
Abstract
Background
The genus Ligusticum belongs to Apiaceae, and its taxonomy has long been a major difficulty. A robust phylogenetic tree is the basis of accurate taxonomic classification of Ligusticum. We herein used 26 (including 14 newly sequenced) plastome-scale data to generate reliable phylogenetic trees to explore the phylogenetic relationships of Chinese Ligusticum.
Results
We found that these plastid genomes exhibited diverse plastome characteristics across all four currently identified clades in China, while the plastid protein-coding genes were conserved. The phylogenetic analyses by the concatenation and coalescent methods obtained a more robust molecular phylogeny than prior studies and showed the non-monophyly of Chinese Ligusticum. In the concatenation-based phylogeny analyses, the two datasets yielded slightly different topologies that may be primarily due to the discrepancy in the number of variable sites.
Conclusions
Our plastid phylogenomics analyses emphasized that the current circumscription of the Chinese Ligusticum should be reduced, and the taxonomy of Ligusticum urgently needs revision. Wider taxon sampling including the related species of Ligusticum will be necessary to explore the phylogenetic relationships of this genus. Overall, our study provided new insights into the taxonomic classification of Ligusticum and would serve as a framework for future studies on taxonomy and delimitation of Ligusticum from the perspective of the plastid genome.
Similar content being viewed by others

Background
Ligusticum L., belonging to the Apiaceae family, has long been known for its medicinal values. The Chinese pharmacopeia [1] records that the dried rhizomes or roots of L. sinense Oliv. or L. jeholense Nakai et Kitag. can dispel wind, disperse cold, remove dampness, and relieve pain, and thus can be used for wild-cold, parietal headache, and rheumatism arthralgia. In addition, the essential oil and supercritical fluid (SFE-CO2) extract of L. pteridophyllum Franch. rhizome have significantly insecticidal properties, and for this reason, can be developed as a more environmentally benignant insecticide [2]. The Ligusticum genus has a broad circumscription where it comprises 40–60 species and is distributed predominantly in Asia, Europe, and North America [3,4,5]. Forty Ligusticum species have been identified (35 species are endemic) in China with most inhabiting the alpine and subalpine belt of Southwestern China, and only a few species distributed in the mountainous areas of Northern China [5, 6].
Ligusticum is one of the most complex genera in Apiaceae, and the taxonomy remains uncertain [5], resulting largely from the diversity of flowers, leaves, bracteoles, and mericarps [5, 7, 8]. Its relationships with allied genera Cnidium, Hymenidium, Pachypleurum, Paraligusticum, Rupiphila, Selinum, Tilingia, and Ligusticopsis are still not elucidated clearly [5]. Ligusticum has long been of interest to many plant taxonomists and numerous studies have been reported, such as on pollen morphology [9], karyological studies [10], cladistic analysis [11], leaf epidermal morphology [12], fruit features [13], and molecular phylogeny [8, 14]. Early years ago, Pu [6] mainly focused on bracteoles, in conjunction with fruits and palynological characters to divide Ligusticum into two sections: L. section Ligusticum L. and L. section Pinnatibracteola Pu. Yet this split has not been adopted by other scholars and is not reflected in later molecular phylogeny [8, 15, 16]. Many molecular phylogenetic studies have implied the non-monophyly of Ligusticum [14,15,16,17,18,19,20,21], and recent studies identified six clades within Ligusticum: Acronema Clade, Conioselinum chinense Clade, Pyramidoptereae, Selineae, Sinodielsia Clade, and East-Asia Clade [8]. So far, all molecular phylogenetic analyses are based on smaller datasets (a single or a few genes), except for Ren et al. [16] using plastome-scale datasets. Nevertheless, few Ligusticum species were involved in this plastid phylogenomics study. Hence, a greater taxon sampling is indispensable to confirm the phylogenetic position of Ligusticum.
Next-generation sequencing technology provides more DNA sequencing data than before and can be employed for phylogenetic studies within angiosperms [22]. Meanwhile, plastome-scale data has been used successfully to address phylogenetic problems at various taxonomic levels. For example, Li et al. [23] used 2881 plastid genomes to construct angiosperm phylogeny and date the origin of the crown angiosperms to the Upper Triassic. Wang et al. [24] constructed the phylogeny of Angelica and demonstrated the power of plastid phylogenomics in resolving the phylogeny of this complex genus. Wen et al. [25] revealed a new backbone relationship of Apioideae from plastid phylogenomic analysis. At present, there are two major methods to construct phylogenetic trees: the concatenation method and the coalescent method. Generally, the coalescent method can construct a phylogenetic tree more accurately than the concatenation method, and the concatenation method may produce spuriously high bootstrap support but topologically incorrect phylogenetic trees with the addition of more data [26, 27]. Recent studies have shown that it is necessary to construct the phylogenetic tree of plastid protein-coding genes by the coalescent method [28,29,30]. Hence, we utilized these two methods to estimate the phylogeny of Ligusticum.
Here, 26 Ligusticum plastomes (including 14 newly sequenced) representing all four currently identified clades in China were used for molecular evolutionary analysis and phylogenetic reconstruction. Our aims were to (1) describe the diversity of plastome characteristics and the evolutionary pattern of plastid protein-coding genes within Ligusticum; (2) obtain a robust Ligusticum phylogeny and assess the power of the plastome-scale data for resolving the phylogeny of this genus; (3) comment on the current taxonomy of Ligusticum in China based on the plastome sequences.
