
Abstract
As with many other diseases, genetic testing in human azoospermia was initially restricted to karyotype analyses (leading to diagnostic chromosome rearrangement tests for Klinefelter and other syndromes). With the advent of molecular biology in the 1980s, genetic screening was broadened to analyses of Y chromosome microdeletions and the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR). Decades later, the emergence of whole-genome techniques has led to the identification of other genetic defects associated with human azoospermia. Although TEX11 and ADGRG2 defects are frequently described in men with azoospermia, most of the causal gene defects found to date are private (i.e. identified in a small number of consanguineous families).
Here, we provide an up-to-date overview of all the types of genetic defects known to be linked to human azoospermia and try to give clinical practice guidelines according to azoospermia phenotype. Along with homozygous mutations, polymorphisms and epigenetic defects are also briefly discussed. However, as these variations predispose to azoospermia, a specific review will be needed to compile data on all the particular genetic variations reported in the literature.
Résumé
Comme pour beaucoup de maladies humaines, les analyses génétiques en cas d’azoospermie étaient initialement limitées à la réalisation d’un caryotype, conduisant au diagnostic de réarrangements chromosomiques comme pour le syndrome de Klinefelter ou autres syndromes. L’avènement de la biologie moléculaire, dans les années 1980, a permis l’élargissement du dépistage génétique à la recherche des microdélétions du chromosome Y et aux anomalies du gène CFTR (cystic fibrosis transmembrane conductance regulator). Il a fallu attendre plusieurs décennies et l’apparition des techniques d’analyses du génome entier pour que soit réalisée l’identification d’autres anomalies génétiques associés à l’azoospermie humaine. Si les anomalies des gènes TEX11 et ADGRG2 sont fréquemment décrites dans la littérature pour les hommes présentant une azoospermie, la plupart des altérations génétiques découvertes à ce jour sont privées, identifiées dans un petit nombre de familles souvent consanguines.
L’objectif dans cette revue est de fournir un aperçu actualisé de toutes les anomalies génétiques décrites dans la littérature et associées à l’azoospermie humaine tout en essayant de fournir des guides de conduite diagnostique en fonction du phénotype de l’azoospermie. En plus des mutations homozygotes et délétères, les polymorphismes et les défauts épigénétiques sont également brièvement abordés. Néanmoins, comme ces variations ne sont que de potentiels facteurs de prédisposition à l’azoospermie, une étude spécifique sera nécessaire pour compiler l’ensemble des données de la littérature pour chaque variant génétique.
Similar content being viewed by others


The World Health Organization (WHO) considers infertility (defined as the inability to conceive after 12 months of sexual intercourse without the use of contraceptives) to be a major health concern. Indeed, infertility affects more than 50 million couples worldwide. In about half of these couples, infertility is of male origin [1].
Semen analysis can often reveal congenital or acquired causes of male infertility. These include quantitative and/or qualitative abnormalities in spermatogenesis, which therefore affect the sperm count, sperm mobility and/or sperm morphology. Azoospermia (defined as the total absence of spermatozoa in the ejaculate in two successive semen examinations) accounts for around 10% of cases of male infertility, and affects about 1% of the men in the general population [2,3,4]. This condition can be classified as non-obstructive azoospermia (NOA, associated with spermatogenesis failure), and obstructive azoospermia (OA, characterized by an obstruction in the seminal tract and normal spermatogenesis). Whereas NOA accounts for 60% of azoospermic patients, OA accounts for around 40% [5, 6].
In almost all cases of azoospermia, the combination of sperm extraction with in vitro fertilization (IVF) and intra-cytoplasmic sperm injection (ICSI) gives these patients an opportunity to father children [7]. A variety of sperm extraction modalities and techniques have been developed, depending on the type of azoospermia. In general, sperm retrieval from the testis or epididymis should be prescribed for azoospermic patients [8]. In patients with OA, percutaneous epididymal aspiration, open fine-needle aspiration, or open surgical procedures (such as microsurgical epididymal sperm aspiration (MESA)) [9, 10] are often used for sperm retrieval. Sperm is successfully retrieved in more than 95% of cases.
However, the clinical management of NOA is more challenging; not all patients have sperm in their testes, and seminiferous tubules with complete spermatogenesis are intermixed with tubules without any germinal cells. In men with NOA, the sperm retrieval rate is around 40 to 50%. As is the case for OA, various sperm extraction techniques have been developed for men with NOA. According to the literature, microdissection testicular sperm extraction (microTESE) in several areas of the testis may be associated with higher sperm retrieval rates and lower postoperative complication rates [11,12,13,14,15].
Three histological phenotypes for NOA can be defined on the basis of the TESE (testicular sperm extraction) results: hypospermatogenesis, Sertoli-cell-only syndrome (SCOS), and maturation arrest (MA) [16, 17]. Thus, TESE also provides information on the infertility phenotype and guides the choice of treatments.
Maturation arrest is defined as incomplete spermatogenesis in which germ cells fail to mature. The condition is subcategorized into early MA, with the presence of spermatogonia or spermatocytes only (i.e. pre-meiotic or meiosis-arrested germ cells) and late MA, in which spermatids can be detected (i.e. post-meiotic arrest). In SCOS, germ cells are completely absent in all seminiferous tubules; only Sertoli cells and Leydig cells can be seen in the seminiferous tubules and the interstitial tissue, respectively [18, 19]. Lastly, hypospermatogenesis is characterized by the presence of all types of germ cell (from spermatogonia to spermatozoa), albeit in small numbers [20]. The degree of this histological phenotype can vary from mild to severe. Although a purely testicular histological phenotype can be found, the mixed pattern, is most frequent observed in azoospermic patients [16].
The many etiologies underlying azoospermia fall into pretesticular, testicular and post-testicular categories (see for review [21, 22]). Pretesticular (central) causes of azoospermia are endocrine abnormalities, and include hypogonadotropic hypogonadism, hyperprolactinemia, and androgen resistance. In contrast, testicular etiologies are characterized by disorders of spermatogenesis inside the testes, such as varicocele-induced testicular damage, undescended testes, testicular torsion, mumps orchitis, gonadotoxic effects of medications, genetic abnormalities, and idiopathic causes. Most cases of NOA have a pretesticular or testicular cause. Lastly, post-testicular etiologies (due to ejaculatory dysfunction or genital tract outflow obstruction) are the major contributors to OA [23, 24]. In the present review, we will not discuss pretesticular etiologies because they correspond to central nervous system defects and not to genital tract disease. Indeed, de novo or familial chromosomal or gene abnormalities constitute well-established genetic causes of azoospermia.
