNA
The 5S rDNA repeat unit was PCR amplified in at least 20 individuals of each wedge clam species, except for D. semistriatus and D. variegatus of which the number of available individuals was low (12 and 8, respectively). The length of the 5S units was about 275–300 bp for D. trunculus, about 450 bp for D. variegatus and around 500 bp for fragments obtained from the other two Donax species with minimal variation (1–12 bp) among clones (Table 2). Taking as reference the 5S rRNA sequences available in other bivalve species [9, 17, 25, 26, 44, 55], the coding region was assigned to 120 bp in the four cases and the non-transcribed spacer (NTS) region to the remaining sequence (Table 2). BLASTn analysis corroborated the identity of the limited region and indicated that no other coding sequence was included in the 5S rDNA repeat unit. The GC content of the repeat units ranged from 38.2 to 43.3% among the wedge clams (Table 2), with higher values in the coding region (53.4–55%) than in the spacer region (33.1–39.1%).
The alignment of all the 5S rDNA wedge clam sequences consisted of 568 pb and showed 130 variable sites of which 108 were parsimony informative, and 342 indels (due mainly to the fact that the sequence of D. trunculus is smaller than for the rest of the species). Almost entirely the variation was located in the spacer region; the sequence corresponding to gene showed 38 variable sites (Additional file 1). Intraindividual variation was minimal within control region and moderate within the spacer region in D. trunculus and D. variegatus. But in D. semistriatus and D. vittatus the sequences displayed considerable variation within the coding and spacer regions. For each species, in D. semistriatus the alignment of 27 clones showed 139 variable sites (17 nucleotide substitutions located in the coding region and 122 in the NTS) and 29 indels located in the NTS. The percentage of differences in pairwise comparisons ranged from 5.2 to 7.5% in intraindividual comparisons and from 6.3 to 7.3% in interindividual comparisons. Global nucleotide diversity was 0.05744 (0.01360 in 5S and 0.07342 in NTS). In D. trunculus the alignment of 20 clones showed 25 variable sites (6 nucleotide substitutions located in the coding region and 19 in the NTS; and 11 indels located in the NTS). The percentage of differences in pairwise comparisons ranged from 2.0 to 2.8% in intraindividual comparisons and from 2.5 to 3.3% in interindividual comparisons. Global nucleotide diversity was 0.02176 (0.01948 in 5S and 0.02359 in NTS). In the case of D. variegatus the alignment of 22 clones presented 13 variable sites (4 nucleotide substitutions located in the coding region and 9 in the NTS) and an indel located in the NTS. Only four clones (Dvar1/5, Dvar1/8, Dvar2/3 and Dvar4/3) displayed a nucleotide substitution in the coding region. The percentage of differences in pairwise comparisons ranged from 0.1 to 0.4% in intraindividual comparisons and from 0.2 to 0.6% in interindividual comparisons. Overall nucleotide diversity was 0.00393 (0.00305 in 5S and 0.00425 in NTS). In D. vittatus the alignment of 15 clones displayed 69 variable sites (15 nucleotide substitutions located in the coding region and 54 in the NTS) and an indel located in the NTS. The percentage of differences in pairwise comparisons ranged from 0.0 to 2.4% in intraindividual comparisons and from 3.2 to 5.3% in interindividual comparisons. Overall nucleotide diversity was 0.05607 (0.04828 in 5S and 0.05881 in NTS). The values of nucleotide divergence (Dxy) and the net number of nucleotide substitutions between groups (Da) with Jukes and Cantor [48] method are shown in Table 3. The values obtained between species are similar, with the highest values being found between D. variegatus and D. vittatus, and lower between D. semistriatus and D. vittatus. These results are in agreement with the 5S phylogenetic tree (see below).
In the four Donax species, the internal control regions (ICRs) described in other organisms were identified. A graphical representation of the 5S internal promoters and their consensus sequences is shown in Fig. 1. The stretches from 3 to 18, from 37 to 44, from 48 to 61, and from 78 to 98 in the alignment displayed high homology with their orthologues ICR I, II, III, and IV of Drosophila melanogaster [88] (12/16, 8/8, 12/14, and 18/21 matches, respectively) (see Fig. 1), and the stretches from 50 to 64, from 67 to 72, and from 80 to 97 were also similar to box A, intermediate element, and box C of Xenopus laevis somatic 5S RNA gene [76] (11/15, 5/6, and 17/18 matches, respectively) (see Fig. 1). Moreover, the NTS region of Donax species contain TATA-like motif recognized at around − 28 nucleotides, other potential transcription control sequences that may be involved in 5S transcription initiation, such as in the silkworm Bombyx mori [62], in Neurospora crassa [98] and D. melanogaster [88]. The NTS sequences of D. semistriatus and D. vittatus retained the complete block TATATA at the 3’end; but not the other species, D. trunculus and D. variegatus, because one insertion T(G)ATATA and a point mutation (TATTTA) occurred within, respectively. Finally, a T-rich stretch was located a few residues downstream of the coding region in the four Donax species, and it is believed that could be related to transcriptional processes, specifically in transcription termination [2, 32, 40].

Graphical representation of the control elements involved in the transcription of 5S rDNA. The top sequences represent a schematic comparison of the ICRs between Donax consensus sequence and D. melanogaster. The bottom sequences represent a schematic comparison of the sequence elements (box A, intermediate element (IE), and box C) between Donax consensus sequence and X. laevis. The asterisks indicate similarities respect to consensus sequences described for D. melanosgaster and X. laevis
ITS1 and ITS2
The ITS region was PCR amplified in at least 20 individuals of D. trunculus and D. vittatus, and 12 individuals of D. semistriatus and 8 individuals of D. variegatus, yielded a single product about 1000 bp for D. vittatus, about 800 bp for D. trunculus and about 900 bp for the rest of species. Table 4 shows the size and GC of the ITS region delimited according to the BLASTn analysis result. The four Donax species showed slight size differences among clones due to the variation occurring in the ITS1 and ITS2, except D. variegatus did not display size differences in the ITS2. The total length of the ITS region was 814–1014 bp with 58–62.4% GC content; ITS1 was 400–542 bp and 59.7–62.6% GC; the 5.8S rRNA gene was 157 bp and 57.3% GC in all clones; and ITS2 was 254–316 bp and 55.7–64.8% GC. The GC content was similar in the four species, with the highest values in the ITS2 followed ITS1 and 5.8S gene, except for D. trunculus that displayed higher content in ITS1 than in ITS2 (Table 4).