Results
Features of Ligusticum plastomes
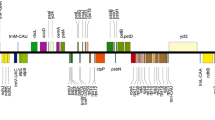
The Illumina NovaSeq sequencing yielded between 32,046,626 (L. tachiroei) and 50,722,538 (L. litangense) clean reads from the 14 newly sequenced species, with the mean base coverage ranging from 278× (L. jeholense_YX) to 1950× (L. nematophyllum) (Table 1). Among the four clades, the Ligusticum plastomes were variable (Table 1, Fig. 1). Selineae and Sinodielsia Clade had similar plastome sizes and IR/SC borders. The total sequence length varied from 146,443 bp (L. pteridophyllum) to 148,608 bp (L. nematophyllum) except for L. tenuissimum (158,500 bp) and L. angelicifolium (163,802 bp). The IR/SC borders were the same except for the IR/LSC borders of the above two plastomes. For Acronema Clade and East-Asia Clade, the plastome sizes and the IR/SC borders were highly similar. The total sequence length varied from 155,455 bp (L. tachiroei) to 157,040 bp (L. weberbauerianum), and the IR/SC borders were identical except for the slightly different IRb/SSC border found in L. tachiroei. Among these 26 plastomes, L. angelicifolium had the longest plastome length (163,802 bp), which is caused by the significant expansion of IR regions (34,719 bp) (Table 1, Fig. 1, Additional file 1: Fig. S1). The LSC/IRb border extended into the petB gene and the IRa/LSC border extended into petB-trnH-GUG in this plastome, whereas the LSC/IRb border extended into ycf2, rpl22, or rps19 gene, and the IRa/LSC border extended into trnL-CAA-trnH-GUG, rps19-trnH-GUG, or rpl2-trnH-GUG for the other Ligusticum species (Fig. 1). All Ligusticum plastomes possessed 128–145 genes, comprising 84–100 protein-coding genes, 36–37 tRNA genes, and eight rRNA genes (Table 1, Additional file 1: Fig. S1). Ligusticum species among the four clades possessed nearly identical GC content not only in whole plastome (37.4–37.6%) but also in LSC (35.7–36.0%) and SSC (30.9–31.4%) (Table 1, Fig. 2). However, the GC content of L. angelicifolium (40.8%) was significantly lower than other species, which may be caused by the longest IR region (Figs. 1, 2).

The total length stacked bar chart of 26 Ligusticum plastomes composed of four regions (LSC, IRb, SSC, and IRa). The numbers on the bar represent the length of the four regions. A–K Represents the genes at IR/SC borders. A ycf2; B petB; C rpl22; D rps19; E ycf1/ndhF; F ycf1; G trnN-GUU-ndhF; H trnL-CAA-trnH-GUG; I petB-trnH-GUG; J rps19-trnH-GUG; K rpl2-trnH-GUG. All the SSC/IRa borders are ycf1, which is indicated by asterisks

Comparison of the GC content (GC%) of 26 Ligusticum plastomes using a radar-plot. From inside to out: SSC GC%, LSC GC%, Total GC%, IR GC%, and rRNA GC%. The background colors of purple, green, blue, and pink represent Selineae, Sinodielsia Clade, Acronema Clade, and East-Asia Clade, respectively
Molecular evolutionary pattern of plastid protein-coding genes
Fifty-three protein-coding sequences (CDSs) of each Ligusticum species were selected to determine the codon usage patterns. Codon usage bias was similar across all Ligusticum species (Additional file 4: Table S2, Fig. 3). We found that 2208–2246 codons (10.4–10.6%) encode Leucine, and 210–222 codons (1.0–1.1%) encode Cysteine, which were the most prevalent and rarest amino acids, respectively. Figure 3 demonstrates that about half of the codons were used more frequently. Specifically, 30 codons were used frequently with RSCU > 1, and all biased codons ended with a purine (A/T) except for TTG (Fig. 3). Within the 53 CDSs, the first and second codon positions had much higher GC content (45.9–46.0% and 38.1–38.3%) than the third codon positions (29.6–30.0%) (Additional file 4: Table S2). We identified 55–60 RNA editing sites for 20–23 protein-coding genes from each Ligusticum species. (Additional file 5: Table S3). Further analysis found that most RNA editing events occurred in the ndh gene (22–24). Although Ligusticum appeared to have a similar pattern of RNA editing, several specific editing sites have been picked out: petD (1 site; only identified in L. involucratum) and rps8 (1 site; only identified in L. jeholense).

The RSCU values of 53 merged protein-coding sequences for 26 Ligusticum plastomes. Color key: the red values indicate higher RSCU values and the blue values indicate lower RSCU values
The ω values of CDSs for 79 plastid protein-coding genes ranged from 0.0001 to 0.9065 (Fig. 4), suggesting conservation of plastid protein-coding genes in Ligusticum. Most genes were under strong purifying selection with a very low ω value (ω < 0.5), yet the ω values in the range of 0.5 to 1.0 (indicating relaxed selection) were observed for seven genes petG, ccsA, rps8, rpl33, psaJ, ycf1, and ycf2 (Fig. 4). However, we found that only three genes (rps8, ycf1, and ycf2) were under relaxed selection due to their significance (P < 0.05) after the likelihood ratio test (LRT) (Additional file 6: Table S4). Nucleotide diversity (Pi) of these 79 CDSs was calculated to assess the sequence divergence level (Additional file 6: Table S4). Among these, Pi values ranged from 0 to 0.02071 (Fig. 4). Six CDSs had relatively higher Pi values, including matK, cemA, ycf1, psbK, ndhF, and atpF genes, which revealed that these genes were more divergent and evolving more rapidly than other genes (Fig. 4). Conversely, CDS of rpl36, psbF, psaI, psbL, psbI, and psbZ genes shared very low Pi values, suggesting that these genes are highly conserved (Fig. 4). Collectively, the low Pi values also indicated that the plastid protein-coding genes were conserved in Ligusticum.