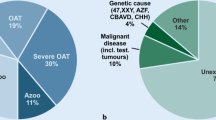
Genetic testing in human azoospermia was initially restricted to karyotype analyses [25,26,27]. With technical progress, genetic screening has been broadened to the analysis of the gene coding for cystic fibrosis transmembrane conductance regulator (CFTR) in patients with OA [28, 29] and Y chromosome microdeletions in patients with NOA [30,31,32,33]. Over the last 5 years, emergence of whole-genome techniques has led to the identification of many other supposedly causal genetic defects – raising the question of which genetic testing techniques should be used to evaluate human azoospermia. Here, we provide an up-to-date overview of all the types of genetic defects known to be linked to human azoospermia, including (i) chromosome abnormalities, (ii) causative gene mutations in OA, (iii) causative gene mutations in NOA, (iv) polymorphisms and (v) epigenetic alterations (Table 1). The last two types of defect are described in less detail.
Chromosome abnormalities
Klinefelter syndrome (KS)
This syndrome was the first chromosomal abnormality to be linked to male infertility. It was first described in 1942 [34], and is the most common genetic etiology of human male infertility. The syndrome is caused by a 47,XXY karyotype [35]. The prevalence of KS is close to 2 per 1000 male births [36, 37]. Eighty percent of cases of KS have a nonmosaic 47,XXY karyotype, whereas the remaining 20% variously show higher-grade chromosome aneuploidies, a 46,XY/47,XXY mosaic, or a structurally abnormal chromosome X [38]. Mosaic KS patients are usually less severely affected than nonmosaic patients are, and few cases of spontaneous paternity have been reported [39, 40]. This situation is not specific to humans; a XXY karyotype is always associated with infertility in various domestic animals (mice, cats, dogs, pigs, cows, horses, etc.) [41,42,43].
The presence of two X chromosomes in a male leads to impaired spermatogenesis and the failure of meiosis because gametogenesis is only possible for 46, XY cells - explaining the presence of gametes in mosaic patients (see for review [44]). Although very few functional tubules may be present in men, focal spermatogenesis enabled the recovery of spermatozoa (using TESE) in almost 50% of cases in a study of 1248 patients; however, none of the tested parameters (including age, testis volume, and levels of FSH, LH and testosterone (T)) had predictive value [45].
In KS, degeneration of the seminiferous tubules starts well before puberty [46] and progresses throughout infancy [47]. A dramatic increase in degeneration frequently occurs at puberty, and often leads to the complete hyalinization of the seminiferous tubules in adulthood [48]. It was initially recommended to cryopreserve testicular tissue as soon as possible in those cases. However, it is now generally acknowledged that TESE in young boys with KS is questionable; germ cells loss probably occurs very early [49], and so may explain the poor results seen for adolescent testicular tissue banking [50].
47,XYY syndrome
This syndrome was first described in 1961 [51], and is associated with a predisposition to infertility ranging from a normal sperm count to azoospermia [52, 53] . In fact, the supernumerary Y chromosome is probably lost in the early stages of spermatogenesis in the great majority of XYY males [54,55,56], thus enabling normal spermatogenesis. However, the supernumerary Y chromosome persists in some XYY males, which results in asynapsed sex chromosomes at the pachytene stage [57, 58]. In this situation, only a trivalent configuration could achieve meiosis [59].
46,XX males
In more than 80% of cases, a 46,X,der(X)t(X;Y)(p22.3;p11.2) karyotype results from an unbalanced de novo X-Y translocation and then the translocation of SRY (sex-determining region of Y chromosome) to the X chromosome. In the remaining 20% of cases, the genetic defect concerns the human sex determination pathway. 46, XX patients often exhibit SCOS [60, 61]. A defect in the SOX9 pathway is most frequently described, with duplication, triplication or balanced chromosomal translocation that overlaps with the so-called RevSex dosage sensitive critical region on chromosome 17q24 [62]. Other defects (like SOX3 duplication [63] and RSPO1 point mutation [64]) are rare but are frequently associated with a syndromic clinical presentation.
Chromosome rearrangements
By comparing infertile men with newborn children, it was found that patients with impaired spermatogenesis have a greater number of chromosome abnormalities and/or rearrangements [65, 66]. Depending on the population studied, the proportion of affected individuals ranged from 2 to 20% [67,68,69], and the frequency of infertility increased with the severity of the impairment in spermatogenesis. Furthermore, it appears that gonosome abnormalities (aneuploidy or balanced translocation) most often result in azoospermia, whereas balanced abnormalities in autosomes tend to result in oligozoospermia.
Chromosome rearrangement appears to impact spermatogenesis through meiotic arrest. Several putative explanations for this association have been suggested. The first hypothesis is based on evidence of an interaction between the human quadrivalent chromosome (the association between the chromosomes involved in the translocation, at the pachytene stage), the acrocentric chromosomes, and the XY body - all of which are located near to the nucleolus [70,71,72]. This leads to an impairment in meiotic sex chromosome inactivation. The second hypothesis relates to the silencing of crucial genes in segments close to the chromosome breakpoints (due to the frequent non-pairing of these autosomal segments) and thus asynapsis. This hypothesis has been confirmed in studies of male mice [73] and boars [74] bearing a translocation.
Y chromosome microdeletions
Frequent observations of Y chromosome rearrangements and large deletions in azoospermic males have suggested that a particular region is required for meiosis (e.g. 46,X,i(Y)(p11); 46,X,r(Y)). Experiments with specific probes have identified various interstitial deletions [75, 76], and have enabled the definition of three regions: AZFa, AZFb, and AZFc (azoospermia factor a, b and c) [77]. The prevalence ranges from 3 to 28%, depending on the type of impairment in spermatogenesis [78]. Although the AZFc phenotype is highly variable, full deletion of AZFa and AZFb always leads to azoospermia (SCOS, and pachytene MA, respectively) [79]. The complete deletion of AZFa and/or AZFb are currently the sole genetic abnormalities that contraindicate TESE.