The alignment of the different Donax ITS sequences consisted of 1048 pb and showed 258 variable sites of which 248 were parsimony informative sites, and 279 indels (see in Additional file 2). The largest differences were found in ITS1 (162 variable sites), followed by ITS2 (90 variable sites) and 5.8 gene (6 variable sites). For each species, in D. semistriatus the alignment of 22 clones showed 33 variable sites (20 nucleotide substitutions located in the ITS1, one in the 5.8 gene and 12 in the ITS2) and 6 and 4 indels located in the ITS1 and ITS2, respectively. The percentage of differences in pairwise comparisons ranged from 0.2 to 2.5% in intraindividual comparisons and from 1.7 to 3.7% in interindividual comparisons. Overall nucleotide diversity was 0.01415 (0.01827 in ITS1, 0.00323 in ITS2 and 0.00058 in 5.8S). In D. trunculus the alignment of 23 clones presented 53 variable sites (21 nucleotide substitutions located in the ITS1, 2 in the 5.8 gene and 30 in the ITS2) and 7 and 17 indels located in the ITS1 and ITS2, respectively. The percentage of differences in pairwise comparisons ranged from 0.0 to 1.3% in intraindividual comparisons and from 0.03 to 2.1% in interindividual comparisons. Global nucleotide diversity was 0.01923 (0.01229 in ITS1, 0.04105 in ITS2 and 0.00314 in 5.8S). In D. variegatus the alignment of 13 clones displayed 28 variable sites (22 nucleotide substitutions located in the ITS1, and 6 in the ITS2) and 4 indels located the ITS1. The percentage of differences in pairwise comparisons ranged from 0.00 to 0.01% in intraindividual comparisons and from 0.04 to 1.80% in interindividual comparisons. Overall nucleotide diversity was 0.01225 (0.01998 in ITS1, 0.00703 in ITS2 and 0.00000 in 5.8S). In D. vittatus the alignment of 12 clones showed 59 variable sites (17 nucleotide substitutions located in the ITS1, one in the 5.8 gene and 41 in the ITS2) and 11 and 38 indels located the ITS1 and ITS2, respectively. The percentage of differences in pairwise comparisons ranged from 0.2 to 2.20% in intraindividual comparisons and from 2.7 to 3.6% in interindividual comparisons. Global nucleotide diversity was 0.02166 (0.01272 in ITS1, 0.05163 in ITS2 and 0.00107 in 5.8S). In addition, the alignment of the four species revealed four stretches of 12, 23, 13, and 59 nucleotides in ITS1 (alignment positions 241–252, 316–338, 387–399 and 499–557, respectively) and four stretches of 17, 33, 22 and 17 nucleotides in ITS2 (alignment positions 715–731, 754–786, 790–811 and 862–878, respectively), all being highly conserved among Donax species (percentages of similarity higher than 96.7%). Sequence similarity of ITS2 (76.4%) was higher than that of ITS1 (70.6%) across species. The values of nucleotide divergence (Dxy) and the net number of nucleotide substitutions between groups (Da) with Jukes and Cantor [48] method are shown in Table 5. The highest values being found between D. trunculus and D. vittatus, and lower between D. semistriatus and D. variegatus. These results are in agreement with the phylogenetic tree derived from the ITS region (see below).
Phylogenetic analyses
Regarding phylogenetic analyses, the results showed well-resolved phylogenies where the four Donax species form a single clade and received high Bayesian support values in nodes (Figs. 2 and 3). However, 5S and ITS tree topologies were not congruent. On the one hand, the BI tree inferred from 5S rDNA sequences of the four Donax species (Fig. 2) shows two groups supported by high posterior probabilities, where D. trunculus + D. variegatus is the sister clade of D. semistriatus + D. vittatus. These results agree with the values of Dxy and Da showed in Table 3, where the highest values being found between D. variegatus and D. vittatus, and lower between D. semistriatus and D. vittatus. On the other hand, the BI tree from ITS sequences (Fig. 3) consisted of two well-supported (with 1.00 posterior probability as branch support) sister clades: one comprising solely D. trunculus sequences, and the other including the remaining Donax ones, where D. semistriatus and D. variegatus appear in the same branch. These results are also in accordance with the values of Dxy and Da showed in Table 5, where the highest values being found between D. trunculus and D. vittatus, and lower between D. semistriatus and D. variegatus.

Bayesian phylogenetic tree inferred from 5S rDNA sequences of Donax semistriatus, Donax trunculus, Donax variegatus and Donax vittatus. The phylogenetic tree was rooted with Cerastoderma edule and Cerastoderma glaucum species. Numbers at the nodes correspond to Bayesian posterior probabilities

Bayesian phylogenetic tree inferred from ITS sequences of Donax semistriatus, Donax trunculus, Donax variegatus and Donax vittatus. The phylogenetic tree was rooted with Cerastoderma edule and Cerastoderma glaucum species. Numbers at the nodes correspond to Bayesian posterior probabilities
Discussion
This work provides the nucleotide sequences of the 5S rDNA and the ITS region of four European Donax species, describes and characterizes for the first time in this group of organisms the general characteristics of these sequences and analyses their variation.
