
Abstract
Background
The genus Ligusticum consists of approximately 60 species distributed in the Northern Hemisphere. It is one of the most taxonomically difficult taxa within Apiaceae, largely due to the varied morphological characteristics. To investigate the plastome evolution and phylogenetic relationships of Ligusticum, we determined the complete plastome sequences of eight Ligusticum species using a de novo assembly approach.
Results
Through a comprehensive comparative analysis, we found that the eight plastomes were similar in terms of repeat sequence, SSR, codon usage, and RNA editing site. However, compared with the other seven species, L. delavayi exhibited striking differences in genome size, gene number, IR/SC borders, and sequence identity. Most of the genes remained under the purifying selection, whereas four genes showed relaxed selection, namely ccsA, rpoA, ycf1, and ycf2. Non-monophyly of Ligusticum species was inferred from the plastomes and internal transcribed spacer (ITS) sequences phylogenetic analyses.
Conclusion
The plastome tree and ITS tree produced incongruent tree topologies, which may be attributed to the hybridization and incomplete lineage sorting. Our study highlighted the advantage of plastome with mass informative sites in resolving phylogenetic relationships. Moreover, combined with the previous studies, we considered that the current taxonomy system of Ligusticum needs to be improved and revised. In summary, our study provides new insights into the plastome evolution, phylogeny, and taxonomy of Ligusticum species.
Similar content being viewed by others


Background
The genus Ligusticum, belonging to the family Apiaceae with approximately 60 species, is distributed throughout Asia, Europe, and North America [1]. It has two distribution centers: one in the Himalayas, and the other in North America [2]. There are 40 species (35 endemics) of this genus in China, most of which are restricted to alpine regions [1].
Ligusticum is one of the most taxonomically difficult genera within Apiaceae, largely due to the varied morphological characteristics of flowers, leaves, bracteoles, and mericarps that make it difficult to distinguish from its neighbors [1, 3, 4]. So far, its phylogenetic relationships with nearby genera are not clear, such as Ligusticopsis, Tilingia, Cnidium, Selinum, Hymenidium, Pachypleurum, Rupiphila, and Paraligusticum [1], especially, merging Tilingia and Ligusticopsis into Ligusticum is still debatable [2]. The diagnostic characters of Tilingia are the distinct calyx teeth and the mericarp bearing a vitta in each furrow [5], which do not distinguish it from Ligusticum. Thus, Tilingia was transferred into Ligusticum [6, 7]. Leute [3] separated Ligusticopsis from Ligusticum based on the prominent calyx teeth. This treatment is not supported, for some Ligusticum species also have this characteristic [2]. Traditional methods to distinguish these species are based on their morphological characteristics, while many above-mentioned species always exhibit similar characteristics leading to extremely difficult species classification and generic delimitation [1, 3, 4]. Previously, a few molecular markers have been used to study the phylogeny of Ligusticum, such as nuclear ribosomal DNA internal transcribed spacer (ITS), plastid DNA rpl16, rps16, and rpoC1 intron [4, 8,9,10,11,12,13,14], yet the DNA fragments fail to recognize Ligusticum as a monophyletic group. Among which, Downie et al. [12] identified five clades within Ligusticum, including Acronema Clade, Conioselinum chinense Clade, Pyramidoptereae, Selineae, and Sinodielsia Clade. Zhou et al. [4] subsequently divided the genus Ligusticum into six clades, and East-Asia (Physospermopsis) Clade was added. It can be seen that the genus Ligusticum is facing a big challenge of taxonomy and phylogeny. Therefore, more genomic resources are needed for reconstructing phylogenetic relationships and re-evaluating the generic limits of Ligusticum.
Additionally, many Ligusticum species are precious traditional herbs with excellent medicinal values. For example, the rhizomes and roots of L. jeholense or L. sinense are used as the traditional Chinese medicine named Gao-ben, which has been widely used to treat colds, headaches, trapped wind, and rheumatic arthralgia [15]. As a result, this herb has been conducted many studies on bioactive, chemical components, or pharmacology [16, 17]. Despite excellent medicinal value, genomic resources are lacking and species authentication is difficult. Thus, it is necessary to develop more DNA barcodes by a comparative plastome method for species authentication to assure medicinal quality.
Plastid is a key organelle for green plants, which participates in the photosynthetic process and provides essential energy for plants [18]. The plastid genome (plastome) is a double-stranded molecule of 115 to 165 kb in most plants [19]. Structural organization, gene arrangement, and gene content of plastome are relatively conserved. Typical plastome contains a large single copy (LSC) region of 82–90 kb, a small single copy (SSC) region of 15–20 kb, and two inverted repeats (IRs) regions of 22–25 kb [19]. It always encodes 110–130 distinct genes, including protein-coding gene(~ 80), transfer RNA (tRNA) gene (~ 30), and ribosomal RNA (rRNA) gene (4) [20]. Moreover, it is usually uniparental inheritance and has low nucleotide substitution rates [21]. For these reasons, the plastome has become useful a tool for plant phylogenetic studies at different taxonomic levels [22,23,24,25]. Currently, the plastid phylogenomics analysis of Ligusticum has not been reported. Meanwhile, the ongoing development of next-generation sequencing and bioinformatics technology makes it cheaper and faster to obtain the complete plastome sequence than ever before. Therefore, we prefer to use the plastomes to infer the phylogenetic relationships for Ligusticum.
Here, we newly sequenced eight plastomes of Ligusticum species. To obtain a comprehensive understanding of phylogenetic relationships, we also used nuclear ITS sequences to construct the phylogenetic tree. Our aims were to (1) infer the plastome evolution of Ligusticum; (2) provide more genomic resources for developing candidate DNA barcodes; (3) test if the plastomes increase resolution than traditional DNA markers; and (4) serve as a reference for subsequent phylogenomics studies of this genus. Overall, the complete plastomes reported here will promote plastome evolution, phylogeny, and taxonomy studies of Ligusticum.