Clinical practice: karyotyping and Y chromosome microdeletion screening are recommended by the latest international guidelines. This approach leads to a diagnosis in more than 15% of cases. Furthermore, a full AZFa and/or AZFb microdeletion diagnosis contraindicates a testicular biopsy.
Causative gene mutations in OA
Some genetic diseases and abnormalities result in OA; they include cystic fibrosis, congenital bilateral absence of the vas deferens (CBAVD), congenital unilateral absence of the vas deferens, congenital bilateral epididymal obstruction and normal vasa, and Young syndrome. According to the literature, some gene mutations are associated with OA. We shall first describe CFTR mutations, and then mutations that have been described in the literature (starting with ADGRG2 mutations).
CFTR
This gene encodes a protein with an essential role in the sodium/chloride balance in cAMP-regulated epithelial secretions. Defects in the CFTR gene lead to the production of sweat with an abnormally high salt content and mucus secretions with an abnormally high viscosity. Complete loss of CFTR protein function leads to the autosomal recessive disease cystic fibrosis (CF) [80, 81]. The most common features of CF are respiratory symptoms, digestive problems, poor growth, short stature, and male sterility (due to CBAVD). The poor prognosis is due to bronchopulmonary involvement. To date, more than 2000 causal mutations are listed in public databases (https://www.re3data.org/repository/r3d100012093; [82]. These mutations are divided into different classes, depending on their effects on the protein and the disease mechanism [83,84,85]. Cystic fibrosis is the most common life-limiting genetic disorder in Caucasian populations. Several different explanations for the high frequency of heterozygotes in Caucasian populations have been suggested. Although greater fertility was initially hypothesized, it appears that heterozygosity for CFTR mutations confers greater resistance to typhoid fever [86], the effects of cholera toxin, and other diarrheal disorders [87]. Other hypotheses include (i) the development of cattle pastoralism, based on similarities in the distributions of lactase persistence and the most common CF mutation (Delta F508) [88], and (ii) possible respiratory advantages during the dusty climate of the last ice age [89].
Cystic fibrosis is caused by the presence of severe mutations (such as ∆F508, the most frequent CFTR mutation in Caucasian population) in both copies of the CFTR gene. This 3 bp deletion leads to the failure of CFTR protein to migrate to the plasma membrane [90]. Nevertheless, combinations of severe/mild mutations and mild/mild mutations lead to CFTR dysfunction that does not meet the diagnostic criteria for CF. These CFTR-related mutations are linked to a “minimal” phenotype that features CBAVD, chronic or recurrent acute pancreatitis, and disseminated bronchiectasis [91].
The incidence of CBAVD is as high as 6% in men with OA [92, 93]. The production of thick mucus in the genital tract associated with the CFTR mutations leads to vas deferens deterioration. Almost 80% of patients with CBAVD carry a CFTR mutation [94], and other etiologies might account for the phenotype in the remaining 20% of cases. Recently, a few genes have been linked to CBAVD as listed below.
Many studies have found a connection between CFTR mutations and impaired spermatogenesis [95]. A body of clinical evidence has highlighted an elevated mutation frequency and/or abnormally low expression of the CFTR gene in men with sperm abnormalities. The CFTR protein seems to be involved in spermatogenesis in rodent Sertoli cells and germ cells, and low CFTR protein expression has been observed in men with NOA [96]. Furthermore, CFTR has a critical role in sperm capacitation by directly or indirectly mediating HCO3− entry, which is essential for this process [97].
ADGRG2
In patients lacking CFTR mutations, hemizygous protein-truncating mutations in the X-linked gene coding for adhesion G-protein-coupled receptor G2 (ADGRG2) were first described in a study of 26 azoospermic men [98], and then in a replication study of an unrelated population of 18 men [98, 99]. ADGRG2 (located in Xp22.3) is expressed in the efferent ducts and epididymis [100]. Moreover, ADGRG2 regulates fluid reabsorption in the efferent ducts through the ADGRG2-Gq/β-arrestin-1/CFTR signaling complex [101,102,103]. All patients with ADGRG2 mutations (Fig. 1) displayed CBAVD only, and no other symptoms of CF (as with certain mild CFTR variants) - indicating a possible similar involvement of both genes in the development of CBAVD [98, 99] In a recent study, an additive nonsense ADGRG2 mutation was described in two brothers with OA from a Pakistani family [104], confirming the involvement of ADGRG2 mutations in OA.

Schematic depiction of the structure of the ADGRG2 protein. Truncating mutations reported in OA patients are indicated. Yellow rectangles represent the seven transmembrane helices. ADGRG2 is also composed of a G-protein-coupled receptor (GPCR) autoproteolysis-inducing (GAIN) domain containing a cysteine-rich GPCR proteolysis site (GPS), and an extracellular STP region (in grey)
SLC9A3
The gene coding for solute carrier family 9 member A3 has also been described as pathogenic in patients with CBAVD. This protein is a Na+/H+ exchanger expressed on the apical membranes of cells in many structures (including the epididymis, vas deferens, and the non-ciliated cells of the efferent duct) [105, 106]. In the male reproductive tract, SLC9A3 is involved in fluid absorption and acidification [107]. It has been reported that loss of SLC9A3 decreases the expression of CFTR protein and causes OA in mice [108]. These findings suggest that SLC9A3 deletion has an impact in patients with CBAVD [109]. Further studies of the SLC9A3 gene’s involvement in CBAVD are required.
PANK2
In a study of gene copy number variations in Asian patients with CBAVD [110], Lee et al. observed the homozygous loss of the PANK2 gene encoding pantothenate kinase 2. This enzyme is the first in the co-enzyme A (CoA) biosynthetic pathway, and catalyzes the ATP (adenosine triphosphate)-dependent phosphorylation of pantothenate. Homozygous male mutants were infertile due to azoospermia [111] but also displayed retinal degeneration with progressive photoreceptor decline. The putative association between CBAVD and PANK2 has not been confirmed to date.
Clinical practice: given that almost 80% of patients with CBAVD carry a CFTR mutation [94], the latter gene should be fully sequenced. If a CFTR mutation is diagnosed, the patient’s spouse should also be tested (given the likelihood of CF in the offspring). If a CFTR mutation is not revealed by full sequencing, the patient could be screened for a possible defect in ADGRG2 – even though this diagnosis would not modify clinical practice.