Regarding the 5S rDNA of the four wedge clams studied show, at least in part, the conventional tandem arrangement, as deduced from successful amplification using contiguous primers with opposite orientation. Moreover, the BLASTn analysis showed that the 5S rRNA genes are separated from one another by a NTS region; the coding region was assigned to 120 bp in the four species and the non-coding spacer to the remaining sequence. The length of the characterized repeat units presented little variation among species (5–32 bp), except D. trunculus with a repeat unit of 277–285 bp and differing from the rest of the Donax species in 170–210 bp. Compared to other bivalve species, the Donax 5S rDNA units are among the shortest with the scallops Aequipecten opercularis (433–465 bp), Mimachlamys varia (453–455 bp), Hinnites distortus (451 bp) and Pecten maximus (463 bp) [42, 55] and the razor clams Ensis arcuatus (420 bp), Ensis siliqua (422 bp), Ensis directus (443 bp) and Ensis macha (434 bp) [24]; although the cockles C. edule and C. glaucum have a repeat unit of 544–546 and 539–568 bp [26, 43], respectively; the oysters about 1100 bp in Crassostrea and 2000 bp in Ostrea [9, 10]; and the mussels Mytilus edulis, Mytilus galloprovincialis and Mytilus trossulus have three types,~260 bp (α band), ~770 bp (β band) and ~1000 bp (γ band) in length, the mussel M. californianus has other three types ~240 bp (small-β band), ~730 bp (β band) and ~980 bp (γ band), and the mussel M. coruscus has a repeat unit of ~300 bp [27]. These differences in size are due to the NTS and have been very useful to differentiate among wedge clams and from other bivalves when morphological criteria are difficult, for instance processed samples, samples without shell or during the larval stage [22, 72]. Despite the fact that Donax species display length differences in the spacer region and a high sequence divergence deduced from the difficulty in obtaining unambiguous sequence alignments, all four species studied show similar GC content (38.20–43.35%), with higher values in the gene (53.40–55.00%) than in the NTS (33.10–39.10%). This difference between coding and spacer region not only occurs in several bivalve species previously studied, such as cockles, mussels and scallops [26, 27, 44, 55], but also in species of crustaceans [70] and fish [85, 90], which have AT rich spacers (73% and > 57%, respectively). On the contrary, the mammalian 5S NTS has been shown to be GC rich (> 60%) [91, 92], and the spacer of the oocyte-type 5S rDNA of Xenopus is AT rich but that of the somatic type is GC rich [73].
The four Donax 5S rDNA units consisted of a coding region linked to a spacer without any other coding sequence associated. This result agree with that observed in cockles [26] and scallops [55], but contrasts with alternative arrangements that have been described, such as the linkage between 5S rDNA and small nuclear RNA (snRNA) in the crustacean Asellus aquaticus [71], the oysters Crassostrea gigas and Crassostrea angulata [9] and the sole Solea senegalensis [56], or the linkage of 5S gene with histone genes in the mussel M. galloprovincialis [16]. Although the use of other PCR amplification conditions or the analysis of a genomic library could reveal 5S rDNA units linked to other multigene families, it is not surprising that the Donax 5S rDNA arrangement differ from other bivalve or species, as the 5S rRNA gene linkages seem to be repeatedly established and lost during the evolution of eukaryotic genomes [14].
The ICRs involved in the transcription of 5S rDNA and the sequence elements box A, IE, and box C were identified in the four Donax species (Fig. 1). Furthermore, all 5S rDNA sequences showed TATA-like motifs upstream of the coding region and they were very similar to that reported in B. mori [62], in N. crassa [98], D. melanogaster [88] and several fish species [59]. Although functional assays are necessary to know the role of these TATA-like motifs, according to the position, they are good candidates for the interaction with TFIIIB for being located near the gene [26]. Recently, Raha et al. [81] have proposed that this region could be involved in RNA pol III transcription together with RNA pol II-like transcriptional factors. Nevertheless, it was less conserved in D. trunculus and D. variegatus because one insertion (T(G)ATATA) and a point mutation (TATTTA) occurred within, respectively, as it happens in cockle, razor clam and scallop species [101].Therefore, as previous authors indicate [101], this could imply that in these molluscan groups i) the 5S rDNA transcription could not precisely be regulated by RNA pol II–like transcriptional factors, ii) they could present lower transcriptional activities, or iii) they do not require the same level of sequence specificity. Moreover, all 5S rDNA sequences displayed a T-rich stretch potentially related to transcription termination [2, 32, 40].
Regarding the ITS region, the lengths determined for both ITS1 (400–542 bp) and ITS2 (254–316 bp) in the four Donax species are in line with those of other bivalve species. Average ITS1 length was 461.9 bp, and GC content was 61.5%, values very similar to those obtained by Chow et al. [5], who studied the ITS1 of several marine animals and reported wide data regarding length and GC content for marine mollusc species (in Mollusca average ITS length was 492.5 bp, and GC content was 55.9%). The ITS1 and ITS2 lengths for the clams Venerupis pullastra, Ruditapes decussatus and Ruditapes philippinarum ranged between 600 and 715 and 316–396 pb, respectively [19]. The ITS1 and ITS2 of four scallops (A. opercularis, M. varia, H. distortus, and P. maximus) are 209–277 and 270–294 bp [45, 103], respectively; and their GC content was 43–49% and 44–49%. They ranged between 367 and 514 and 317–446 bp in the Unionoidea species Unio pictorum, Unio tumidus, Unio crassus, Anodonta anatina, Anodonta cygnea, Pseudanodonta complanata, and Margaritifera margaritifera [49]. In the Veneridae species Meretrix meretrix, Cyclina sinensis, Mercenaria mercenaria, Protothaca jedoensis, Dosinia corrugata and R. philippinarum, ITS1 and ITS2 length were 522–900 and 281–412 bp, respectively; and their GC content were 57.66–65.62% and 65.21–67.87% [3]. In the two cockles C. edule and C. glaucum, ITS1 and ITS2 length ranged between 226 and 251 and 305–325 bp, and their GC content was 52–62% and 61–63%, respectively [28]. Data on ITS1 and ITS2 length in the razor shell E. directus ranged between 484 and 510 and 295–299 bp, and their average GC content were 58.9% for ITS1 and 63% for ITS2 [100]. Thus, Donax species ITS length and GC content were similar to those found in other bivalve species. Just as in the clams V. pullastra, R. decussatus and R. philippinarum [19] and other Veneridae species [3], the ITS1 Donax spacer is longer than ITS2, while in other bivalves the two spacers differ by < 100 bp [28, 49, 51]. As bivalve data accumulate, it seems that there are few restrictions that affect the variation in spacer length, since any type of the following situations may occur: ITS1 and ITS2 of similar size, ITS1 longer than ITS2 and ITS2 longer than ITS1 [28]. ITS GC content in Donax species is similar to that in venerids, E. directus and Cerastoderma species, as would be expected considering that scallops are Pteriomorphia bivalves, and venerids, Ensis and Cerastoderma species are Heterodonta. The high GC content of the ITS1 and ITS2 contrasted with the very low GC content of the NTS. This could be due to the fact the NTS region is not transcribed or folded into a secondary structure, whereas both ITS1 and ITS2 are transcribed and have known secondary structures. Maybe the high GC content is related to secondary structure stability. The length showed here for the 5.8S gene (157 bp) was previously described for the ocean quahog Arctica islandica [11], the four scallops studied by Insua et al. [45], and the six Veneridae species studied by Cheng et al. [3]; although sizes of 158 bp [19], 156 bp [51], and 158–161 bp [28] were reported in some species, but all of them are in line with the average length of eukaryote 5.8S rRNA of about 160 bp deduced from direct sequencing [66]. As expected for a high conserved sequence, the GC content of the 5.8S gene did not show variation (57.3%) and the values correspond to those observed in bivalves [28] and other animals [68, 94, 104].