Results
Characteristics of Ligusticum plastomes
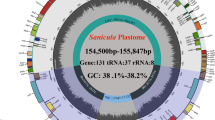
After quality control, 5.76 Gb (L. scapiforme) to 7.47 Gb (L. delavayi) clean reads were generated for the eight Ligusticum species (Table 1), then we obtained eight complete plastome sequences by a de novo assembly. The determined complete plastome sequences of the eight Ligusticum species ranged from 146,443 bp in L. pteridophyllum to 155,623 bp in L. delavayi (Table 1). All of them were highly conserved in structure compared to most angiosperms, sharing the typical quadripartite structure with two copies of IR regions (18,166–26,908 bp), SSC regions (16,741–17,591 bp), and LSC regions (85,066–93,363 bp). The overall GC content was between 37.3–37.6%, while the IR regions were higher (42.5–44.8%) than that of the LSC (35.7–36.0%) and SSC (30.9–31.2%) regions (Table 1). The eight plastomes contained about 129–133 genes, including 85–88 protein-coding genes, 36–37 tRNA genes, and eight rRNA genes (Fig. 1, Table 1, Additional file 2: Table S1). L. delavayi contained four more genes (ycf2, rpl23, rpl2, and trnI-CAU) than seven other Ligusticum species in IRa (Fig. 1, Additional file 2: Table S1). The rps12 gene was trans-spliced with the 5’end and the duplicated 3’end were located in the LSC and IR regions, respectively (Fig. 1). The trnK-UUU had the longest intron (2485–2543 bp) containing the matK gene (Fig. 1). The GC content of four rRNA (rrn16, rrn23, rrn4.5, and rrn5) genes was high (55.1–55.3%) (Fig. 1).

Gene map of eight Ligusticum plastomes. The genes shown outside of the circle are transcribed clockwise, while those inside are transcribed counterclockwise. The genes belonging to different functional groups are color-coded. The innermost darker gray represents the GC content of the plastome
Codon usage and RNA editing sites
The total sequence sizes of the protein-coding genes for codon analysis were 67,905–68,268 bp in the eight Ligusticum plastomes. These protein sequences encoded 22,635–22,756 codons, which are summarized in Additional file 3: Table S2. Leu was encoded by the highest number of codons (2382-2419), whereas Cys was the least (232–241). The RSCU values of all codons in the form of a heatmap are shown in Fig. 2. The red values indicate higher RSCU values and the blue values indicate lower RSCU values. The heatmap showed that about half of the codons were used more frequently. Specifically, 30 codons were used frequently with RSCU > 1, and all biased codons ended with a purine (A/T) except TTG (Fig. 2, Additional file 3: Table S2). The mean values of GC content of the first, second, and third codon positions were 46.0, 38.3, and 29.7%, respectively (Additional file 4: Table S3). This GC content also implied that the plastome of Ligusticum has a strong bias towards A/T at the third codon position. The usage of two codons (ATG and TGG) had no bias (RSCU = 1) (Additional file 3: Table S2).

The RSCU values of all merged protein-coding genes for eight Ligusticum plastomes. Color key: the red values indicate higher RSCU values and the blue values indicate lower RSCU values
Additionally, potential RNA editing sites were identified for 35 genes of the eight Ligusticum plastomes. A total of 469 RNA editing sites were identified, in which the number of editing sites ranged from 55 (L. delavayi) to 67 (L. likiangense) (Additional file 5: Table S4). The ndhB gene had the highest number of RNA editing sites (10) in all of the eight Ligusticum plastomes, whereas the rps8 gene also had 10 RNA editing sites in L. likiangense. All of the identified RNA editing sites were Cytosine to Uracil (C-U) conversion and most of them were situated in the second codon position (40–51), followed by the first codon position (12–16), but no sites situated in the third codon position (Additional file 1: Figure S1). The amino acid conversion Serine to Leucine (S-L) occurred most frequently. Furthermore, a mass of RNA editing sites (420) caused amino acid changes for hydrophobic products, such as Leucine (L; 219), Phenylalanine (F; 63), Isoleucine (I; 58), Tyrosine (Y; 25), Methionine (M; 21), Tryptophan (W; 17), and Valine (V; 17) (Additional file 5: Table S4).
Repeat element analysis
Forward, palindromic, reverse, and complementary repeats were detected in the eight Ligusticum plastomes. In all, we detected 308 repeats with 30–82 bp long (Additional file 6: Table S5). The number of forward repeats (176) was higher than that of palindromic repeats (116), reverse repeats (10), and palindromic repeats (6). L. likiangense contained the most repeats (49), while L. thomsonii contained the least (25) (Fig. 3). According to the length, we artificially divided the repeats into four categories: 30–45 bp, 45–60 bp, 60–75 bp, and > 75 bp (Fig. 3). Among them, most of the repeats (85%) were 30–45 bp long. The majority of the repeats were located in intergenic or intron regions (70.5%), and a minority were located in gene regions (29.5%). 603 simple sequence repeats (SSRs) were detected, but the number of SSRs differed among eight Ligusticum species (Additional file 1: Figure S2, Additional file 7: Table S6). L. scapiforme contained the most SSRs (82), while L. delavayi and L. hispidum contained the least (68). The most abundant were mononucleotide repeats (54.9%), followed by dinucleotides (25.5%), tetranucleotides (12.3%), trinucleotides (3.0%), and pentanucleotides (3.0%). Hexanucleotides are very rare across the plastomes. SSRs were distributed mainly in the LSC (68.3%), with less in the IRs (16.6%) and SSC (15.1%) (Additional file 7: Table S6).

Analysis of repeat sequences in the eight Ligusticum plastomes. a Total numbers of four repeat types. b Number of repeats divided by length
Comparisons of border and sequence identity
The differences between inverted repeat and single-copy (IR/SC) borders among eight Ligusticum plastomes were examined (Fig. 4). Besides L. delavayi, seven other Ligusticum species were conserved in terms of the gene order and gene content at the IR/SC borders. For L. delavayi, the LSC/IRb border was rps19/rpl2 genes and the IRa/LSC border was rpl2/trnH genes. For the other Ligusticum species, LSC/IRb borders extended 576–701 bp into the ycf2 gene. The SSC/IRb borders extended 3–140 bp into the ycf1 genes, where the smallest and largest extensions occurred in L. thomsonii (3 bp) and L. involucratum (140 bp). The ndhF gene in L. capillaceum overlapped with the SSC/IRb border by 59 bp. The ycf1 genes, crossing the SSC/IRa borders, were located at the SSC and IRa regions with 3514–3574 bp and 1886–2057 bp. The trnL and trnH genes were 1033–1871 bp and 6–881 bp away from the IRa/LSC borders.