The dN/dS (ω) and nucleotide diversity (Pi) of the 79 protein-coding sequences within 26 Ligusticum plastomes
Phylogenetic relationships
We performed a series of phylogenetic analyses using two datasets (complete plastome sequences and 76 common CDSs) and two methods (concatenation and coalescent-based analyses) for 66 species of Apiaceae (Additional file 7: Table S5). The aligned two datasets were 124,230 bp and 65,979 bp long, with 23,598 and 8932 variable sites, and proportions of 18.99% and 13.54%, respectively. As expected, our analyses obtained robust support at most nodes. All phylogenetic analyses produced largely identical tree topologies, the incongruence mainly occurred in the interspecific relationships within clades, and the relationship between the clades was congruent except for the systematic position of Cachrys Clade (Figs. 5, 6; Additional file 2: Fig. S2). Cachrys Clade was resolved as sister to Sinodielsia Clade + Selineae ((Selineae, Sinodielsia Clade), Cachrys Clade) in the ML tree based on dataset-2 (BS = 54), while it was sister to Apieae in the other four phylogenetic trees with moderate-to-high support (Figs. 5, 6; Additional file 2: Fig. S2). For Ligusticum, it was still a non-monophyletic taxon, and the clades of these species were consistent with previous studies. Although we have enriched the plastome data of Ligusticum, the systematic position of L. pteridophyllum is still unclear in this study. L. pteridophyllum belonged to Sinodielsia Clade based on dataset-1 (BS = 98, PP = 1), while it was resolved as sister to Sinodielsia Clade + Selineae ((Selineae, Sinodielsia Clade), (L. pteridophyllum, L. pteridophyllum_DL)) in the other three phylogenetic trees (BS = 100, PP = 1, LPP = 1) (Fig. 6, Additional file 2: Fig. S2). Given that the variation level of the 76 CDSs and the incongruent topologies of dataset-2 (76 CDSs) obtained by different analyses (ML and BI), as well as the positions of L. pteridophyllum and Cachrys Clade were distinct from dataset-1, we then used 76 CDSs to perform a phylogenetic analysis according to the multi-species coalescent model by ASTRAL v5.7.3 [45] (Fig. 6). Thus, we used this coalescent-based phylogeny and concatenation-based phylogeny (dataset-1) as the basis in this study (Figs. 5, 6).

Phylogenetic relationships inferred from Maximum likelihood (ML) and Bayesian inference (BI) analyses based on 66 complete plastomes within Apiaceae. The bootstrap support values (BS) and posterior probabilities (PP) are listed at each node

Phylogenetic relationships of 66 Apiaceae species inferred from 76 common protein-coding sequences based on the coalescent-based approach using ASTRAL. The local posterior probabilities (LPP) are listed at each node
Compared to the results from concatenation-based phylogeny (dataset-1), the phylogenetic relationship among the clades in the coalescent-based phylogeny was identical, but the interspecific relationships within clades had a few discrepancies (Figs. 5, 6). The 26 Ligusticum accessions were distributed in four clades (Acronema Clade, Selineae, Sinodielsia Clade, and East-Asia Clade) (Figs. 5, 6). Two species (L. tachiroei and L. delavayi) fell into Acronema Clade and formed a clade with strong supports (BS = 100, PP = 1, LPP = 1). L. weberbauerianum (= H. weberbaueriana) and L. litangense fell into East-Asia Clade (BS = 100, PP = 1, LPP = 1). The Sinodielsia Clade is more complicated. In the concatenation-based phylogeny, eight Ligusticum accessions clustered with C. officinale and A. sinensis fell into Sinodielsia Clade with high support (BS = 98, PP = 1). However, Sinodielsia Clade was not recovered as a monophyletic group, because two L. pteridophyllum accessions were resolved as sister to Sinodielsia Clade + Selineae ((Selineae, Sinodielsia Clade), (L. pteridophyllum, L. pteridophyllum_DL)) in the coalescent-based phylogeny (LPP = 1). Most Ligusticum accessions were within Selineae, while they did not form a clade. Two L. thomsonii accessions clustered with S. divaricate, P. praeruptorum, and P. japonicum, and L. angelicifolium clustered with M. pimpinelloideum. Eleven other Ligusticum accessions formed a clade with strong support (BS = 100, PP = 1, LPP = 1).
Discussion
The diversity of plastome characteristics
By combining the 14 newly sequenced plastome sequences with the 12 published sequences of Ligusticum, we can represent all four currently identified clades in China. The Ligusticum plastomes were variable among the four clades, as well as the plastomes of Selineae and Sinodielsia Clade were significantly different from that of Acronema Clade and East-Asia Clade, which might have phylogenetic and taxonomic significance. Previous studies concluded that the total length of angiosperm plastomes is usually influenced by the contraction and expansion of the IRs [31, 32]. Similarly, we noticed that the longer total lengths of six Ligusticum (L. angelicifolium, L. tenuissimum, L. litangense, L. weberbauerianum, L. delavayi, and L. tachiroei) plastomes were determined by the expansion of IRs. We detected four types of LSC/IRb border (ycf2, petB, rpl22, and rps19 genes) and four types of LSC/IRa border (petB-trnH-GUG, trnL-CAA-trnH-GUG, rps19-trnH-GUG, and rpl2-trnH-GUG), which were also reported in Apiaceae and other plant lineages [25, 28, 33,34,35]. Compared to the dynamically shifted LSC/IR border, the SSC/IR border was more conserved, as most SSC/IR borders were ycf1 genes with a few exceptions in the 26 Ligusticum plastomes. In addition to IR border shifts, IR has been significantly increased, reduced, or even eliminated, such as in Pelargonium × hortorum [36], Cephalotaxus oliveri [37], some species of Erodium and Pinaceae [38, 39]. Gene content of Ligusticum was not conserved, mainly due to the increase of gene number caused by the expansion of IRs [40]. For example, L. angelicifolium possessed the most genes. The rpl22, rps3, rpl16, rpl14, rps8, infA, rpl36, rps11, rpoA, and petD genes located in LSC regions of other Ligusticum species, have moved to IR regions to become double-copy genes in L. angelicifolium. GC content of Ligusticum plastomes was close to other Apiaceae [25, 41]. High GC content was observed in IRs, which is probably due to the presence of the four rRNA genes [42, 43] as they had a GC content of up to 54.9–55.3% (Fig. 2).