The four Donax species showed intraindividual variation mainly in the spacers, ITS1 being more variable than ITS2 in D. semistriatus and D. variegatus, as evidenced by the number of variable sites in the sequence alignments and the distance values in pairwise comparisons. This is in line with that observed in other organisms such as Drosophila [89], Similium damnosum [93], and Cerastoderma [28]. By the contrast, ITS2 being more variable than ITS1 in D. trunculus and D. vittatus, as in scallop species [45] and with similar values to that described in ITS2 in the pearl oysters Pinctada martensi, Pinctada maxima, Pinctada margaritifera, Pinctada chemnitzi, Pinctada nigra, and Pteria penguin [36]. Nevertheless, intraindividual variation of the ITS sequences for D. variegatus was minimal or almost nonexistent as in the M. varia and P. maximus scallop species [45]. Therefore, it seems that intradindividual variation in D. variagatus is more moderate than that showed in the other three Donax species and that described in some other animal species [29, 68, 93, 104]. Globally, both ITS1 and ITS2 show sequence variation among wedge clams, with sequence similarity of ITS2 higher than that of ITS1 across species. However, blocks highly conserved across the Donax species were distinguished both in ITS1 and ITS2, which may suggest that they play a role in rRNA processing.
Overall, 5S and ITS sequences show higher values of nucleotide diversity (D. trunculus: 0.019–0.022; D. vittatus: 0.022–0.056) than other nuclear (18S, 28S and H3) and mitochondrial (16S and Cytb) markers in the same Donax species (D. trunculus: ~0.005, [23]; D. vittatus: 0–0.007, [23]), possibly due to the high variability of NTS and ITS, even though 5S and 5.8S genes present high conservation degree through species [14, 38], but also they are of smaller length. These results are in line with other marine species such as Hexaplex trunculus [33] where 5S was more variable than mitochondrial sequences (12S, 16S and COI).
The phylogenetic analyses inferred from 5S rDNA sequences provides a similar tree to that based on the 13 protein-coding genes of mitochondrial genome of the same species [20], the phylogeny based on several mitochondrial (16S, COI and Cytb) and nuclear (18S, 28S and H3) genes [21], and the phylogenetic tree derived from the mitochondrial COI gene [30]. This is in accordance with other bivalve studies where phylogenies have been successfully reconstructed by using the 5S region (e.g. [55, 101]). On the other hand, ITS phylogeny displays a different topology, but in all cases D. semistriatus and D. vittatus species are grouped in the same clade when markers of different nature are used [20, 21, 30]. In a previous study carried out by Chow et al. [5] who studied the ITS1 in several marine animals was reported that ITS1 has a limited utility for phylogenetic analysis. Anyway, phylogenies based on larger genetic regions, for instance mitogenomes, should thus be preferred.
Due to the variation observed in the 5S rDNA and ITS region among Donax species, these sequences have allowed the identification of reliable molecular markers that have been used to differentiate these wedge clams [22]. In this way, it has possible to develop a proper tool, based on multiplex PCR, which could be easily implemented by the government or private entities to guarantee the correct identification and authentication of commercial seafood products avoiding the unintentional substitution of different wedge clam, or detecting and avoiding fraud, to ensure composition and safety of commercial marine products, to protect consumers’ rights and to achieve other quality objectives, such as a certificate of origin [22]. Furthermore, this technique could be useful for conservation of these marine resources and species differentiation to obtain seed with correct identity [22]. In fact, the use of the 5S rDNA and ITS have been reported to be useful for discrimination of several bivalve species with commercial value, such as clams [19, 39], cockles [26], mussels [13, 37, 86, 97], oysters [10], razor clams [24], scallops [54] and wedge clams [72]. Additionally, these sequences could be studied to provide other genetic resources allowing to undertake further molecular and cytogenetic studies of this important bivalve species. For instance, 5S and ITS sequences could be used as probes in fluorescent in situ hybridization (FISH) experiments to study the possibility of hybridization in four Donax species studied here due to the fact that these species can live on the same beds. These sequences have been studied in the oysters Pinctada fucata and Pinctada maculata [60] and in the clams R. decussatus and R. philippinarum [39] for this same purpose.
Conclusions
This is not only a basic research work, where we describe and characterize, for the first time, the 5S rDNA and the ITS regions in four bivalve molluscs belonging to the genus Donax, but also new data and new knowledge is provided for the scientific community about Donax species. Moreover, sequences provided here have allow to develop a method for authentication of the four European Donax species, and they will allow to undertake further genetic studies.
Abbreviations
- COI:
-
Cytochrome c oxidase subunit I
- ICRs:
-
Internal control regions
- ITS:
-
Internal transcribed spacer
- NTS:
-
Non-transcribed spacer
- RFLPs:
-
Restriction Fragment Length Polymorphisms
References
Bejega García V, González Gómez de Agüero E, Fernández Rodríguez C, Álvarez García JC (2010) Los concheros de O Neixón (Boiro, A Coruña) y Punta Atalaia (San Cibrao, Lugo): Una propuesta de muestreo y excavación de depósitos de la Edad del Hierro y época romana en Galicia, en González Gómez de Agüero, E., Bejega García, V., Fernández Rodríguez, C. y Prieto Fuertes, N. (coord.), Actas de la I Reunión de Arqueomalacología de la Península Ibérica. Spain: Férvedes. 33–42.