Comparison of the border regions of the eight Ligusticum plastomes. LSC (large single copy), SSC (small single copy), and IR (inverted repeat) regions are indicated. This figure is not to scale
The mVISTA program was used to conduct a sequence identity analysis using L. delavayi as a reference. The results are revealed in Fig. 5, of which coding regions showed more sequence conservation than non-coding regions. 766 SNPs and 351 Indels were detected among the eight Ligusticum plastomes (Additional file 8: Table S7). The majority of SNPs and Indels (786) were from non-coding regions, while a minority (331) were from coding regions. We also identified the average percentage of variation for 149 regions (66 coding regions, 64 intergenic spacers, and 19 introns) (Fig. 6, Additional file 9: Table S8). Among these regions, the average percentage of variation for non-coding regions (18.5%) was higher than that (3.2%) for coding regions (Additional file 9: Table S8). Twelve non-coding regions exhibited high variation: trnH-GUG/psbA, psbA/trnK-UUU, trnK-UUU/rps16, rps16/trnQ-UUG, psbK/psbI, atpF/atpH, trnE-UUC/trnT-GGU, accD/psaI, ycf4/cemA, trnW-CCA/trnP-UGG, ycf2/trnL-CAA, and ndhF/rpl32 (the percentage of variation > 30%). Eight coding regions exhibited high variation: matK, rps3, ycf2, ycf1 × 2, ndhF, rpoA, and ccsA (the percentage of variation > 5%). Seven other Ligusticum species showed sequence differences when L. delavayi was a reference in the mVISTA plot (Fig. 5). Therefore, we calculated the percentages of variable characters for coding and non-coding region of seven Ligusticum species (not including L. delavayi), as well as the genetic distance of the eight Ligusticum plastomes. The pairwise genetic distance ranged from 0.0010 to 0.0239 with an overall average was 0.0092 (Additional file 10: Table S9). However, the values of pairwise genetic distance between L. delavayi and seven other Ligusticum species were higher: 0.0234 (L. capillaceum), 0.0236 (L. scapiforme), 0.0234 (L. likiangense), 0.0237 (L. hispidum), 0.0239 (L. involucratum), 0.0221 (L. pteridophyllum), and 0.0232 (L. thomsonii). Figure 6 shows that the sequence difference of seven other Ligusticum species was lower without L. delavayi. In a word, our results demonstrated that L. delavayi showed a higher sequence difference than the rest seven Ligusticum species (Fig. 6, Additional file 10: Table S9).

VISTA-based sequence identity plot of the eight Ligusticum plastomes using L. delavayi as a reference. The vertical scale represents the percentage of identity ranging from 50 to 100%. Coding and non-coding regions are marked in purple and pink, respectively

Percentage of variable characters in aligned Ligusticum plastomes. a Coding region. b Non-coding region. The blue and orange lines show the eight Ligusticum and seven Ligusticum (not including L. delavayi). These regions are oriented according to their locations in the plastome
Selective pressure in plastid genes
The dN/dS ratios of the 79 common protein-coding genes were calculated to estimate selective pressures (Additional file 11: Table S10). The dN/dS ratios of the most genes in our results were less than 0.5, suggested that they were under the purifying selection. Despite this, we also detected an increase in dN/dS, indicating relaxed selection in nine genes (0.5 < dN/dS < 1.0). Unexpectedly, none but four genes were significant (P < 0.05) after the likelihood ratio test (LRT). Therefore, the analyses presented here demonstrated that four genes were under relaxed selection, namely ccsA, rpoA, ycf1, and ycf2 (Additional file 11: Table S10). Meanwhile, only one gene with dN/dS > 1.0 (psaJ), but the LRT was not significant (P > 0.05).
Phylogenetic relationships
39 complete plastomes and 80 nuclear ITS sequences were used to carry out the phylogenetic analyses (Additional file 12: Table S11). The plastome tree and ITS tree produced incongruent tree topologies, while they all inferred the non-monophyly of Ligusticum species (Fig. 7, Additional file 1: Figure S3). In the plastome tree, the L. capillaceum, L. scapiforme, L. likiangense, L. hispidum, L. involucratum, and L. thomsonii belonged to Selineae. However, five other Ligusticum species formed a clade, L. thomsonii clustered with S. divaricata, L. seseloides, and P. praeruptorum. L. tenuissimum and L. sinense belonged to Sinodielsia Clade, but they did not form a clade. L. sinense was more closely related to C. officinale, then they clustered with L. tenuissimum. L. delavayi always clustered with P. neurophyllum, belonged to Acronrma Clade. L. pteridophyllum belonged to Sinodielsia Clade [4], while it was resolved as sister to Sinodielsia Clade + Selineae. In the ITS tree, the L. capillaceum, L. scapiforme, L. likiangense, L. hispidum, L. involucratum, and L. thomsonii formed a clade and belonged to Selineae. L. tenuissimum was resolved as sister to Selineae with weak support (BS = 54%, PP = 0.8). L. sinense was still more closely related to C. officinale. The systematic position of L. delavayi was in line with the plastome tree. The ITS tree topologies resulting from ML and BI analysis were some different. For example, L. pteridophyllum clustered with L. sinense and C. officinale by BI analysis (PP = 0.57), whereas it was the parallel branch’s relationships with L. sinense and C. officinale by ML analysis (BB = 57%). In Selineae, six Ligusticum and seven Angelica falled within this tribe. Like Ligusticum, Angelica also showed polyphyly, and M. pimpinelloideum and G. littoralis embedded in it. Chamaesium Clade was the basal taxa of the Apioideae rather than Bupleureae. Sinodielsia Clade was not a monophyletic group in our phylogenetic analyses.

Phylogenetic tree reconstruction of the 39 species inferred from Maximum likelihood (ML) and Bayesian inference (BI) analyses based on the complete plastomes. The bootstrap support values and posterior probability values are listed at each node
Discussion
Plastome evolution
The plastomes are highly conserved in genome structure, gene order, and gene content [19, 21, 26]. Nevertheless, genome rearrangement, gene loss (pseudogenization or deletion), differences in structure and size, expansions and contractions of IR have been confirmed to occur many times during plastome evolution [20, 27,28,29,30]. In this study, the Ligusticum plastomes were low conservation, exhibiting striking differences in terms of genome size (146,443–155,623 bp), gene number (129–133), and IR/SC borders. L. delavayi had the longest plastome length, whereas its LSC and SSC regions were shorter than that of seven other Ligusticum species. The differences in length reflect the extensions of IRs and the contractions of two SCs. It had four more genes (ycf2, rpl23, rpl2, and trnI-CAU) than seven other Ligusticum species in IRa, which was likely ascribed to the gene duplication resulting from the extensions of IRs. The varied gene number in the congeneric species has been found in other angiosperm plastomes [31, 32]. For L. delavayi, the IRa/LSC border was rpl2/trnH genes and the LSC/IRb border was rps19/rpl2 genes, which were different from seven other Ligusticum species (trnL/trnH and ycf2). The IRa region extended into the ycf1 gene was a common feature in plastome evolution [31, 33, 34]. The IR regions had a higher GC content of 42.5–44.8% possibly caused by the high GC content of four rRNA genes [34].