The evolutionary conservation of plastid protein-coding genes
Codon usage bias is an important evolutionary feature in the genome that can be influenced by many evolutionary processes [44]. Therefore, codon usage bias provides useful information for studying molecular evolution. GC content is generally the product of directional mutation pressure and is a critical factor affecting codon usage [38, 44, 45]. All 26 Ligusticum plastomes had a strong bias toward A/T at the third codon position as observed in other angiosperm species [46, 47]. High AT content in plastomes is the major reason for bias codons ending with A/T [48]. RNA editing is one of the posttranscriptional maturation processes of primary transcripts, which allows nucleotide insertion/deletion and conversion to alter transcripts [49, 50]. The first chloroplast RNA editing was discovered in maize rpl2 transcript, in which an initiation codon ACG changes to AUG [51]. After that, RNA editing has been found in a growing number of higher plant chloroplasts. The ndh genes encode subunits of the plastid NDH (NADH dehydrogenase-like) complex, which contained most RNA editing sites for Ligusticum species. The ndh genes play an important role in mediating cyclic electron flow around photosystem I and facilitating chlororespiration [52]. Therefore, RNA editing on the ndh genes is more likely to ensure the physiological and biochemical processes of the plant. Similar codon usage and RNA editing patterns for 26 Ligusticum plastomes possibly because of the evolutionary conservation of plastomes among angiosperms.
The synonymous and non-synonymous nucleotide substitution pattern is a major indicator in the study of gene evolution. The ratio (ω) of dN/dS is generally interpreted as: purifying selection (ω < 1, especially less than 0.5), positive selection (ω > 1), neutral evolution (ω = 1), whereas ω value close to 1 indicates relaxed selection [53, 54]. All protein-coding genes held low ω and Pi values, suggesting the conservation of plastid genes in Ligusticum. Three genes (rps8, ycf1, and ycf2) were under relaxed selection. The rps8 is one of the genes that encodes a protein for the small ribosomal subunits, therefore essential for the plastid ribosome [55]. The rps8 gene was under positive selection in Curcuma [56]. The plastid gene rps8 RNA editing defect accounted for the low-temperature sensitivity in rice and maize [57, 58]. This indicated that rps8 gene is very important for plant adaptability. The ycf1 gene, the second-largest gene in the plastome, is indispensable for photosynthetic protein import and is therefore vital for plant viability [59]. Positive selection or relaxed selection on ycf1 have been observed in Bulbophyllum [60] and Lennoaceae [61]. ycf2 is a conserved open reading frame with the exact function still unknown, although its putative gene product is a protein of 2280 amino acids [55, 62]. Ligusticum is mainly distributed in the alpine and subalpine regions of Southwest China. Consequently, we speculated that the possible relaxed selection pressure on these three genes may be related to adapting to high-altitude living environments.
Phylogenetic relationships and taxonomic implications
Early studies have shown that the genus Ligusticum was a non-monophyletic group [14,15,16,17,18,19,20,21], and was divided into six clades: Acronema Clade, Conioselinum chinense Clade, Pyramidoptereae, Selineae, Sinodielsia Clade, and East-Asia Clade [8]. Here, the Ligusticum plastomes of 20 species (26 accessions) representing all four currently recognized clades in China were used to reconstruct the phylogenetic trees. We used different datasets and methods to perform phylogenetic analyses to obtain robust phylogenetic relationships of this genus, which revealed that the plastome‐scale data is a promising tool for resolving the phylogeny of the controversial taxon. In the concatenation-based phylogeny analysis, dataset-1 and dataset-2 yielding slightly different topologies may be primarily due to the discrepancy in the number of variable sites. On consideration, we finally decided to use coalescent-based phylogeny and concatenation-based phylogeny (dataset-1) as the basis to explore the phylogeny of Ligusticum.
Among the four clades of Ligusticum, only Sinodielsia Clade was not monophyletic, owing to the two accessions of L. pteridophyllum not being clustered with other Sinodielsia Clade species in the coalescent-based result. This was also observed in previous studies [16]. In fact, the systematic position of L. pteridophyllum has not been correctly described. L. pteridophyllum was once placed in the Selineae [14] or Sinodielsia Clade [8]. Zhou et al. [8] involved more Ligusticum species in their study, and the results were more reliable. Morphologically, L. pteridophyllum does not share several general characteristics of Ligusticum in Selineae, such as pinnate bracteoles, and with fibrous remnant sheaths at the stem bases (Additional file 8: Table S6). Taken together, we agreed that L. pteridophyllum belongs to Sinodielsia Clade, while more species in Sinodielsia Clade should be involved to verify this result. Six other accessions of Ligusticum clustered with C. officinale and formed a clade in Sinodielsia Clade, with C. officinale being closely related to L. sinense. C. officinale has been referred to as L. officinale [8], and the strong cross-hybridization of genomes was found between C. officinale and L. sinense [63].
Selineae contained most Ligusticum species, whereas they did not group together in this tribe. Two accessions of L. thomsonii and L. angelicifolium were not clustered with eleven other accessions of Ligusticum, which may be explained by morphology. The most obvious difference among them is bracteole and fruit. L. thomsonii and L. angelicifolium have linear or lanceolate bracteoles, as well as the prominent dorsal and intermediate ribs, winged lateral ribs. However, the eleven other accessions of Ligusticum have pinnate bracteoles, as well as raised dorsal and intermediate ribs, winged lateral ribs (Additional file 8: Table S6). The genus Ligusticopsis with 14 species was separated from Ligusticum because of its prominent calyx teeth [7], however, these 14 species did not form a monophyletic group. Pimenov [64] proposed several new nomenclarural combinations in Ligusticopsis, which included seven species (L. brachylobum, L. capillaceum, L. daucoides, L. hispidum, L. involucratum, L. likiangense, and L. scapiforme) analysed in this study. We approved this conclusion and suggested that L. oliverianum should be also incorporated into the genus Ligusticopsis based on molecular and morphological evidence (Figs. 5, 6; Additional file 2: Fig. S2; Additional file 8: Table S6).
L. weberbauerianum (= H. weberbaueriana) and L. litangense fell into East-Asia Clade. The two accessions of H. weberbaueriana (= L. weberbauerianum) were sisters in the phylogenetic trees, thus we agreed with this treatment that L. weberbauerianum is a synonym of H. weberbaueriana [65, 66]. Several studies have shown that L. litangense should be placed in Hansenia rather than Ligusticum [64]. L. litangense was related to H. phaea within Hansenia, which implied that L. litangense should be merged into Hansenia [8, 64, 67].