Causative gene mutations in NOA
The above-listed chromosome defects are observed in 15% of cases of azoospermia. Hence, one can reasonably hypothesize that most of the genetic causes of male infertility have yet to be characterized - probably because of the large number of genes involved [112]. Given that no more than 20% of men with NOA have chromosomal abnormalities, other spermatogenesis-related gene mutations are probably located elsewhere on the genome. To date, gene mutations have been discovered through studies of inbred families, which have confirmed the great genetic heterogeneity of this pathology. Furthermore, many azoospermic murine models have been described in the literature. A large number of possibly causal single-gene mutations have been reported for patients with the testicular phenotype of NOA (Table 1). Below, we briefly profile a number of candidate genes as a function of the testicular phenotype. We first describe mutations in TEX11 (the gene most frequently cited in the literature) and then list other genes in alphabetical order.
TEX11
This gene (coding for testis expressed 11) on Xq13.1 appears to be the prime gene of interest in NOA. Initially, a 90 kb deletion (encompassing exons 9, 10, and 11) in one isoform of TEX11 was identified (using a chromosome micro-array) in two azoospermic patients with homogeneous or mixed meiotic arrest [113]. This deletion resulted in the loss of 79 amino acids from the TEX11 protein’s meiosis-specific sporulation (Spo22) domain. Additional TEX11 mutations (missense and splice mutations) were found in 2.4% of the azoospermic patients. In line with the phenotype of male Tex11−/− mice, a histological analysis evidenced meiotic arrest and low levels of TEX11 protein expression in patients bearing these mutations. The TEX11 mutations reported to date (Fig. 2) are strongly associated with the occurrence of NOA due to testicular meiotic arrest [114]. In fact, TEX11 gene abnormalities are the sole defects recurrently described in the literature and in sporadic patients. The genes described below have been linked to azoospermia in consanguineous families.

Schematic diagram of the location of TEX11 variants in isoform 2, as detected in patients with azoospermia. Brackets indicate the TEX11 protein’s interaction domains (the SPO22 sporulation domain and the TPR tetratricopeptide repeat-containing domain), according to the TEX11–203 transcript in the Ensembl database (https://www.ensembl.org/index.html). Orange boxes represent exons, and black lines represent introns. Missense mutations are shown in red, with splice site mutations in blue, silent mutations in green, frameshift mutations in grey, intronic mutations in pink, and deletions in purple
DMC1
DMC1 is essential for meiotic recombination in various organisms. Whole-exome sequencing of DNA (deoxyribonucleic acid) samples from two members of a consanguineous Chinese family (a man with NOA and a woman with premature ovarian insufficiency) enabled the identification of a homozygous missense mutation in the DMC1 gene [115]. A detailed analysis evidenced MA at the zygotene stage in the seminiferous tubules of the patient with NOA.
DNAH6
A rare, nonsynonymous mutation in the dynein axonemal heavy chain 6 (DNAH6) gene has been reported in azoospermic brothers from a consanguineous family [116]. DNAH6 protein is strongly expressed in testis, and DNAH6 is important for meiosis [117] and ciliary beating. Mutations in DNAH6 have also been linked to primary ciliary dyskinesia and sperm head anomaly [118], as well as to NOA.
MAGEB4
The analysis of a consanguineous Turkish family led to the identification of a novel nonstop mutation in the X-linked gene MAGEB4 (coding for melanoma antigen family B4) that segregated with an azoospermic and oligozoospermic phenotype [119]. In the testis, MAGEB4 is specifically expressed during germ cell differentiation [120].
MCM8
[121] reported a homozygous mutation in the MCM8 gene (coding for minichromosome maintenance complex component 8 and located on chromosome 20p12.3) in a consanguineous family in which a male with a 22q11.2 microdeletion presented azoospermia and a female had primary amenorrhea. Both individuals presented mild mental retardation. The complex formed by the MCM8 and MCM9 proteins has a key role in homologous-recombination (HR)-mediated DNA repair [122,123,124]. MCM8−/− mice display infertility, a blockage in meiotic HR-mediated double-strand break (DSB) repair, and the absence of post-meiotic cells - confirming the importance of this gene in the meiotic stage of spermatogenesis [124].
MEIOB
A homozygote non-synonymous mutation in the MEIOB gene has been identified in members of one family [116]. One of the brothers showed a meiotic arrest, as observed in Meiob knock-out mice [125, 126]. The mutation occurred in the MEIOB protein’s replication protein A1 DNA binding domain, and might have altered the meiotic recombination process. These studies highlight MEIOB’s role in meiosis (DSB repair and complete synapsis) and fertility in both humans and mice.
MEI1
A homozygous missense mutation in the MEI1 gene (coding for meiotic double-stranded break formation protein 1) has been described in two azoospermic brothers from a consanguineous family [127]. Meiotic arrest at the pachytene stage was confirmed in one brother. The mutation affecting the MEI1 gene was found to co-segregate with the family’s NOA phenotype, and was heterozygous or absent in the other (fertile) family members. Meiotic double-stranded break formation protein 1 is overexpressed in testis, and is necessary for pairing of meiotic chromosomes. It may also be involved in the formation of meiotic DSBs in gonocytes. Mutant mice were infertile, due to meiotic arrest [128]. Consequently, defects in this gene are thought to disrupt the meiotic process. It has been reported that polymorphic alleles of the human MEI1 are associated with human azoospermia caused by meiotic arrest [129].
NPAS2
Using whole-exome sequencing, [130] identified a damaging non-synonymous mutation in NPAS2 in three brothers with NOA from a consanguineous family. NPAS2 (expressed in testis and cerebral cortex) encodes a member of the basic helix-loop-helix/PAS family of transcription factors, with functions in circadian rhythms and fertility.
PSMC3IP
PSMC3 interacting protein has several functions, including the co-activation of ligand-dependent transcription mediated by nuclear hormone receptors, and the activation of DMC1 and RAD51 during meiotic recombination [131]. Recently, Al-Agha et al. identified a homozygous stop gain mutation in exon 6 of the PSMC3IP gene in an azoospermic man from a consanguineous family. This mutation was also present in his four sisters – all of whom suffered from primary ovarian insufficiency [132]. PSMC3IP is strongly expressed in testis of humans and mice. Null-mutant mice exhibit meiotic arrest at the spermatocyte I stage, and the failure of synaptonemal complex formation.