Bogenhagen DF, Brown DD. Nucleotide sequences in Xenopus 5S DNA required for transcription terminator. Cell. 1981;24(1):261–70.
Cheng HL, Meng XP, Ji HJ, Dong ZG, Chen SY. Sequence analysis of the ribosomal DNA internal transcribed and 5.8S ribosomal RNA gene in representatives of the clam family Veneridae (Mollusca: Bivalvia). J Shellfish Res. 2006;25(3):833–9.
Chícharo L, Chícharo A, Gaspar M, Alves F, Regala J. Ecological characterization of dredged and non-dredged bivalve fishing areas off South Portugal. J Mar Biol Assoc UK. 2002;82:41–50.
Chow S, Ueno Y, Toyokawa M, Oohara I, Takeyama H. Preliminary analysis of length and GC content variation in the ribosomal first internal transcribed spacer (ITS1) of marine animals. Mar Biotechnol. 2009;11(3):301–6.
Coleman AW, Vacquier VD. Exploring the phylogenetic utility of ITS sequences for animals: a test case for abalone (Haliotis). J Mol Evol. 2002;54:246–57.
Consellería do Mar, Xunta De Galicia. (2018). In Pesca de Galicia - Plataforma tecnolóxica da pesca. http://www.pescadegalicia.gal/estadisticas/. Accessed 5 July 2018.
Cornet M, Soulard C. Chromosome number and karyotype of Donax trunculus L. (Mollusca, Bivalvia, Tellinacea). Genetica. 1990;82:93–7.
Cross I, Rebordinos L. 5S rDNA and U2 snRNA are linked in the genome of Crassostrea angulata and Crassostrea gigas oysters: does the (CT)n.(GA)n microsatellite stabilize this novel linkage of large tandem arrays? Genome. 2005;48(6):1116–9.
Cross I, Rebordinos L. Species identification of Crassostrea and Ostrea oysters by polymerase chain reaction amplification of the 5S rRNA gene. J AOC Int. 2006;89(1):144–8.
Dahlgren TG, Weinberg JR, Halanych KM. Phylogeography of the ocean quahog (Arctica islandica): influences of paleoclimate on genetic diversity and species range. Mar Biol. 2000;137(3):487–95.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Dixon DR, Sole-Cava AM, Pascoe PL, Holland PWH. Periostracal adventitious hairs on spat of the mussel Mytilus edulis. J Mar Biol Assoc UK. 1995;75(2):363–72.
Drouin G, Moniz de Sá M. The concerted evolution of 5S ribosomal genes linked to the repeat unit of other multigene families. Mol Biol Evol. 1995;12(3):481–93.
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Thierer T, Wilson A. Geneious v4.8.5. 2009. Available at: http://www.geneious.com/
Eirín-López JM, Ruiz F, González-Tizón AM, Martínez A, Sánchez L, Méndez J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: first record of a tandemly repeated unit of five histone genes containing an H1 subtype with “orphon” features. J Mol Evol. 2004;58(2):131–44.
Fang BL, De Baere R, Vandenberghe A, De Wachter R. Sequences of the three molluscan 5S ribosomal RNAs confirm the validity of a dynamic secondary structure model. Nucleic Acids Res. 1982;10(15):4679–85.
FAO-FIGIS: Fisheries Global Information System. 2018. Available at: http://www.fao.org/fishery/statistics/global-capture-production/query/en. Accessed 30 Mar 2018.
Fernández A, García T, Asensio L, Rodríguez MA, González I, Hernández PE, Martín R. PCR-RFLP analysis of the internal transcribed spacer (ITS) region for identification of 3 clams species. J Food Sci. 2001;66(5):657–61.
Fernández-Pérez J, Nantón A, Ruiz-Ruano FJ, Camacho JPM, Méndez J. First complete female mitochondrial genome in four bivalve species genus Donax and their phylogenetic relationship within the Veneroida order. PLoS One. 2017a;12:e0184464.
Fernández-Pérez J, Froufe E, Nantón A, Gaspar MB, Méndez J. Genetic diversity and population genetic analysis of Donax vittatus (Mollusca: Bivalvia) and phylogeny of the genus with mitochondrial and nuclear markers. Estuar Coast Shelf Sci. 2017b;197:126–35.
Fernández-Pérez J, Nantón A, Méndez J. An alternative method for rapid and specific authentication of four European Donax species, including D. trunculus, a commercially-important bivalve. Eur Food Res Technol. 2018a;244:1815–20.
Fernández-Pérez J, Nantón A, Arias-Pérez A, Freire R, Martínez-Patiño D, Méndez J. Mitochondrial DNA analyses of Donax trunculus (Mollusca: Bivalvia) population structure in the Iberian Peninsula, a bivalve with high commercial importance. Aquat Conserv Mar Freshwat Ecosyst. 2018b. https://doi.org/10.1002/aqc.2929.
Fernández-Tajes J, Méndez J. Identification of the razor clam species Ensis arcuatus, E. siliqua, E. directus, E. macha, and Solen marginatus using PCR-RFLP analysis of the 5S rDNA region. J Agric Food Chem. 2007;55(18):7278–82.
Fernández-Tajes J, Méndez J. Two different size classes of 5S rDNA units coexisting in the same tandem array in the razor clam Ensis macha: is this region suitable for phylogeographic studies? Biochem Genet. 2009;47(11–12):775–88.
Freire R, Insua A, Méndez J. Cerastoderma glaucum 5S ribosomal DNA: characterization of the repeat unit, divergence with respect to Cerastoderma edule, and PCR-RFLPs for the identification of both cockles. Genome. 2005;48(3):427–42.
Freire R, Arias A, Insua AM, Méndez J, Eirín-López JM. Evolutionary dynamics of the 5S rDNA gene family in the mussel Mytilus: mixed effects of the birth-and-death and concerted evolution. J Mol Evol. 2010a;70(5):413–26.