Two species (L. tachiroei and L. delavayi) that were in Acronema Clade formed a clade with strong support. However, the two species should be transferred from Ligusticum to another genus according to prior research [64]. It is worth mentioning that the generic type of Ligusticum (namely, Ligusticum scoticum) was placed in Acronema Clade [8, 14]. In the present study, most of the Ligusticum species were not fell into Acronema Clade, except for L. tachiroei and L. delavayi. Moreover, Zhou et al. [8] found that L. scoticum and L. scoticum subsp. hultenii occurred in Acronema Clade formed a monophyletic group with high support, and they were separated from L. tachiroei and L. delavayi. Consequently, in the light of the plastome’s results, we concluded that the current circumscription of the Chinese Ligusticum should be reduced, which is consistent with Zhou et al.’s [8] study based on ITS sequences.
Ligusticum is one of the most taxonomically difficult taxa within Apiaceae, largely due to the diversity of flowers, leaves, bracteoles, and mericarps. Ligusticum is described as a dustbin genus, as it contains several species that cannot be classified correctly [8]. In addition, fruit is the most important taxonomic character of Ligusticum, yet most species of the genus grow at high elevations with late fruiting. As a perennial herb, the genus sometimes does not blossom and bear fruit in a year, which was encountered many times during our field sampling. These factors make it difficult to sample the fruits of Ligusticum, resulting in a lack of unique taxonomic characters. Thus, the fruits of Ligusticum should also be collected to provide a morphological foundation for the taxonomic revision of Ligusticum. Together with the molecular phylogenetic analyses, including the use of traditional molecular markers and plastome-scale data [8, 14,15,16,17,18,19,20,21], we therefore strongly argue that a revision of Ligusticum taxonomy is necessary. Further studies will require more taxa of Ligusticum and its allied genera, as well as combine molecular and morphological evidences to resolve the taxonomy and delimitation of Ligusticum. Overall, our study provides new insights into the taxonomic classification of Ligusticum and will serve as a framework for future studies on the taxonomy and delimitation of Ligusticum from the perspective of the plastid genome.
Materials and methods
Taxon sampling and DNA extraction
We newly sequenced 14 plastomes, including 13 species covering four clades of Ligusticum. We also recovered 12 plastomes from the NCBI database (https://www.ncbi.nlm.nih.gov/). In total, we sampled 20 species (26 accessions) within Ligusticum (Additional file 3: Table S1). Fresh leaves from adult plants of each newly sequenced species were collected in the field and immediately dried with silica gel for future DNA extraction. These plants are not protected, therefore permission is not required for sample collection. The species identification of the plant material was undertaken by Xingjin He (Sichuan University, Chengdu, China). Voucher specimens were deposited at the herbarium of Sichuan University (Chengdu, China) (Additional file 3: Table S1). Total genomic DNA was extracted from silica-dried leaves with a CTAB protocol [68]. The quality and concentration of the DNA products were assessed using 1% agarose gel electrophoresis and a Quant-iT PicoGreen dsDNA Assay Kit.
Illumina sequencing, assembly, and annotation
The DNA library with an insert size of 400 bp was constructed using the TruSeq DNA Sample Preparation Kits (Illumina) according to the manufacturer’s protocol. The DNA library was sequenced using Illumina NovaSeq platform with an average paired-end read length of 150 bp at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China). The quality of the newly generated sequencing data was assessed using the FastQC v0.11.9 software [69]. The obtained raw reads were adapter-trimmed and quality-filtered by AdapterRemoval v2 (trimwindows = 5 and minlength = 50) [70], yielding at least 5 GB clean reads for each species. Clean reads were then used to perform a de novo assembly by NOVOPlasty v2.6.2 (K-mer = 39) [71]. The seed sequence was the rbcL gene from the reference plastome sequence of L. delavayi (NC_049052) [16]. The annotation of the 14 plastomes was completed using GeSeq [72], and we manually adjusted the positions of start and stop codons and the exon/intron boundaries in Geneious v9.0.2 (Biomatters Ltd., Auckland, New Zealand) against its congeneric species. The 14 newly obtained plastome sequences are available at the GenBank (Accession numbers: MZ532560–MZ532573). The circle plastome map was generated using the online program OrganellarGenomeDRAW (OGDRAW) [73].
Molecular evolutionary analysis
To identify the codon usage patterns, MEGA6 [74] was employed for the codon usage bias analyses using protein-coding genes with CDS lengths greater than 300 bp to avoid sampling bias [75]. The heatmap was drawn using TBtools [76]. The total GC content and the GC content for the first, second, and third codon positions of these CDSs were also calculated by MEGA6 [74]. To reveal the composition and characteristics of RNA editing, the potential RNA editing sites in protein-coding genes of 26 Ligusticum plastomes were predicted using the PREP-Cp program [77] with a cutoff value of 0.8.
To explore the selection patterns on the plastid protein-coding genes, the nonsynonymous (Ka) and synonymous (Ks) nucleotide substitution rates of 79 protein-coding genes were calculated using a site-specific model implemented in Codeml program (seqtype = 1, model = 0, NSsites = 0, 1, 2, 3, 7, 8) [78] of PAML4.9 software [79]. Codon frequencies were determined using the F3 × 4 model and gapped regions were excluded with the parameter “cleandata = 1” option. For PAML analyses, the ML tree constructed using RAxML v8.2.8 [80] based on 79 plastid protein-coding genes was used as the input treefile. Likelihood ratio test (LRT) with a Chi-square distribution was used to confirm the model fit. The Bayes Empirical Bayes (BEB) analysis was used to statistically identify selected sites with posterior probabilities ≥ 95%. The nucleotide diversity (Pi) of the CDSs of 79 protein-coding genes was also calculated using DnaSP v5.1 [81].