SPINK2
SPINK2 is an acrosomal protein that targets acrosin in sperm and has an essential role in spermiogenesis. It is located in the acrosomal vesicle in round spermatids, and persists in mature spermatozoa. Researchers identified a homozygous splice mutation in the SPINK2 gene in two brothers from a consanguineous family [133]. One of the two brothers had a low round spermatid count in a testicular biopsy. Studies of knock-out mice also confirmed the involvement of SPINK2 in NOA, with spermiogenesis arrest at the round spermatid stage. This arrest was due to Golgi fragmentation and the failure of acrosome biogenesis in the absence of SPINK2 protein.
STX2
Nakamura et al. identified a homozygous frameshift mutation in the syntaxin-2 (STX2) gene [134] in just one member of a population of 131 Japanese men with NOA. Histological analysis of the patient’s testis revealed MA and multinucleated spermatocytes. Furthermore, this gene is located within the 58.4 Mb genomic region with loss of heterozygosity, suggesting that the parents were consanguineous. In view of the phenotype seen in mice [135], it has been suggested that NOA may be caused by STX2 mutations in a small proportion of patients.
SYCE1
A pathogenic splice site mutation in the SYCE1 gene (coding for synaptonemal complex central element 1) was identified in two azoospermic brothers with complete meiotic arrest from a consanguineous family [136]. This mutation disrupted the acceptor site of intron 3, and as a result, no SYCE1 protein could be detected in the patient’s seminiferous tubules. SYCE1 is one of the four components of the synaptonemal complex required for chromosome pairing. Its absence leads to the disruption of synapsis in mice [137].
TAF4B
A homozygous mutation in the TAF4B gene (coding for TATA box-binding protein-associated factor 4B) resulted in NOA in two unrelated consanguineous families [138]. In the first family, the three affected brothers were homozygous for the same nonsense mutation in TAF4B; the resulting truncated protein lacked the histone fold domain (which is important for the DNA-binding activity of TAFs) and the TAF12 interaction domain). This gene is a transcriptional regulator enriched in human and mouse testis. However, TAF4B variants were not associated with NOA in a recent study of a Han population in north-east China [139]. Null mutant mice become infertile by the age of 3 months, with an absence of germ cells in the seminiferous tubules and an impairment in spermatogonial stem cell proliferation [140].
TDRD7
A recent study of a consanguineous Chinese family reported two novel homozygous loss-of-function mutations in the TDRD7 gene in individuals with congenital cataract and NOA [141]. One of the patients displayed a post-meiotic arrest in spermatogenesis, with the absence of mature spermatozoa in the seminiferous tubules. However, a TDRD7 mutation is not a common cause for NOA because variants were not found in cohorts of patients with NOA alone or with congenital cataract alone. The researchers then confirmed the mutations’ impact in a mouse model, where the phenotype was similar to that seen in the two patients. TDRD7 encodes a Tudor family protein required for the remodeling of dynamic ribonucleoprotein particles in chromatid bodies during spermatogenesis [142]. Furthermore, the encode protein repressed LINE1 retrotransposons in the male germline - highlighting its importance in spermatogenesis and male fertility.
TDRD9
The Tudor-domain containing 9 protein (TDRD9) is a member of the DEAD-box helicase family. It represses transposable elements and prevents their mobility via the piwi-interacting RNA (piRNA) metabolic process [143]. A 4 bp deletion frameshift mutation in TDRD9 has been identified in five infertile azoospermic men from a large consanguineous family; the mutation led to the loss of all the known functional domains [144]. Tdrd9−/− male mice were sterile, with activation of retrotransposon line-1 and chromosomal synapsis failure [143].
TEX14
TEX14 is considered to be a novel causative gene for NOA because its expression is abnormally low in men with NOA [145]. TEX14 protein is exclusively expressed in testis, especially during meiosis [146]. TEX14 has a major role in spermatogenesis, where it is thought to be required for the formation of intercellular bridges in germ cells during meiosis [147]. A recent study of two azoospermic brothers from a consanguineous family revealed a 10 bp frameshift deletion, which resulted in an early stop codon [116]. Azoospermia or infertility has also been observed in pigs [148] and mice with Tex14 mutations [147].
TEX15
In studies of two different families, mutations in the TEX15 gene (required for meiotic recombination in spermatocytes) segregated with the NOA phenotype [149, 150]. In the first study, two brothers with NOA had a compound-heterozygote nonsense mutation. In the second, a homozygous nonsense mutation was identified in three Turkish brothers with azoospermia. Observations in a mouse model confirmed the patients’ infertility phenotype, since loss of the Tex15 gene disrupted the DSB repair process and induced sterility (in males only) with meiotic arrest in the testis [151]. Two association studies of TEX15 single-nucleotide polymorphisms (SNPs) gave contradictory results; a link to spermatogenetic failure was observed in one study [152] but not the other [153].
XRCC2
Recently, Yang et al. identified a point mutation in the XRCC2 gene (coding for X-ray repair cross-complementing protein 2 homolog, a RAD51 paralog) in two brothers with meiotic arrest and azoospermia from a consanguineous family [154]. The XRCC2 gene’s product is involved in HR (homologous-recombination)-mediated DSB repair. Recreation of this mutation in mice using Crispr-Cas9 (clustered regularly interspaced short palindromic repeats associated proteins 9) technology also induced meiotic arrest and infertility, and thus confirmed its involvement in the patients’ phenotype. Another study identified a mutation in XRCC2 that causes NOA and premature ovarian insufficiency [155]. One can therefore conclude that XRCC2 is an essential for the progression of meiosis, and that a mutation in this gene could cause infertility in humans. Polymorphisms in XRCC2 homologs 1, 5, 6 and 7 have been linked to male infertility [156,157,158].