Freire R, Arias A, Méndez J, Insua A. Sequence variation of the internal transcribed spacer (ITS) region of ribosomal DNA in Cerastoderma species (Bivalvia: Cardiidae). J Molluscan Stud. 2010b;76(1):77–86.
Gandolfi A, Bonilauri P, Rossi V, Menozzi P. Intraindividual and intraspecies variability of ITS 1 sequences in the ancient asexual Darwinula stevensoni (Crustacea: Ostracoda). Heredity. 2001;87(4):449–55.
García-Souto D, Pérez-García C, Pasantes JJ. Are Pericentric inversions reorganizing wedge shell genomes? Genes. 2017;8:370.
Gaspar MB, Santos MN, Vasconcelos P, Monteiro CC. Shell morphometric relationships of the most common bivalve species (Mollusca: Bivalvia) of the Algarve coast (southern Portugal). Hydrobiologia. 2002;477:73–80.
Geiduschek EP, Tocchini-Valentini GP. Transcription by RNA polymerase III. Annu Rev Biochem. 1988;57:873–914.
González-Tizón A, Fernández-Moreno M, Vasconcelos P, Gaspar MB, Martínez-Lage A. Genetic diversity in fishery-exploited populations of the banded murex (Hexaplex trunculus) from the southern Iberian Peninsula. J Exp Mar Biol Ecol. 2008;363:35–41.
González-Tizón A, Martínez-Lage A, Mariñas L, Freire R, Cornudella L, Méndez J. Cytogenetic characterization of Donax trunculus (Mollusca, Bivalvia). Cytogenet Cell Genet. 1998;81:109.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
He M, Huang L, Shi J, Jiang Y. Variability of ribosomal DNA ITS-2 and ITS utility in detecting genetic relatedness of pearl oyster. Mar Biotechnol. 2005;7(1):40–5.
Heath DD, Rawson PD, Hilbish TJ. PCR-based nuclear markers identify alien blue mussel (Mytilus spp.) genotypes on the west coast of Canada. Can J Fish Aquat Sci. 1995;52(12):2621–7.
Hillis DM, Dixon MT. Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol. 1991;66:411–53.
Hurtado SN, Pérez-García C, Morán P, Pasantes JJ. Genetic and cytological evidence of hybridization between native Ruditapes decussatus and introduced Ruditapes philippinarum (Mollusca, Bivalvia, Veneridae) in NW Spain. Aquaculture. 2011;311:123–8.
Huang Y, Maraia RJ. Comparison of the RNA polymerase III transcription machinery in Schizosaccharomyces pombe, Saccharomyces cerevisiae and human. Nucleic Acids Res. 2001;29(13):2675–90.
Huang X, Hu J, Hu X, Zhang C, Zhang L, Wang S, Lu W, Bao Z. Cytogenetic characterization of the bay scallop, Argopecten irradians irradians, by multiple staining techniques and fluorescence in situ hybridization. Genes Genet Syst. 2007;82:257–63.
Insua A, López-Piñón MJ, Méndez J. Characterization of Aequipecten opercularis (Bivalvia: Pectinidae) chromosomes by different staining techniques and fluorescent in situ hybridization. Genes Genet Syst. 1998;73(4):193–200.
Insua A, Freire R, Méndez J. The 5S rDNA of the bivalve Cerastoderma edule: nucleotide sequence of the repeat unit and chromosomal location relative to 18S-28S rDNA. Genet Select Evol. 1999;31:509–18.
Insua A, Freire A, Ríos J, Méndez J. The 5S rDNA of mussels Mytilus galloprovincialis and M. edulis: sequence variation and chromosomal location. Chromosom Res. 2001;9(6):495–505.
Insua A, López-Piñón MJ, Freire R, Méndez J. Sequence analysis of the ribosomal DNA internal transcribed spacer in some scallop species (Mollusca: Bivalvia: Pectinidae). Genome. 2003;46(4):595–604.
Insua A, López-Piñón MJ, Freire R, Méndez J. Karyotype and chromosomal location of 18S-28S and 5S ribosomal DNA in the scallops Pecten maximus and Mimachlamys varia (Bivalvia: Pectinidae). Genetica. 2006;126:291–301.
Jansen G, Devaere S, Weekers PHH, Adriaens D. Phylogenetic relationships and divergence time estimate of African anguilliform catfish (Siluriformes: Clariidae) inferred from ribosomal gene and spacer sequences. Mol Phylogenet Evol. 2006;38(1):65–78.
Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN. Mammalian protein metabolism. Nueva York: Academic press; 1969. p. 21–123.
Källersjö M, Von Proschwitz T, Lundberg S, Eldenäs P, Erséus C. Evaluation of ITS rDNA as a complement to mitochondrial gene sequences for phylogenetic studies in freshwater mussels: an example using Unionidae from North-Western Europe. Zoologica Scripta. 2005;34(4):415–24.
Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9(4):286–98.
Kenchington E, Bird CJ, Osborne J, Reith M. Novel repeat elements in the nuclear ribosomal RNA operon of the flat oysters Ostrea edulis C. Linnaeus, 1758 and O. angasi Sowerby, 1978. J Shellfish Res. 2002;21:679–705.
Librado P, Rozas J. DnaSP v5: software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Long EO, Dawid IB. Repeated genes in eukaryotes. Annu Rev Biochem. 1980;49:727–64.
López-Piñón MJ, Insua A, Méndez J. Identification of four scallop species using PCR and restriction analysis of the ribosomal DNA internal transcribed spacer region. Mar Biotechnol. 2002;4(5):495–502.
López-Piñón MJ, Freire R, Insua A, Méndez J. Sequence characterization and phylogenetic analysis of the 5S ribosomal in some scallop (Bivalvia: Pectinidae). Hereditas. 2008;145(1):9–19.
Manchado M, Zuasti E, Cross I, Merlo A, Infante C, Rebordinos L. Molecular characterization and chromosomal mapping of the 5S rRNA gene in Solea senegalensis: a new linkage to the U1, U2 and U5 small nuclear RNA genes. Genome. 2006;49:79–86.
Marie AD, Lejeusne C, Karapatsiou E, Cuesta JA, Drake P, Macpherson E, Bernatchez L, Rico C. Implications for management and conservation of the population genetic structure of the wedge clam Donax trunculus across two biogeographic boundaries. Sci Rep. 2016;6:39152.