Phylogenetic analysis
Sixty-six species of Apiaceae were used to infer the phylogeny of Ligusticum, among which, two Bupleurum species served as the outgroups (Additional file 7: Table S5). Both concatenation and coalescent-based analyses were carried out. For the concatenation-based approach, two datasets were used to conduct the phylogenetic analysis: dataset-1 was the complete plastomes (excluding one inverted repeat region); dataset-2 encompassed the 76 common protein-coding sequences (CDSs) (Genes list used in the phylogenetic analyses was provided in Additional file 7: Table S5). The number of variable sites of the two datasets was calculated by MEGA6 [74]. To avoid duplicate regions increasing the phylogenetic signal, the second IR was removed from the first dataset. Sequence alignment was achieved using the MAFFT v7.221 [82] and ambiguously aligned areas were removed using Gblocks v0.91b [83] with the default setting. The nucleotide sequences of the 76 common CDSs were extracted and then concatenated into a supermatrix using PhyloSuite v1.2.1 [84]. The maximum likelihood (ML) analysis was conducted in RAxML v8.2.8 [80] with 1000 bootstrap replicates and GTRGAMMA model. Bayesian inference (BI) was carried out using MrBayes v3.1.2 [85] with the best-fitting evolutionary model determined by Modeltest v3.7 [86]. The selected models for complete plastomes and 76 common CDSs in BI analyses were TVM + I + G and GTR + I + G, respectively. Markov chain Monte Carlo (MCMC) algorithm was run for 5,000,000 generations, with one tree sampled every 1000 generations. The MCMC reached stationarity when the average standard deviation of the split frequencies was less than 0.01. The initial 25% of the sampled data was discarded as burn-in, and the consensus tree was generated using the remaining trees.
Given that the variation level of different genes (Additional file 7: Table S5), and to provide the best estimate of the phylogeny of Ligusticum, we also undertook a coalescent-based analysis using ASTRAL v5.7.3 [87]. This approach inferred a species tree using individual gene trees. The gene trees were separately generated for 76 CDSs using RAxML v8.2.8 [80] with 500 bootstraps and GTRGAMMA model. The 76 RAxML best ML gene trees were used as input for ASTRAL v5.7.3 [87] to estimate a species tree with local posterior probability (LPP) [88].
Conclusions
In this study, we integrated 26 plastomes (including 14 newly sequenced plastomes) to perform molecular evolutionary analysis and phylogenetic reconstruction. These plastid genomes exhibited diverse plastome characteristics. The analyses of codon usage, RNA editing, dN/dS, and nucleotide variability (Pi), have demonstrated the conservation of the protein-coding genes in Ligusticum. The phylogenetic analyses obtained a more robust molecular phylogeny than prior studies and showed the non-monophyly of Ligusticum containing four clades. Our results emphasized that the current circumscription of the Chinese Ligusticum should be reduced. Wider taxon sampling including related species of Ligusticum will be necessary to explore the phylogenetic relationships of Ligusticum. Overall, our study provided new insights into the phylogenetic relationships of Ligusticum and would serve as a framework for the taxonomy and delimitation studies of this genus.
Availability of data and materials
The newly sequenced plastid genome sequences were deposited into GenBank (MZ532560–MZ532573).
Abbreviations
- BEB:
-
Bayes empirical Bayes
- BI:
-
Bayesian inference
- bp:
-
Base pair
- BS:
-
Branch support
- CDS:
-
Protein-coding sequences
- CTAB:
-
Cetyl trimethylammonium bromide
- IR:
-
Inverted repeat
- LPP:
-
Local posterior probability
- LRT:
-
Likelihood ratio test
- LSC:
-
Large single copy
- MCMC:
-
Markov chain Monte Carlo
- ML:
-
Maximum likelihood
- PP:
-
Posterior probability
- rRNA:
-
Ribosomal RNA
- RSCU:
-
Relative synonymous codon usage
- SSC:
-
Small single copy
- tRNA:
-
Transfer RNA
References
Chinese Pharmacopeia Commission. Pharmacopoeia of the People’s Republic of China Part I. Beijing: China Medical Science Press; 2015.
Qi XJ, Pang X, Cao JQ, Du SS. Comparative analysis on bioactivity against three stored insects of Ligusticum pteridophyllum Franch. rhizomes essential oil and supercritical fluid (SFE-CO2) extract. Environ Sci Pollut R. 2020;27(13):15584–91.
Leute GH. Untersuchungen über den Verwandtschaftskreis der Gattung Ligusticum L. (Umbelliferae) II. Teil. Ann Naturhist Mus Wien. 1970;74:457–519.
Kadereit JW, Bittrich V. Flowering plants. Eudicots: Aapiales, Gentianales (except Rubiaceae). In: The families and genera of vascular plants. Vol. 15. Berlin: Springer; 2018.
Pu FD, Watson MF. Ligusticum L. Flora of China, vol. 14. Beijing: Science Press; 2005. p. 140–51.
Pu FD. A revision of the genus Ligusticum L. (Umbelliferae) in China. Acta Phytotax Sin. 1991;29:385–93.
Leute GH. Untersuchungen über den Verwandtschaftskreis der Gattung Ligusticum L. (Umbelliferae) Teil I. Ann Naturhist Mus Wien. 1969;73:55–98.
Zhou J, Gao YZ, Wei J, Liu ZW, Downie SR. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: rampant polyphyly, placement of the Chinese endemic species, and a much-reduced circumscription of the genus. Int J Plant Sci. 2020;181(3):306–23.
Wang PL, Pu FT, Ma JS. Pollen morphology of the genus Ligusticum from China and its systematic significance. J Syst Evol. 1991;29(3):235–45.
Zhou J, Pu F, Peng H, Pan Y, Gong X. Karyological studies of ten Ligusticum species (Apiaceae) from the Hengduan Mountains Region of China. Caryologia. 2008;61(4):333–41.
Sun N, He XJ, Zhou SD. Morphological cladistic analysis of Ligusticum (Umbelliferae) in China. Nord J Bot. 2008;26(1–2):118–28.
Sun N, He XJ, Zhou SD. Epidermal morphology of Ligusticum (Apiaceae) from China. Ann Bot Fenn. 2010;47(4):261–79.