SSRs have been used widely in plant population genetics and evolutionary studies [35, 36]. The most abundant SSRs were mononucleotide in the eight Ligusticum plastomes, followed by dinucleotide, tetranucleotide, trinucleotide, pentanucleotide, and hexanucleotide repeats. This phenomenon has been reported in Primula [31] and Allium [37]. The most probable explanation for the largest amount of SSRs in the LSC is that LSC is longer than SSC and IRs. The majority of the SSRs contained A/T motifs, causing the AT richness of the overall plastome [34]. The cpSSRs reported here are informative sources for developing molecular markers for genetic diversity studies of Ligusticum species.
Codons with a higher AT content are usually used in plastomes, and the trend is more striking for A/T use in the third codon positions [38]. The bias also showed in the eight Ligusticum plastomes. Leucine was encoded by the highest number of codons, and the order of codon preference was TTA > CTT > TTG > CTA > CTC > CTG, which following most Geraniaceae species [20]. RNA editing is an important process to regulate the gene expression of posttranscriptional in plant organelles [39]. The events occur in all major lineages of land plants, except Marchantia polymorpha and some green algae [40]. RNA editing can correct DNA mutations at the RNA level, thus recovers conserved amino acid residues to maintain functions of encoded proteins [41,42,43]. Most of the editing sites occurred at the second codon position and no sites occurred at the third codon position, the distribution pattern was also found in Forsythia suspensa [44]. Like many other plants, the ndhB gene had the most RNA editing sites [40, 45], which suggests that ndhB gene is critical in regulating plant physiological and biochemical processes.
Large repeat sequences are considered to be the major cause to promote plastome rearrangement and sequence divergence [46,47,48]. Among the identified 308 repeats, short repeat with 30–45 bp (85.4%) was the most, which was consistent with many unrearranged plastomes [49, 50]. Moreover, non-coding regions distributed more SNPs and Indels and had a higher average percentage of variation than that of coding regions. Consequently, our study showed that non-coding regions were less conservative than coding regions. The distribution of repeats is correlated with mutational events, and repeats may play a role in inducing mutations [30, 51, 52]. Our results also indicated that the distribution of repeats was relevant to mutational events, for repeats located predominantly in highly variable non-coding regions (70.5%) instead of coding regions (29.5%). DNA barcodes are defined as the short DNA sequences with adequate variations to identify species in the given taxonomic group [53]. Eight coding regions and 12 non-coding regions with the highest percentage of variation have been described. Thereinto, several regions have been ascertained in other angiosperms, such as matK, ndhF, rps3, ycf2, ycf1 × 2, rpoA, trnH-GUG/psbA, ndhF/rpl32, trnK-UUU/rps16, psbK/psbI, ycf4/cemA, accD/psaI, ycf2/trnL-CAA, rps16/trnQ-UUG, and trnE-UUC/trnT-GGU [22, 49, 50, 54,55,56]. For the herbal medicinal genus Ligusticum, these regions could serve as candidate DNA barcodes for species authentication to assure medicinal quality.
We estimated the selective pressures of 79 common protein-coding genes in Ligusticum plastomes. Most of them were under purifying selection, which reflected the typically evolutionary conservation of plastid genes in plants [57, 58]. Four genes (ycf1, ycf2, ccsA, and rpoA) were under relaxed selection. The ycf1 and ycf2 genes, the largest and the second-largest genes in the plastome, have been proved to be absent or pseudogenized in many prior works [21, 59]. Relaxed selection on the two genes also has been observed in Corallorhiza striata, Lennoa madreporoides, and Pholisma arenarium [60, 61]. The genes rpoA and ccsA encode an α subunit of RNA polymerase and a protein required for heme attachment to C-type cytochrome, respectively [62, 63]. They usually present in land plants, whereas they are absent from the plastome of Physcomitrella patens [63]. Nevertheless, the plastomes of parasitic plants generally are the best model systems to study the effect of relaxed selection on photosynthetic function [64, 65]. Indeed, some parasitic plants harbor drastically reduced plastome size and gene content resulting from the relaxed selection on photosynthesis-related genes [60, 64, 66]. Therefore, further studies are necessary to investigate the important role of relaxed selection in Ligusticum plastid genes. Overall, these findings shed new lights on the plastid genes of Ligusticum species.
Phylogenetic relationships
Until now, molecular phylogenetic studies based on a few molecular markers do not support the monophyly of Ligusticum species [4, 8,9,10,11,12,13,14]. Here, we performed phylogenetic analyses for Ligusticum using complete plastomes and ITS sequences. Unfortunately, we still failed to recognize Ligusticum as a monophyletic group. The plastome tree and ITS tree produced incongruent tree topologies. L. capillaceum, L. scapiforme, L. likiangense, L. hispidum, and L. involucratum share some similar morphological characteristics (e.g., bracteole pinnate and stem bases clothed in fibrous remnant sheaths), and they formed a clade (BS = 100%, PP = 1) in the plastome tree. However, L. thomsonii clustered with the above five species to form a clade in the ITS tree. L. tenuissimum belonged to Sinodielsia Clade in the plastome tree, which in accord with the results of Zhou et al. [4], whereas it was resolved as sister to Selineae with weak support in the ITS tree (BS = 54%, PP = 0.8). The incongruence between nuclear and plastome phylogenies has been commonly observed in other plant lineages [67,68,69]. This incongruence was likely the result of different inherited background and mutation rates of ITS and plastid DNA [70, 71]. The nuclear ITS is biparentally inherited and has a higher mutation rate, whereas the plastid DNA is maternally inherited and has a lower mutation rate [70, 71]. Moreover, the hybridization and incomplete lineage sorting (ILS) may be responsible for the inconsistent relationships between ITS- and plastome-based phylogenies [72, 73]. L. sinense was more closely related to C. officinale, which can be possibly explained well using the cross-hybridization of genomes [74]. L. pteridophyllum clustered with L. sinense and C. officinale in ITS tree by BI analysis, which was consistent with the results of Zhou et al. [4]. These together suggested that Ligusticum species may have experienced a complex evolutionary history. The polyphyly of Angelica, as well as Glehnia and Melanosciadium embedded in it, have been documented by earlier studies [12, 14, 75]. Sinodielsia Clade was not recovered as a monophyletic that has been observed in other work [14]. Chamaesium Clade was the basal taxa of the Apioideae as a recent study based on 3351 single-copy genes [76].