ZMYND15
In three azoospermic brothers with MA at the spermatid stage, a homozygous mutation in the gene coding for ZMYND15 (zinc finger MYND-containing protein 15) led to amputation of the proline-rich domain (essential for cytoskeleton binding and signal transduction) [138]. ZMYND15 is involved in spermiogenesis and acts as a histone deacetylase-dependent transcriptional repressor. When ZMYND15 was inactivated, male mice displayed infertility and a low late spermatid count [159].
Clinical practice: with the exception of TEX11 defects (recurrent but rare in NOA), the other mutations seems to be private. So, whole-exome sequencing might be of diagnostic value, given that most gene defects are associated with meiotic arrest and thus rule out the retrieval of any spermatozoa. A number of points must to be considered: (i) the need for pedigree studies to identify consanguineous patients, (ii) the practical difficulty of analyzing genomic DNA samples, (iii) the time and cost of whole-exome sequencing, (iv) the absence of specific therapies, (v) the patient’s gratitude upon receipt of an etiologic diagnosis for his infertility. At present, whole-exome sequencing appears to have been restricted to clinical research. Hence, only TEX11 screening should be considered because defects are associated with meiotic arrest. However, the development of genetic analysis software and emergence of new genetic therapies (e.g. induced pluripotent stem cells [160]) might modify the diagnosis of NOA.
Polymorphisms and related variations associated with azoospermia
Gene-targeted sequencing and candidate gene approaches have enabled the identification of a large number of SNPs and heterozygous mutations linked to azoospermia or which might predispose to impairments of spermatogenesis. Most of these studies were carried out on a small numbers of azoospermic patients and controls. We searched the PubMed database with the following keywords: (((((azoospermia[MeSH Major Topic]) or azoospermia[Title/Abstract]) AND (polymorphism[Title/Abstract] OR polymorphisms[Title/Abstract]))) NOT review[Publication Type]) NOT meta-analysis[Title]) AND English[Language], and then (((((azoospermia[MeSH Major Topic]) or azoospermia[Title/Abstract]) AND (mutation[Title/Abstract] OR mutation[Title/Abstract]))) NOT review[Publication Type]) NOT meta-analysis[Title]) AND English[Language]. The search yielded a list of more than 600 publications. After selecting only publications dealing with polymorphisms, SNP or heterozygote mutations, we found that 182 genes have been highlighted in azoospermic or oligo/azoospermic populations. The most frequently studied gene was MTHFR, in 19 different publications. Few genome-wide association studies have been performed in this field; a few loci have been identified but their association with male infertility has yet to be confirmed. We did not find any clear methodological proposals in the literature on how to use SNPs associated with spermatogenesis failure.
Clinical practice: screening polymorphism does not currently appear to be of great value because a diagnosis wouldn’t influence the patient’s treatment. Only MTHFR screening could be considered [161], despite the present lack of a randomized, placebo-controlled study.
Epigenetic alterations in azoospermia
Along with genetic defects, epigenetic alterations (i.e. heritable alterations in gene function that do not affect the basic DNA sequence [162]) are now being increasing studied in the field of human infertility [163,164,165,166]. Epigenetics has an essential role during sperm production, sperm function, and fertilization. Sperm cells are epigenetically programmed through histone-protamine replacement, DNA methylation (> 80%), chromatin remodeling, genomic imprinting, and the involvement of small non-coding RNAs (piRNAs [167] and microRNAs (miRNA) [168, 169]). Hence, many studies have evidenced epigenetic changes in cases of azoospermia.
It was recently shown that mRNA and protein expression levels of the KDM3A gene (coding for lysine demethylase 3A) were abnormally low in testicular biopsies from patients with meiotic arrest at the round spermatid level or with SCOS, relative to samples from patients with OA [170]. Lysine demethylase 3A is a histone demethylase that is dynamically expressed in male germ cells. It regulates the expression of genes required for the packaging and condensation of sperm chromatin, such as PRM1 and TNP1 [166, 167, 171,172,173]. Furthermore, elevated histone H4 acetylation (essential for spermiogenesis) was observed in the nuclei of Sertoli cells in testicular biopsies from patients with SCOS, relative to controls [174]. Earlier, Sonnack et al. had observed low levels of H4 acetylation in the spermatids of patients with azoospermia; this contrasted with the hyperacetylation of this histone seen in spermatids from fertile patients [175].
In 2009, the methylation status of the promoter region of the MTHFR gene (coding for a regulatory enzyme involved in re-methylation reactions, DNA synthesis and the process of folate metabolism) was performed in patients with NOA and OA [176]. Relative to fertile controls, MTHFR was hypermethylated in DNA obtained from testicular biopsies (but not from peripheral blood) in men with NOA. It has been suggested that aberrant methylation of the MTHFR promoter reduces the expression and enzymatic activity of the encoded protein, leading to the development of azoospermia in these patients.
Genome-wide DNA methylation was subsequently assessed in testicular tissues from 94 azoospermic patients with OA or NOA and either positive or negative TESEs. The OA and NOA differed significantly with regard to the DNA methylation profile at over 9000 CpG sites. Accordingly, patients could be classified as having OA or NOA by considering the 212 CpG sites with the greatest methylation differences [177]. Fourteen of these 212 CpG sites were located in genes with a specific testicular function - suggesting the presence of epigenetic differences between types of azoospermia.
The association between DNA methylation and azoospermia has been extensively explored [178, 179]. For example, more than 30% of gene promoters differed in their DNA methylation status in men with NOA vs. fertile controls [180]. In particular, a hypermethylated DDR1 gene (coding for discoidin domain receptor 1, a subfamily of receptor tyrosine kinases expressed in human postmeiotic germ cells) displayed an abnormal expression profile; it was overexpressed in 25% of the patients and underexpressed in 16%. The protein was not found in the testis of patients with SCOS.
Most recently, Li et al. have sought to identify methylation-regulated genes involved in NOA [181]. In a microarray analysis, a hypermethylated, down-regulated gene coding for zinc-finger CCHC-type containing 13 (ZCCHC13) was found to have low protein expression in NOA testis. The ZCCHC13 protein upregulates the AKT/MAPK/c-MYC signaling pathway. Hypermethylation of ZCCHC13 might induce c-MYC lower expression and therefore act on cell differentiation and proliferation by altering the expression of c-MYC’s target genes.