Chen JP, Liu S, Ma XG, He XJ. Fruit features of twenty species of Ligusticum (Apiaceae) and their taxonomic significance. Acta Botan Boreali-Occiden Sin. 2015;35(8):1574–86.
Downie SR, Spalik K, Katz-Downie DS, Reduron JP. Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant Divers Evol. 2010;128(1):111–36.
Zhou J, Peng H, Downie SR, Liu ZW, Gong X. A molecular phylogeny of Chinese Apiaceae subfamily Apioideae inferred from nuclear ribosomal DNA internal transcribed spacer sequences. Taxon. 2008;57(2):402–16.
Ren T, Li ZX, Xie DF, Gui LJ, Peng C, Wen J, et al. Plastomes of eight Ligusticum species: characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol. 2020;20(1):1–14.
Downie SR, Ramanath S, Katz-Downie DS, Llanas E. Molecular systematics of Apiaceae subfamily Apioideae: phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacer and plastid rpoC1 intron sequences. Am J Bot. 1998;85(4):563–91.
Katz-Downie DS, Valiejo-Roman CM, Terentieva EI, Troitsky AV, Pimenov MG, Lee B, et al. Towards a molecular phylogeny of Apiaceae subfamily Apioideae: additional information from nuclear ribosomal DNA ITS sequences. Plant Syst Evol. 1999;216(3):167–95.
Downie SR, Katz-Downie DS, Watson MF. A phylogeny of the flowering plant family Apiaceae based on chloroplast DNA rpl16 and rpoC1 intron sequences: towards a suprageneric classification of subfamily Apioideae. Am J Bot. 2000;87(2):273–92.
Downie SR, Watson MF, Spalik K, Katz-Downie DS. Molecular systematics of Old World Apioideae (Apiaceae): relationships among some members of tribe Peucedaneae sensu lato, the placement of several island-endemic species, and resolution within the apioid superclade. Can J Bot. 2000;78(4):506–28.
Zhou J, Gong X, Downie SR, Peng H. Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Mol Phylogenet Evol. 2009;53(1):56–68.
Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425(6960):798–804.
Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 2019;5(5):461–70.
Wang M, Wang X, Sun J, Wang Y, Ge Y, Dong W, et al. Phylogenomic and evolutionary dynamics of inverted repeats across Angelica plastomes. BMC Plant Biol. 2021;21(1):1–12.
Wen J, Xie DF, Price M, Ren T, Deng YQ, Gui LJ, et al. Backbone phylogeny and evolution of Apioideae (Apiaceae): new insights from phylogenomic analyses of plastome data. Mol Phylogenet Evol. 2021;161:107183.
Kubatko LS, Degnan JH. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst Biol. 2007;56(1):17–24.
Liu L, Yu L, Kubatko L, Pearl DK, Edwards SV. Coalescent methods for estimating phylogenetic trees. Mol Phylogenet Evol. 2009;53(1):320–8.
Zhang X, Deng T, Moore MJ, Ji Y, Lin N, Zhang H, et al. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 2019;19(1):1–10.
Goncalves DJ, Shimizu GH, Ortiz EM, Jansen RK, Simpson BB. Historical biogeography of Vochysiaceae reveals an unexpected perspective of plant evolution in the Neotropics. Am J Bot. 2020;107(7):1004–20.
Xiao TW, Xu Y, Jin L, Liu TJ, Yan HF, Ge XJ. Conflicting phylogenetic signals in plastomes of the tribe Laureae (Lauraceae). PeerJ. 2020;8:e10155.
Green BR. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011;66(1):34–44.
Weng ML, Ruhlman TA, Jansen RK. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017;214(2):842–51.
Dong WL, Wang RN, Zhang NY, Fan WB, Fang MF, Li ZH. Molecular evolution of chloroplast genomes of orchid species: insights into phylogenetic relationship and adaptive evolution. Int J Mol Sci. 2018;19(3):716.
Li B, Zheng Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci Rep. 2018;8(1):1–11.
Ren T, Yang Y, Zhou T, Liu ZL. Comparative plastid genomes of Primula species: sequence divergence and phylogenetic relationships. Int J Mol Sci. 2018;19(4):1050.
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, et al. The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;23(11):2175–90.
Yi X, Gao L, Wang B, Su Y, Wang T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 2013;5(4):688–98.
Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28(1):583–600.
Wu CS, Lin CP, Hsu CY, Wang RJ, Chaw SM. Comparative chloroplast genomes of Pinaceae: insights into the mechanism of diversified genomic organizations. Genome Biol Evol. 2011;3:309–19.
Zhu A, Guo W, Gupta S, Fan W, Mower JP. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016;209(4):1747–56.
Downie SR, Jansen RK. A comparative analysis of whole plastid genomes from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst Bot. 2015;40(1):336–51.
Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE. 2013;8:e57607.
Mower JP, Ma PF, Grewe F, Taylor A, Michael TP, VanBuren R, et al. Lycophyte plastid genomics: extreme variation in GC, gene and intron content and multiple inversions between a direct and inverted orientation of the rRNA repeat. New Phytol. 2019;222(2):1061–75.
Mitreva M, Wendl MC, Martin J, Wylie T, Yin Y, Larson A, et al. Codon usage patterns in Nematoda: analysis based on over 25 million codons in thirty-two species. Genome Biol. 2006;7(8):1–19.
Knight RD, Freeland SJ, Landweber LF. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001;2(4):1–13.
Yang Y, Zhu J, Feng L, Zhou T, Bai G, Yang J, et al. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Front Plant Sci. 2018;9:82.
Gu C, Ma L, Wu Z, Chen K, Wang Y. Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 2019;19(1):1–19.
Tian S, Lu P, Zhang Z, Wu JQ, Zhang H, Shen H. Chloroplast genome sequence of Chongming lima bean (Phaseolus lunatus L.) and comparative analyses with other legume chloroplast genomes. BMC Genomics. 2021;22(1):1–14.
Freyer R, Kiefer-Meyer MC, Kossel H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc Natl Acad Sci USA. 1997;94(12):6285–90.
Tsudzuki T, Wakasugi T, Sugiura M. Comparative analysis of RNA editing sites in higher plant chloroplasts. J Mol Evol. 2001;53(4):327–32.