The plastome tree obtained moderate-to-high support, conversely the ITS tree obtained lower support and more parallel branches. Therefore, our results highlight the advantage of plastome with mass informative sites in resolving phylogenetic relationships. This study is also the first to support the polyphyly of Ligusticum based on plastomes. Further studies that include greater taxon sampling are necessary to confirm the polyphyletic position of the Ligusticum. Moreover, combined with the previous studies [4], we considered that the current taxonomy system of Ligusticum needs to be improved and revised. In a word, our study provided useful information for future phylogeny, taxonomy, and evolutionary history studies of the Ligusticum species.
Methods
Taxa sampling and DNA extraction
Fresh green leaves from adult plants of eight species were sampled from the field, and then immediately dried with silica gel for the next step. Permission is not required to sample these plants because they are not key protected plants. Total genomic DNA was extracted from silica-dried leaves with a modified CTAB protocol [77]. The formal identification of the plant material was undertaken by Xingjin He (Sichuan University). Voucher specimens were deposited at the herbarium of Sichuan University (Chengdu, China) (Additional file 13: Table S12). For ITS analyses, we newly sequenced 17 ITS have been submitted into NCBI (accession numbers: MT974009-MT974025) (Additional file 12: Table S11).
Genome sequencing, assembly, and annotation
The raw reads of the eight newly sequenced species were generated from an Illumina HiSeq X Ten platform (paired-end, 150 bp) at Novogene (Tianjin, China). Quality control of the raw reads was performed using fastP version v0.15.0 (−n 10 and -q 15) [78], yielding at least 5GB clean reads for each species. Then clean reads were used to perform a de novo assembly by NOVOPlasty v2.6.2 [79] with the default parameters. The seed sequence is the rbcL gene from the reference genome sequence of L. tenuissimum (NC_029394). The program DOGMA [80] was used to annotate the genes of the eight plastomes, and adjusted manually in Geneious v9.0.2 (Biomatters Ltd., Auckland, New Zealand) based on comparisons with its congeneric species. All of the eight newly generated complete plastomes were available in NCBI (accession numbers: MT409612-MT409619) (Table 1). The circle plastome map was drawn using the online program OrganellarGenomeDRAW (OGDRAW) [81].
Codons, RNA editing sites, and repeat sequences
The protein-coding genes were extracted from the eight Ligusticum plastomes for codon analysis. All overlapping genes were removed, and the final dataset included 80 protein-coding genes for each species. Codon usage and relative synonymous codon usage (RSCU) [82] values were calculated using the CodonW v1.4.2 program [83]. The heatmap from all RSCU of the eight plastomes was produced using TBtools [84]. The base compositions for protein-coding genes were calculated by MEGA6 [85]. The online program Predictive RNA Editor for Plants suite [86] with a cutoff value of 0.8 was used to predict the potential RNA editing sites.
The online REPuter program [87] was used to identify repeat sequences, including forward, palindromic, reverse, and complementary repeats. According to the following parameters: (1) a repeat size of more than 30 bp; (2) more than 90% sequence identity between the two repeats; and (3) Hamming distance = 3. All overlapping repeat sequences were removed. The Perl script MISA (http://pgrc.ipk-gatersleben.de/misa/) was used to exploit simple sequence repeats (SSRs). The minimum number of SSRs was set to 10, 5, 4, 3, 3, and 3, for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively.
Sequence divergence
The whole-genome alignment of the eight Ligusticum plastomes was generated and visualized using the mVISTA [88] using L. dilavayi as a reference. Eight Ligusticum plastomes were aligned in Geneious v9.0.2 (Biomatters Ltd., Auckland, New Zealand) with MAFFT v7.221 [89], subsequently, Indels and SNPs were counted and positioned using the “Find Variations/SNPs”. The percentages of variable characters for coding and non-coding regions were calculated based on the method of Zhang et al. [90]. The genetic distance of the eight Ligusticum plastomes was calculated using MEGA6 [85].
Selective pressure analysis
Selective pressures were analyzed for common 79 protein-coding genes among ten Ligusticum species (including 2 published plastomes). The ratio (ω) of non-synonymous to synonymous nucleotide substitution rates (dN/dS) was calculated using the Codeml program in PAML4.9 with the site-specific model (seqtype = 1, model = 0, NSsites = 0, 1, 2, 3, 7, 8) [91, 92]. The codon frequencies were determined by the F3 × 4 model. We compared three sets: M0 vs M3, M1 vs M2, and M7 vs M8 to detect selected sites. The likelihood ratio test (LRT) was used to confirm the quality of the three sets. Bayes Empirical Bayes (BEB) analysis was used to statistically identify selected sites with posterior probabilities ≥95%. We classified genes as evolving under positive selection (dN/dS > 1.0), relaxed selection (0.5 < dN/dS < 1.0), and purifying selection (dN/dS < 0.5) [61, 93].
Phylogenetic analysis
Earlier molecular systematic studies identified five clades within Ligusticum, including Acronema Clade, Conioselinum chinense Clade, Pyramidoptereae, Selineae, and Sinodielsia Clade [12]. More recently, the genus Ligusticum has been divided into six clades, and East-Asia (Physospermopsis) Clade was added [4]. Here, we used 39 complete plastomes and 80 nuclear ITS sequences to infer the phylogenetic relationships of Ligusticum. Sequence alignment was achieved using the MAFFT v7.221 [89]. The aligned sequence was then manually examined and corrected. Maximum likelihood (ML) and Bayesian inference (BI) methods were used to infer phylogenetic relationships. RAxML v8.2.8 [94] was used to perform the ML analysis with 1000 replicates and GTRGAMMA model as suggested (see RAxML manual). MrBayes v3.2.7 [95] was used to perform the Bayesian inference with the best substitution model was determined by Modeltest v3.7 [96]. The selected models for complete plastomes and ITS sequences in BI analyses were TVM + I + G and GTR + I + G, respectively. Markov chain Monte Carlo (MCMC) algorithm was run for two million generations, with one tree sampled every 100 generations. The MCMC convergence was determined by calculating the average standard deviation of split frequencies (ASDSF), which fell below 0.01. The first 25% of the trees were discarded as burn-in and the consensus tree generated using the remaining trees. The ITS trees were visualized and edited using Interactive Tree of Life (iTOL) [97]: nodes under 50% bootstrap support were collapsed.