Similarly, a study of the methylation status of the paternally imprinted H19 gene and the maternally imprinted MEST gene in spermatogenic cells from azoospermic patients with either complete or incomplete MA revealed the presence of imprinting errors [182]. Low levels of H19 gene methylation were observed in primary spermatocytes and elongated spermatids, and MEST methylation errors were found in spermatocytes [182]. These results are in line with previous reports of gene imprinting errors in azoospermia [183].
These epigenetic alterations might be valuable biomarkers for male infertility in general and idiopathic azoospermia in particular. For example, it has been suggested that miRNAs (essential for spermatogenesis and possibly involved in the regulation of gene expression) are diagnosis biomarkers for azoospermia. Indeed, miRNA expression was altered in patients, relative to controls [179,180,181,182, 184,185,188]. A recent comparison of men with OA and men with NOA evidenced differences in miRNA expression in spermatogonia, spermatocytes and round spermatids, and thus suggested the presence of epigenetic dysregulation in NOA [189]. A comparison of subgroups of NOA patients with a positive vs. negative TESE gave similar results [190].
Clinical practice: in summary, it is clear that dynamic epigenetic processes are essential for normal spermatogenesis, and are being increasingly investigated in men with NOA. This research may open up perspectives for diagnosis and treatment.
Conclusion
After the description of the Klinefelter syndrome karyotype (in 1959) and various chromosome rearrangements, it was several decades before the emergence of new genomic techniques initiated a new age for molecular studies of the etiology, mechanism, and diagnosis of azoospermia. Therapeutic approaches may even emerge in the near future. Genetic causes of azoospermia are not limited to gene alterations alone; epigenetic variations, SNPs and other polymorphisms have an impact on spermatogenesis.
Experiments in animal models will probably be needed to characterize all the pathways involved in spermatogenesis and (from a therapeutic perspective) circumvent defects in this process. New technologies (such as Crispr-Cas9) may make it possible to perform genome editing in animal models and thus confirm the causes of spermatogenesis failure.
Ideally, genetic studies of azoospermia should include a large number of patients with a defined phenotype, and a control group matched for ethnicity. Nevertheless, studies of consanguineous families may also generate new strategies that could be extended to all types of azoospermia.
Lastly, the following question arises; does it really make sense to restrict the genetic evaluation of azoospermia to karyotyping, CFTR testing and screening for chromosome Y microdeletions?
General guidelines:
Genetic screening in NOA: patients should be karyotyped and screened for Y chromosome microdeletions; these analyses lead to a diagnosis in more than 15% of cases, and contraindicate a testicular biopsy when a full AZFa and/or AZFb microdeletion is present. Depending on the geneticist’s experience, whole-exome sequencing could also be performed (together with a family segregation study). It should be borne in mind that guidelines on new gene defects are lacking, and that (with the exception of TEX11 defects) most gene defects are private.
Genetic screening in OA: with a view to avoiding CF in the offspring, patients with CBAVD should undergo whole gene sequencing. If mutations are detected, the patient’s spouse should also undergone this sequencing. Although screening might detect defects in ADGRG2, this observation would not change clinical practice.
Abbreviations
- ATP:
-
Adenosine triphosphate
- AZF:
-
Azoospermia factor
- CBAVD:
-
Congenital bilateral absence of the vas deferens
- CF:
-
Cystic fibrosis
- Crispr-Cas9:
-
Clustered regularly interspaced short palindromic repeats associated proteins 9
- DNA:
-
Deoxyribonucleic acid
- DSB:
-
Double-strand break
- HR:
-
Homologous-recombination
- ICSI:
-
Intra-cytoplasmic sperm injection
- IVF:
-
In vitro fertilization
- KS:
-
Klinefelter syndrome
- MA:
-
Maturation arrest
- MESA:
-
Microsurgical epididymal sperm aspiration
- micro-TESE:
-
Microdissection testicular sperm extraction
- miRNA:
-
Micro RNA
- NOA:
-
Non-obstructive azoospermia
- OA:
-
Obstructive azoospermia
- piRNA:
-
Piwi-interacting RNA
- SCOS:
-
Sertoli-cell-only syndrome
- SNP:
-
Single-nucleotide polymorphism
- TESE:
-
Testicular sperm extraction
- WHO:
-
World Health Organization
References
Agarwal A, Mulgund A, Hamada A, Chyatte MR. A unique view on male infertility around the globe. Reprod Biol Endocrinol RBE. 2015;13:37. https://doi.org/10.1186/s12958-015-0032-1.
Jarow JP, Espeland MA, Lipshultz LI. Evaluation of the azoospermic patient. J Urol. 1989;142:62–5. https://doi.org/10.1016/S0022-5347(17)38662-7.
Foresta C, Ferlin A, Bettella A, Rossato M, Varotto A. Diagnostic and clinical features in azoospermia. Clin Endocrinol. 1995;43:537–43.
Willott GM. Frequency of azoospermia. Forensic Sci Int. 1982;20:9–10.
Thonneau P, Marchand S, Tallec A, Ferial ML, Ducot B, Lansac J, et al. Incidence and main causes of infertility in a resident population (1,850,000) of three French regions (1988-1989). Hum Reprod Oxf Engl. 1991;6:811–6.
Matsumiya K, Namiki M, Takahara S, Kondoh N, Takada S, Kiyohara H, et al. Clinical study of azoospermia. Int J Androl. 1994;17:140–2. https://doi.org/10.1111/j.1365-2605.1994.tb01233.x.
Donoso P, Tournaye H, Devroey P. Which is the best sperm retrieval technique for non-obstructive azoospermia? A systematic review. Hum Reprod Update. 2007;13:539–49. https://doi.org/10.1093/humupd/dmm029.
Esteves SC, Miyaoka R, Agarwal A. Surgical treatment of male infertility in the era of intracytoplasmic sperm injection – new insights. Clinics. 2011;66:1463–77. https://doi.org/10.1590/S1807-59322011000800026.
Kovac JR, Lehmann KJ, Fischer MA. A single-center study examining the outcomes of percutaneous epididymal sperm aspiration in the treatment of obstructive azoospermia. Urol Ann. 2014;6:41–5. https://doi.org/10.4103/0974-7796.127026.
Esteves SC, Miyaoka R, Orosz JE, Agarwal A. An update on sperm retrieval techniques for azoospermic males. Clinics. 2013;68:99–110. https://doi.org/10.6061/clinics/2013(Sup01)11.