Hoch B, Maier RM, Appel K, Igloi GL, Kossel H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature. 1991;353(6340):178–80.
Martín M, Sabater B. Plastid ndh genes in plant evolution. Plant Physiol Biochem. 2010;48(8):636–45.
Kimura M. The neutral theory of molecular evolution and the world view of the neutralists. Genome. 1989;31(1):24–31.
Ivanova Z, Sablok G, Daskalova E, Zahmanova G, Apostolova E, Yahubyan G, et al. Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front Plant Sci. 2017;8:204.
Wicke S, Schneeweiss GM, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–97.
Liang H, Zhang Y, Deng J, Gao G, Ding C, Zhang L, et al. The complete chloroplast genome sequences of 14 Curcuma species: insights into genome evolution and phylogenetic relationships within Zingiberales. Front Genet. 2020;11:802.
Cui X, Wang Y, Wu J, Han X, Gu X, Lu T, et al. The RNA editing factor DUA 1 is crucial to chloroplast development at low temperature in rice. New Phytol. 2019;221(2):834–49.
Zhang J, Guo Y, Fang Q, Zhu Y, Zhang Y, Liu X, et al. The PPR-SMR protein ATP4 is required for editing the chloroplast rps8 mRNA in rice and maize. Plant Physiol. 2020;184(4):2011–21.
Kikuchi S, Bédard J, Hirano M, Hirabayashi Y, Oishi M, Imai M, et al. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science. 2013;339(6119):571–4.
Zavala-Páez M, Vieira LDN, Baura VAD, Balsanelli E, Souza EMD, Cevallos MC, et al. Comparative plastid genomics of neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front Plant Sci. 2020;11:799.
Schneider AC, Braukmann T, Banerjee A, Stefanović S. Convergent plastome evolution and gene loss in Holoparasitic Lennoaceae. Genome Biol Evol. 2018;10(10):2663–70.
Drescher A, Ruf S, Calsa T Jr, Carrer H, Bock R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000;22(2):97–104.
Lee SH, Choi HW, Sung JS, Bang JW. Inter-genomic relationships among three medicinal herbs: Cnidium officinale, Ligusticum chuanxiong and Angelica polymorpha. Genes Genom. 2010;32(1):95–101.
Pimenov MG. Updated checklist of Chinese Umbelliferae: nomenclature, synonymy, typification, distribution. Turczaninowia. 2017;20(2):106–239.
Pimenov MG, Kljuykov EV, Ostroumova TA. Reduction of Notopterygium to Hansenia (Umbelliferae). Willdenowia. 2008;38(1):155–72.
Pimenov MG, Kljuykov EV. New nomenclatural combinations for Chinese Umbelliferae. Feddes Repert. 1999;110(7–8):481–91.
Tan JB, Jia SB, He XJ, Ma XG. Accommodating Haplosphaera in Hansenia (Apiaceae) based on morphological and molecular evidence. Phytotaxa. 2020;464(3):207–16.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 2 March 2021.
Schubert M, Lindgreen S, Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes. 2016;9(1):1–7.
Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4): e18.
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, et al. GeSeq-versatile and accurate annotation of organelle genomes. Nucl Acids Res. 2017;45(W1):W6-11.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47(W1):W59-64.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Wright F. The effective number of codons’ used in a gene. Gene. 1990;87(1):23–9.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Mower JP. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes, and user-defined alignments. Nucleic Acids Res. 2009;37:W253–9.
Yang ZH, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19(6):908–17.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77.
Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20(1):348–55.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Boil. 2012;61(3):539–42.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8.
Mirarab S, Warnow T. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 2015;31(12):i44-52.
Sayyari E, Mirarab S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol Biol Evol. 2016;33(7):1654–68.
Acknowledgements
We are grateful to Jun Wen and Sheng-Bin Jia for their help in the collection of plant material. We thank Shanghai Personal Biotechnology company for sequencing.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32070221, 31872647), National Herbarium of China, National Herbarium resources teaching specimen database (Grant No. 2020BBFK01), the fourth national survey of traditional Chinese medicine resources (Grant No. 2019PC002).
Author information
Authors and Affiliations
Contributions
TR and XH conceived and designed the work. TR, DX, and CP performed bioinformatics analyses of the sequence data. CP and LG provided the materials/analysis tools. TR wrote the manuscript. MP, SZ, and XH revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Our plant materials were collected from the wild. These plants are not protected, therefore permission is not required for sample collection. In addition, the place of sample collection is not a protected area, therefore no any legal authorization/license is required.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Gene map of Ligusticum angelicifolium plastomes. The genes shown outside of the circle are transcribed clockwise, while those inside are transcribed counterclockwise. The genes belonging to different functional groups are color-coded. The innermost darker gray represents the GC content of the plastome.
Additional file 2: Figure S2.
Phylogenetic relationships of 66 Apiaceae species inferred from 76 common protein-coding sequences based on Maximum likelihood (ML) and Bayesian inference (BI) analyses. The bootstrap support values (BS) and posterior probabilities (PP) are listed at each node.
Additional file 3: Table S1.
Information regarding the 26 Ligusticum accessions used in this study.
Additional file 4: Table S2.
Codon and base compositions for protein-coding sequences of 53 plastid genes in the 26 Ligusticum plastomes.
Additional file 5: Table S3.
RNA editing sites analyses of the 26 Ligusticum plastomes.
Additional file 6: Table S4.
The ω (dN/dS) and Pi values for protein-coding sequences of 79 plastid genes in the 26 Ligusticum plastomes.
Additional file 7: Table S5.
List of 66 species and 76 common protein-coding sequences used in the phylogenetic analyses.
Additional file 8: Table S6.
Comparison of the morphology of 20 Ligusticum species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ren, T., Xie, D., Peng, C. et al. Molecular evolution and phylogenetic relationships of Ligusticum (Apiaceae) inferred from the whole plastome sequences. BMC Ecol Evo 22, 55 (2022). https://doi.org/10.1186/s12862-022-02010-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-022-02010-z