Martínez A, Mariñas L, González-Tizón A, Méndez J. Cytogenetic characterization of Donax trunculus (Bivalvia: Dionacidae) by means of karyotyping, fluorochrome banding and fluorescent in situ hybridization. J Molluscan Stud. 2002;68:393–6.
Martins C, Galetti PM. Two 5S rDNA arrays in Neotropical fish species: is it a general rule for fishes? Genetica. 2001;111:439–46.
Masaoka T, Kobayashi T. Natural hybridization between Pinctada fucata and Pinctada maculata inferred from internal transcribed spacer regions of nuclear ribosomal genes. Fish Sci. 2005;71:829–36.
Molares J, Parada JM, Navarro-Pérez E, Fernández A. Variabilidad interanual de las ventas de los principales recursos marisqueros de Galicia y su relación con las condiciones ambientales. Rev Gal Rec Mar. 2008;2:1–42.
Morton DG, Sprague KU. In vitro transcription of a silkworm 5S RNA gene requires an upstream signal. Proc Natl Acad Sci U S A. 1984;81(17):5519–22.
Nantón A, Arias-Pérez A, Freire R, Fernández-Pérez J, Nóvoa S, Méndez J. Microsatellite variation in Donax trunculus from the Iberian Peninsula, with particular attention to Galician estuaries (NW Spain). Estuar Coast Mar Sci. 2017;197:27–34.
Nantón A, Arias-Pérez A, Méndez J, Freire R. Characterization of nineteen microsatellite markers and development of multiplex PCRs for the wedge clam Donax trunculus (Mollusca: Bivalvia). Mol Biol Rep. 2014;41:5351–7.
Nantón A, Freire R, Arias-Pérez A, Gaspar MB, Méndez J. Identification of four Donax species by PCR-RFLP analysis of cytochrome c oxidase subunit I (COI). Eur Food Res Technol. 2015a;240:1129–33.
Nazar RN. The ribosomal 5.8S RNA: eukaryotic adaptation or processing variant? Can J Biochem Cell Biol. 1984;62:311–20.
Nei M. Molecular evolutionary genetics. New York: Columbia Univ. Press; 1987.
Odorico DM, Miller DJ. Variation in the ribosomal internal transcribed spacers and 5.8S rDNA among five species of Acropora (Cnidaria: Scleractinia): patterns of variation consistent with reticulate evolution. Mol Biol Evol. 1997;14(5):465–73.
Özden Ö, Erkan N, Deval MC. Trace mineral profiles of the bivalve species Chamelea gallina and Donax trunculus. Food Chem. 2009;113:222–6.
Pelliccia F, Barzotti R, Volpi EV, Bucciarelli E, Rocchi A. Nucleotide sequence and chromosomal mapping of the 5S rDNA repeat of the crustacean Proasellus coxalis. Genome. 1998;41(1):129–33.
Pelliccia F, Barzotti R, Bucciarelli E, Rocchi A. 5S ribosomal and U1 small nuclear RNA genes: a new linkage type in the genome of a crustacean that has 3 different tandemly repeated units containing 5S ribosomal sequences. Genome. 2001;44:331–5.
Pereira AM, Fernández-Tajes J, Gaspar MB, Méndez J. Identification of the wedge clam Donax trunculus by a simple PCR technique. Food Control. 2012;23(1):268–70.
Peterson RC, Doering JL, Brown DD. Characterization of two Xenopus somatic 5S DNAs and one minor oocyte-specific 5S DNA. Cell. 1980;20(1):131–41.
Petrović V, Pérez-García C, Pasantes JJ, Satović E, Prats E, Plohl M. A GC-rich satellite DNA and karyology of the bivalve mollusk Donax trunculus: a dominance of GC-rich heterochromatin. Cytogenet Genome Res. 2009;124:63–71.
Petrović V, Plohl M. Sequence divergence and conservation in organizationally distinct subfamilies of Donax trunculus satellite DNA. Gene. 2005;362:37–43.
Pieler T, Hamm J, Roeder RG. The 5S gene internal control region is composed of three distinct sequence elements, organized as two functional domains with variable spacing. Cell. 1987;48(1):91–100.
Pinhal D, Yoshimura TS, Araki CS, Martins C. The 5S rDNA family evolves through concerted and birth-and-death evolution in fish genomes: an example from freshwater stingrays. BMC Evol Biol. 2011;11:151.
Plohl M, Cornudella L. Characterization of a complex satellite DNA in the mollusc Donax trunculus: analysis of sequence variations and divergence. Gene. 1996;169:157–64.
Plohl M, Cornudella L. Characterization of interrelated sequence motifs in four satellite DNAs and their distribution in the genome of the mollusc Donax trunculus. J Mol Evol. 1997;44:189–98.
Plohl M, Prats E, Martínez-Lage A, González-Tizón A, Méndez J, Cornudella L. Telomeric localization of the vertebrate-type hexamer repeat, (TTAGGG)(n), in the wedgeshell clam Donax trunculus and other marine invertebrate genomes. J Biol Chem. 2002;277:19839–46.
Raha D, Wan Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M. Close association of RNA polymerase II and many transcription factors with pol III genes. Proc Natl Acad Sci U S A. 2010;107:3639–44.
Rico C, Cuesta JA, Drake P, Macpherson E, Bernatchez L, Marie AD. Null alleles are ubiquitous at microsatellite loci in the wedge clam (Donax trunculus). PeerJ. 2017;5:e3188.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: effective Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Rufino MM, Gaspar MB, Pereira AM, Maynou F, Monteiro CC. Ecology of megabenthic bivalve communities from sandy beaches on the south coast of Portugal. Sci Mar. 2010;74(1):163–78.
Sajdak SL, Reed KM, Phillips RB. Intraindividual and interspecies variation in the 5S rDNA of coregonid fish. J Mol Evol. 1998;46(6):680–8.
Santaclara FJ, Espiñeira M, Cabado AG, Aldaroso A, Gonzalez-Lavín N, Vieites JM. Development of a method for the genetic identification of mussel species belonging to Mytilus, Perna, Aulacomya, and other genera. J Agric Food Chem. 2006;54(22):8461–70.