Conclusions
In this study, we determined the complete plastome sequences of eight Ligusticum species using a de novo assembly approach. Through a comprehensive comparative analysis, we observed that compared with the other seven species, L. delavayi exhibited striking differences in genome size, gene number, IR/SC borders, and sequence identity. We performed the phylogenetic analyses for Ligusticum using 39 complete plastomes and 80 nuclear ITS sequences and found that Ligusticum was not monophyletic as presented in previous studies. The hybridization and incomplete lineage sorting may be responsible for the inconsistent relationships between ITS- and plastome-based phylogenies. The phylogenetic analyses highlighted the advantage of using plastome with mass informative sites in resolving phylogenetic relationships. Our study enriches the data on the plastomes of Ligusticum and serves as a reference for subsequent phylogenomics studies of this genus.
Availability of data and materials
Eight annotated plastomes and newly sequenced 17 ITS have been submitted into NCBI (https://www.ncbi.nlm.nih.gov) with accession numbers: MT409612-MT409619 and MT974009-MT974025, respectively.
Abbreviations
- ASDF:
-
Average standard deviation of split frequencies
- BEB:
-
Bayes empirical bayes
- BI:
-
Bayesian inference
- bp:
-
Base pair
- BS:
-
Branch support
- CDS:
-
Protein-coding sequences
- CTAB:
-
Cetyl trimethylammonium bromide
- ILS:
-
Incomplete lineage sorting
- IR:
-
Inverted repeat
- ITS:
-
Internal transcribed spacer
- LRT:
-
Likelihood ratio test
- LSC:
-
Large single copy
- MCMC:
-
Markov chain Monte Carlo
- ML:
-
Maximum Likelihood
- PP:
-
Posterior probability
- rRNA:
-
Ribosomal RNA
- RSCU:
-
Relative synonymous codon usage
- SSC:
-
Small single copy
- SSR:
-
Simple sequence repeat
- tRNA:
-
Transfer RNA
References
Pu FD, Watson MF. Ligusticum L. Flora of China. Science Press: Beijing, China. 2005;14:140–51.
Pu FD. A revision of the genus Ligusticum L. (Umbelliferae) in China. Acta Phytotax Sin. 1991;29:385–93.
Leute GH. Untersuchungen über den Verwandtschaftskreis der Gattung Ligusticum L. (Umbelliferae). Teil I. Ann Naturhist Mus Wien. 1969;73:55–98.
Zhou J, Gao YZ, Wei J, Liu ZW, Downie SR. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: rampant polyphyly, placement of the Chinese endemic species, and a much-reduced circumscription of the genus. Int J Plant Sci. 2020;181(3):306–23.
Regel E, Tiling H. Florula ajanensis. Nouveaux Mémoires de la Société Impériale des Naturalistes de Moscou. 1858;1:97.
Kozo-Poljansky BM. Sciadopnytorum systematis lineamenta. Bull Soc Nat Mosc. 1916;29:93–222.
Hiroe M, Constance L. Umbelliferae of Japan. Univ Calif Publ Bot. 1958;230:1–444.
Downie SR, Ramanath S, Katz-Downie DS, Llanas E. Molecular systematics of Apiaceae subfamily Apioideae: phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacer and plastid rpoC1 intron sequences. Am J Bot. 1998;85(4):563–91.
Katz-Downie DS, Valiejo-Roman CM, Terentieva EI, Troitsky AV, Pimenov MG, Lee B, et al. Towards a molecular phylogeny of Apiaceae subfamily Apioideae: additional information from nuclear ribosomal DNA ITS sequences. Plant Syst Evol. 1999;216(3):167–95.
Downie SR, Katz-Downie DS, Watson MF. A phylogeny of the flowering plant family Apiaceae based on chloroplast DNA rpl16 and rpoC1 intron sequences: towards a suprageneric classification of subfamily Apioideae. Am J Bot. 2000;87(2):273–92.
Downie SR, Watson MF, Spalik K, Katz-Downie DS. Molecular systematics of Old World Apioideae (Apiaceae): relationships among some members of tribe Peucedaneae sensu lato, the placement of several island-endemic species, and resolution within the apioid superclade. Can J Bot. 2000;78(4):506–28.
Downie SR, Spalik K, Katz-Downie DS, Reduron JP. Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant Divers Evol. 2010;128(1):111–36.
Zhou J, Peng H, Downie SR, Liu ZW, Gong X. A molecular phylogeny of Chinese Apiaceae subfamily Apioideae inferred from nuclear ribosomal DNA internal transcribed spacer sequences. Taxon. 2008;57(2):402–16.
Zhou J, Gong X, Downie SR, Peng H. Towards a more robust molecular phylogeny of Chinese Apiaceae subfamily Apioideae: additional evidence from nrDNA ITS and cpDNA intron (rpl16 and rps16) sequences. Mol Phylogenet Evol. 2009;53(1):56–68.
Commission SP. Pharmacopoeia of the People’s republic of China part I. Beijing: China Medical Science and Technology Press; 2015.
Donkor PO, Chen Y, Ding L, Qiu F. Locally and traditionally used Ligusticum species - a review of their phytochemistry, pharmacology and pharmacokinetics. J Ethnopharmacol. 2016;194:530–48.
Zhang X, Han B, Feng Z, Jiang J, Yang Y, Zhang P. Bioactive thionic compounds and aromatic glycosides from Ligusticum chuanxiong. Acta Pharm Sin. 2018;8(5):818–24.
Raven JA, Allen JF. Genomics and chloroplast evolution: what did cyanobacteria do for plants? Genome Biol. 2003;4(3):209.
Ravi V, Khurana JP, Tyagi AK, Khurana P. An update on chloroplast genomes. Plant Systemat Evol. 2008;271(1):101–22.
Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28(1):583–600.
Wicke S, Schneeweiss GM, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–97.
Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7(1):84.
Zhang SD, Jin JJ, Chen SY, Chase MW, Soltis DE, Li HT, et al. Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 2017;214(3):1355–67.
Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 2019;5(5):461–70.
Xie DF, Tan JB, Yu Y, Gui LJ, Su DM, Zhou SD, et al. Insights into phylogeny, age, and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann Bot. 2020;125(7):1039–55.
Jansen RK, Raubeson LA, Boore JL. dePamphilis CW, Chumley TW, Haberle RC, et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Method Enzymol. 2005;395:348–84.
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, et al. The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;23(11):2175–90.
Martín M, Sabater B. Plastid ndh genes in plant evolution. Plant Physiol Biochem. 2010;48(8):636–45.
Wicke S, Müller KF, de Pamphilis CW, Quandt D, Wickett NJ, Zhang Y, et al. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell. 2013;25(10):3711–25.
Yi X, Gao L, Wang B, Su Y, Wang T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 2013;5(4):688–98.
Ren T, Yang Y, Zhou T, Liu ZL. Comparative plastid genomes of Primula species: sequence divergence and phylogenetic relationships. Int J Mol Sci. 2018;19(4):1050.
Feng S, Zheng K, Jiao K, Cai Y, Chen C, Mao Y, et al. Complete chloroplast genomes of four Physalis species (Solanaceae): lights into genome structure, comparative analysis, and phylogenetic relationships. BMC Plant Biol. 2020;20(1):1–14.
Li ZH, Ma X, Wang DY, Li YX, Wang CW, Jin XH. Evolution of plastid genomes of Holcoglossum (Orchidaceae) with recent radiation. BMC Evol Biol. 2019;19(1):63.
Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One. 2013;8:e57607.
Powell W, Morgante M, Andre C, McNicol JW, Machray GC, Doyle JJ, et al. Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr Biol. 1995;5:1023–9.
Roullier C, Rossel G, Tay D, McKey D, Lebot V. Combining chloroplast and nuclear microsatellites to investigate origin and dispersal of new world sweet potato landraces. Mol Ecol. 2011;20:3963–77.
Huo Y, Gao L, Liu B, Yang Y, Kong S, Sun Y, et al. Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci Rep. 2019;9(1):1–14.
Morton BR. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J Mol Evol. 1998;46(4):449–59.
Wakasugi T, Tsudzuki T, Sugiura M. The genomics of land plant chloroplasts: gene content and alteration of genomic information by RNA editing. Photosynth Res. 2001;70(1):107–18.
Freyer R, Kiefer-Meyer MC, Kossel H. Occurrence of plastid RNA editing in all major lineages of land plants. Proc Natl Acad Sci U S A. 1997;94(12):6285–90.
Yang Y, Lv J, Gui B, Yin H, Wu X, Zhang Y, et al. A-to-I RNA editing alters less-conserved residues of highly conserved coding regions: implications for dual functions in evolution. RNA. 2008;14(8):1516–25.
Takenaka M, Zehrmann A, Verbitskiy D, Härtel B, Brennicke A. RNA editing in plants and its evolution. Annu Rev Genet. 2013;47(1):335–52.
Wang W, Zhang W, Wu Y, Maliga P, Messing J. RNA editing in chloroplasts of Spirodela polyrhiza, an aquatic monocotelydonous species. PLoS One. 2015;10(10):e0140285.
Wang W, Yu H, Wang J, Lei W, Gao J, Qiu X, et al. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int J Mol Sci. 2017;18(11):2288.
Krüger M, Abeyawardana OA, Juříček M, Krüger C, Štorchová H. Variation in plastid genomes in the gynodioecious species Silene vulgaris. BMC Plant Biol. 2019;19(1):1–15.
Ogihara Y, Terachi T, Sasakuma T. Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc Natl Acad Sci U S A. 1988;85(22):8573–7.
Weng ML, Blazier JC, Govindu M, Jansen RK. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31(3):645–59.
Timme RE, Kuehl JV, Boore JL, Jansen RK. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am J Bot. 2007;94:302–12.
Yang Y, Zhou T, Duan D, Yang J, Feng L, Zhao G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front Plant Sci. 2016;7:959.
Zhang X, Zhou T, Kanwal N, Zhao Y, Bai G, Zhao G. Completion of eight Gynostemma B.L. (Cucurbitaceae) chloroplast genomes: characterization, comparative analysis, and phylogenetic relationships. Front Plant Sci. 2017;8:1583.
Ahmed I, Biggs PJ, Matthews PJ, Collins LJ, Hendy MD, Lockhart PJ. Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol. 2012;4(12):1316–23.
Abdullah MF, Shahzadi I, Ali Z, Islam M, Naeem M, Mirza B, et al. Correlations among oligonucleotide repeats, nucleotide substitutions and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J Syst Evol. 2020. https://doi.org/10.1111/jse.12585.
Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S. Plant DNA barcoding: from gene to genome. Biol Rev Camb Philos Soc. 2015;90(1):157–66.
Fan WB, Wu Y, Yang J, Shahzad K, Li ZH. Comparative chloroplast genomics of Dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front Plant Sci. 2018;9:689.
Wang X, Zhou T, Bai G, Zhao Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci Rep. 2018;8(1):1–12.
Xie DF, Yu HX, Megan P, Xie C, Deng YQ, Chen JP, et al. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front Plant Sci. 2019;10:460.
Lee-Yaw JA, Grassa CJ, Joly S, Andrew RL, Rieseberg LH. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 2019;221(1):515–26.
Zhang X, Deng T, Moore MJ, Ji Y, Lin N, Zhang H, et al. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae). BMC Plant Biol. 2019;19(1):1–10.
Timme RE, Kuehl JV, Boore JL, Jansen RK. A comparison of the first two sequenced chloroplast genomes in Asteraceae: lettuce and sunflower. Lawrence Berkeley National Laboratory. (LBNL), Berkeley, CA (United States). 2009. https://escholarship.org/uc/item/2kd25122.
Barrett CF, Davis JI. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. Am J Bot. 2012;99(9):1513–23.
Schneider AC, Braukmann T, Banerjee A, Stefanović S. Convergent plastome evolution and gene loss in Holoparasitic Lennoaceae. Genome Biol Evol. 2018;10(10):2663–70.
Xie Z, Merchant S. The plastid-encoded ccsA gene is required for heme attachment to chloroplast c-type cytochromes. J Biol Chem. 1996;271(9):4632–9.