Satovic E, Plohl M. Tandem repeat-containing MITEs in the clam Donax trunculus. Genome Biol Evol. 2013;5:2549–59.
Sharp SJ, Garcia AD. Transcription of the Drosophila melanogaster 5S RNA gene requires an upstream promoter and four intragenic sequence elements. Mol Cell Biol. 1988;8(3):1266–74.
Schlötterer C, Tautz D. Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Curr Biol. 1994;4(9):777–83.
Sola L, Rossi AR, Annesi F, Gornung E. Cytogenetic studies in Sparus auratus (Pisces, Perciformes): molecular organization of 5S rDNA and chromosomal mapping of 5S and 45S ribosomal genes and of telomeric repeats. Hereditas. 2003;139(3):232–6.
Suzuki H, Moriwaki K, Sakurai S. Sequences and evolutionary analysis of mouse 5S rDNAs. Mol Biol Evol. 1994;11(4):704–10.
Suzuki H, Sakurai S, Matsuda Y. Rat 5S rDNA spacer sequences and chromosomal assignment of the genes to the extreme terminal region of chromosome 19. Cytogenet Cell Genet. 1996;72:1–4.
Tang J, Toè L, Back C, Unnasch TR. Intra-specific heterogeneity of the rDNA internal transcribed spacer in the Simulium damnosum (Diptera: Simuliidae) complex. Mol Biol Evol. 1996;13:244–52.
Tautz D, Hancock JM, Webb DA, Tauzt C, Dover G. Complete sequences of the rRNA genes of Drosophila melanogaster. Mol Biol Evol. 1988;5(4):366–76.
Thébaud O, Verón G, Fifas S. Incidences des épisodes d’efflorescences de micro algues toxiques sur les écosystèmes et sur les pêcheries de coquillages en baie de Douarnenez. In: Rapport Ifremer R.INT.DCB/DEM - DCB/STH/UDPP 05-010- Brest, France; 2005. p. 88.
Theologidis I, Fodelianakis S, Gaspar MB, Zouros E. Doubly uniparental inheritance (DUI) of mitochondrial DNA in Donax trunculus (Bivalvia: Donacidae) and the problem of its sporadic detection in Bivalvia. Evolution. 2008;62:959–70.
Toro JE. Molecular identification of four species of mussels from southern Chile by PCR-based nuclear markers: the potential use in studies involving planktonic surveys. J Shellfish Res. 1998;17:1203–5.
Tyler BM. Transciption of Neurospora crassa 5S rRNA genes requires a TATA box and three internal elements. J Mol Evol. 1987;196:801–11.
Vierna J, González-Tizón A, Martínez-Lage A. Long-term evolution of 5S ribosomal DNA seems to be driven by birth-and-death processes and selection in Ensis razor shells (Mollusca: Bivalvia). Biochem Genet. 2009;47:635–44.
Vierna J, Martínez-Lage A, González-Tizón AM. Analysis of ITS1 and ITS2 sequences in Ensis razor shells: suitability as molecular markers at the population and species levels, and evolution of these ribosomal DNA spacers. Genome. 2010;53(1):23–34.
Vizoso M, Vierna J, Gónzález-Tizón AM, Martínez-Lage A. The 5S rDNA gene family in mollusks: characterization of transcriptional regulatory regions, prediction of secondary structures, and long-term evolution, with special attention to Mytilidae mussels. J Heredity. 2011;102(4):433–47.
Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–13.
Wang S, Zhang L, Zhan A, Wang X, Liu Z, Hu J, Bao Z. Patterns of concerted evolution of the rDNA family in a natural population of zhikong scallop, Chlamys farreri. J Mol Evol. 2007;65:660–7.
Wesson DM, Porter CH, Collins FH. Sequence and secondary structure comparisons of ITS rDNA in mosquitoes (Diptera: Culicidae). Mol Phylogenet Evol. 1992;1:253–69.
Yao H, Song J, Liu C, Luo K, Han J, Li Y, Pang X, Xu H, Zhu Y, Xiao P, Chen S. Use of ITS2 region as the universal DNA barcode for plants and animals. PLoS One. 2010;5(10):e13102.
Zeichen MM, Agnesi S, Mariani A, Maccaroni A, Ardizzone GD. Biology and population dynamics of Donax trunculus L. (Bivalvia: Donacidae) in the south Adriatic coast (Italy). Estuar Coast Shelf Sci. 2002;54:971–82.
Acknowledgements
We would like to thank Dra D. Martínez Patiño and S. Nóvoa from Centro de Cultivos Marinos de Ribadeo – CIMA (Xunta de Galicia) and Dr. M. Gaspar from Instituto Português do Mar e da Atmosfera – IPMA (Portugal) for supplying the samples. This work was supported by the Ministerio de Economía y Competitividad (Spain) through project AGL2016-75288-R AEI/FEDER, UE.
Funding
This work was funded by the Ministerio de Economía y Competitividad (Spain) through project AGL2016–75288-R AEI/FEDER, UE. This funding source had no role in the design of this study and collection, and will not have any role during its execution, analyses, interpretation of the data, writing the manuscript, or decision to submit results.
Availability of data and materials
Nucleotide sequences are available without restriction and samples could be requested directly from the authors. All data generated or analysed during this study are included in this article.
Author information
Authors and Affiliations
Contributions
JM conceived the idea and JFP and AN designed the study. JFP conducted laboratory work, developed the analysed the data and wrote the paper. All authors discussed the results, read and approved the final version of the manuscript for publishing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Compliance with ethical standards. Field work was conducted in accordance with local legislation and with regulations and guidelines established by the University of A Coruña. No endangered or protected species were involved.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Alignment of the 5S rDNA sequences of the four European Donax species. (FAS 49 kb)
Additional file 2:
Alignment of the ITS sequences of the four European Donax species. (FAS 74 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fernández-Pérez, J., Nantón, A. & Méndez, J. Sequence characterization of the 5S ribosomal DNA and the internal transcribed spacer (ITS) region in four European Donax species (Bivalvia: Donacidae). BMC Genet 19, 97 (2018). https://doi.org/10.1186/s12863-018-0684-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-018-0684-x