Sugiura C, Kobayashi Y, Aoki S, Sugita C, Sugita M. Complete chloroplast DNA sequence of the moss Physcomitrella patens: evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 2003;31(18):5324–31.
Ravin NV, Gruzdev EV, Beletsky AV, Mazur AM, Prokhortchouk EB, Filyushin MA, et al. The loss of photosynthetic pathways in the plastid and nuclear genomes of the non-photosynthetic mycoheterotrophic eudicot Monotropa hypopitys. BMC Plant Biol. 2016;16(3):153–61.
Wicke S, Müller KF. dePamphilis CW, Quandt D, Bellot S, Schneeweiss GM. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc Natl Acad Sci U S A. 2016;113(32):9045–50.
Barrett CF, Freudenstein JV, Li J, Mayfield-Jones DR, Perez L, Pires JC, et al. Investigating the path of plastid genome degradation in an early transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol Biol Evol. 2014;31(12):3095–112.
Pérez-Escobar OA, Balbuena JA, Gottschling M. Rumbling orchids: how to assess divergent evolution between chloroplast endosymbionts and the nuclear host. Syst Biol. 2016;65(1):51–65.
Hu H, Hu Q, Al-Shehbaz IA, Luo X, Zeng T, Guo X, Liu J. Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front Plant Sci. 2016;7:1826.
Jiang Y, Yang Y, Lu Z, Wan D, Ren G. Interspecific delimitation and relationships among four Ostrya species based on plastomes. BMC Genet. 2019;20(1):33.
Wolfe KH, Li WH, Sharp PM. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 1987;84(24):9054–8.
Koch M, Haubold B, Mitchell-Olds T. Molecular systematics of the Brassicaceae: evidence from coding plastidic matK and nuclear Chs sequences. Am J Bot. 2001;88(3):534–44.
Pelser PB, Kennedy AH, Tepe EJ, Shidler JB, Nordenstam B, Kadereit JW, et al. Patterns and causes of incongruence between plastid and nuclear Senecioneae (Asteraceae) phylogenies. Am J Bot. 2010;97(5):856–73.
Suh A, Smeds L, Ellegren H. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of Neoavian birds. PLoS Biol. 2015;13(8):e1002224.
Lee SH, Choi HW, Sung JS, Bang JW. Inter-genomic relationships among three medicinal herbs: Cnidium officinale, Ligusticum chuanxiong and Angelica polymorpha. Genes Genom. 2010;32(1):95–101.
Liao C, Downie SR, Li Q, Yu Y, He X, Zhou B. New insights into the phylogeny of Angelica and its allies (Apiaceae) with emphasis on east Asian species, inferred from nrDNA, cpDNA, and morphological evidence. Syst Bot. 2013;38(1):266–81.
Wen J, Yu Y, Xie DF, Peng C, Liu Q, Zhou SD, et al. A transcriptome-based study on the phylogeny and evolution of the taxonomically controversial subfamily Apioideae (Apiaceae). Ann Bot. 2020;125(6):937–53.
Doyle JJ. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):e18.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47(W1):W59–64.
Sharp PM, Li WH. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24(1):28–38.
Peden JF. Analysis of codon usage. PhD thesis. Nottingham: University of Nottingham; 1999.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Mower JP. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes, and user-defined alignments. Nucleic Acids Res. 2009;37:W253–9.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucl Acids Res. 2001;29(22):4633–42.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(suppl_2):W273–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Zhang Y, Ma P, Li D. High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS One. 2011;6:e20596.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91.
Yang ZH, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19(6):908–17.
Zhang X, Zhou T, Yang J, Sun J, Ju M, Zhao Y, et al. Comparative analyses of chloroplast genomes of Cucurbitaceae species: lights into selective pressures and phylogenetic relationships. Molecules. 2018;23(9):2165.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Boil. 2012;61(3):539–42.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8.
Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–9.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32070221, 31872647), National Specimen Information Infrastructure, Educational Specimen Sub- Platform (Grant No. 2005DKA21403-JK), the fourth national survey of traditional Chinese medicine resources (Grant No. 2019PC002). The funders were not involved in the design of the study, collection, analysis and interpretation of data, and manuscript writing.
Author information
Authors and Affiliations
Contributions
TR and X-JH conceived and designed the work. TR, Z-XL, and D-FX analyzed the sequence data. L-JG and CP provided the materials/analysis tools. TR wrote the manuscript. JW and X-JH revised the manuscript. All authors gave final approval of the paper.
Authors’ information
Key Laboratory of Bio-Resources and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, 610065, Chengdu, China
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Number of RNA editing sites in the eight Ligusticum plastomes. Figure S2. Analysis of simple sequence repeats (SSRs) in the eight Ligusticum plastomes. Figure S3. Phylogenetic tree reconstruction of the 39 species inferred from Maximum likelihood (ML) and Bayesian inference (BI) analyses based on nuclear internal transcribed spacer (ITS) sequences. The bootstrap support values and posterior probability values are listed at each node.
Additional file 2: Table S1.
List of genes present in the eight Ligusticum plastomes.
Additional file 3: Table S2.
Codon usage and relative synonymous codon usage (RSCU) values of protein-coding genes of the eight Ligusticum plastomes.
Additional file 4: Table S3.
Base compositions of protein-coding genes for the eight Ligusticum plastomes.
Additional file 5: Table S4.
RNA editing sites analyses of the eight Ligusticum plastomes.
Additional file 6: Table S5.
The repeat sequences distribution in the eight Ligusticum plastomes.
Additional file 7: Table S6.
Simple sequence repeats (SSRs) distribution in the eight Ligusticum plastomes.
Additional file 8: Table S7.
The Indel and SNP in the eight Ligusticum plastomes.
Additional file 9: Table S8.
Percentages of variable characters in coding and non-coding regions.
Additional file 10: Table S9.
Genetic distance of the eight Ligusticum plastomes.
Additional file 11: Table S10.
Results of selective pressure analysis in Paml with the site-specific model.
Additional file 12: Table S11.
List of species and their accession numbers in GenBank included in the phylogenetic analysis.
Additional file 13: Table S12.
Collection locality and voucher information are provided for eight sequenced plastomes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ren, T., Li, ZX., Xie, DF. et al. Plastomes of eight Ligusticum species: characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol 20, 519 (2020). https://doi.org/10.1186/s12870-020-02696-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-02696-7