Schlegel PN. Testicular sperm extraction: microdissection improves sperm yield with minimal tissue excision. Hum Reprod Oxf Engl. 1999;14:131–5.
Amer M, Ateyah A, Hany R, Zohdy W. Prospective comparative study between microsurgical and conventional testicular sperm extraction in non-obstructive azoospermia: follow-up by serial ultrasound examinations. Hum Reprod Oxf Engl. 2000;15:653–6.
Okada H, Dobashi M, Yamazaki T, Hara I, Fujisawa M, Arakawa S, et al. Conventional versus microdissection testicular sperm extraction for nonobstructive azoospermia. J Urol. 2002;168:1063–7. https://doi.org/10.1097/01.ju.0000025397.03586.c4.
Tsujimura A. Microdissection testicular sperm extraction: prediction, outcome, and complications. Int J Urol Off J Jpn Urol Assoc. 2007;14:883–9. https://doi.org/10.1111/j.1442-2042.2007.01828.x.
Franco G, Scarselli F, Casciani V, De Nunzio C, Dente D, Leonardo C, et al. A novel stepwise micro-TESE approach in non obstructive azoospermia. BMC Urol. 2016;16. https://doi.org/10.1186/s12894-016-0138-6.
McLachlan RI, Rajpert-De Meyts E, Hoei-Hansen CE, de Kretser DM, Skakkebaek NE. Histological evaluation of the human testis--approaches to optimizing the clinical value of the assessment: mini review. Hum Reprod Oxf Engl. 2007;22:2–16. https://doi.org/10.1093/humrep/del279.
Robin G, Boitrelle F, Leroy X, Peers M-C, Marcelli F, Rigot J-M, et al. Assessment of azoospermia and histological evaluation of spermatogenesis. Ann Pathol. 2010;30:182–95. https://doi.org/10.1016/j.annpat.2010.03.015.
Tournaye H, Camus M, Vandervorst M, Nagy Z, Joris H, Van AS, et al. Surgical sperm retrieval for intracytoplasmic sperm injection. Int J Androl. 1997;20(Suppl 3):69–73.
Tsujimura A, Matsumiya K, Miyagawa Y, Tohda A, Miura H, Nishimura K, et al. Conventional multiple or microdissection testicular sperm extraction: a comparative study. Hum Reprod. 2002;17:2924–9. https://doi.org/10.1093/humrep/17.11.2924.
Dohle GR, Elzanaty S, van Casteren NJ. Testicular biopsy: clinical practice and interpretation. Asian J Androl. 2012;14:88–93. https://doi.org/10.1038/aja.2011.57.
Hamada AJ, Esteves SC, Agarwal A. A comprehensive review of genetics and genetic testing in azoospermia. Clin Sao Paulo Braz. 2013;68(Suppl 1):39–60.
Cocuzza M, Alvarenga C, Pagani R. The epidemiology and etiology of azoospermia. Clinics. 2013;68:15–26. https://doi.org/10.6061/clinics/2013(Sup01)03.
Fisch H, Lambert SM, Goluboff ET. Management of ejaculatory duct obstruction: etiology, diagnosis, and treatment. World J Urol. 2006;24:604–10. https://doi.org/10.1007/s00345-006-0129-4.
Clements KM, Shipley CF, Coleman DA, Ehrhart EJ, Haschek WM, Clark SG. Azoospermia in an 8-month-old boar due to bilateral obstruction at the testis/epididymis interface. Can Vet J Rev Veterinaire Can. 2010;51:1130–4.
Jalbert P, Servoz-Gavin M, Amblard F, Pison H, Augusseau S, Jalbert H, et al. Role of karyotype in studying male infertility. J Gynecol Obstet Biol Reprod (Paris). 1989;18:724–8.
Hazama M, Nakano M, Shinozaki M, Fujisawa M, Okamoto Y, Oka N, et al. Male infertility with chromosomal abnormalities. III. 46, XYq. Hinyokika Kiyo. 1988;34:1063–8.
Diaz-Castaños LR, Rivera H, Gonzalez-Montes RM, Diaz M. Translocation (Y;19)(q12;q13) and azoospermia. Ann Genet. 1991;34:27–9.
Meschede D, Keck C, De Geyter C, Eigel A, Horst J, Nieschlag E. Mutation in the cystic fibrosis transmembrane-regulator gene in bilateral congenital ductus deferens aplasia. Dtsch Med Wochenschr 1946. 1993;118:661–4. https://doi.org/10.1055/s-2008-1059376.
Stuppia L, Antonucci I, Binni F, Brandi A, Grifone N, Colosimo A, et al. Screening of mutations in the CFTR gene in 1195 couples entering assisted reproduction technique programs. Eur J Hum Genet EJHG. 2005;13:959–64. https://doi.org/10.1038/sj.ejhg.5201437.
Quilter CR, Svennevik EC, Serhal P, Ralph D, Bahadur G, Stanhope R, et al. Cytogenetic and Y chromosome microdeletion screening of a random group of infertile males. Fertil Steril. 2003;79:301–7.
Bor P, Hindkjær J, Ingerslev HJ, Kølvraa S. Genetics: multiplex PCR for screening of microdeletions on the Y chromosome. J Assist Reprod Genet. 2001;18:291–8. https://doi.org/10.1023/A:1016618418319.
Bardoni B, Zuffardi O, Guioli S, Ballabio A, Simi P, Cavalli P, et al. A deletion map of the human Yq11 region: implications for the evolution of the Y chromosome and tentative mapping of a locus involved in spermatogenesis. Genomics. 1991;11:443–51.
Henegariu O, Hirschmann P, Kilian K, Kirsch S, Lengauer C, Maiwald R, et al. Rapid screening of the Y chromosome in idiopathic sterile men, diagnostic for deletions in AZF, a genetic Y factor expressed during spermatogenesis. Andrologia. 1994;26:97–106.
Klinefelter HF, Reifenstein EC, Albright F. Syndrome characterized by gynecomastia, aspermatogenesis without A-Leydigism, and increased excretion of follicle-stimulating hormone. J Clin Endocrinol. 1942;2:615–27. https://doi.org/10.1210/jcem-2-11-615.
Jacobs PA, Strong JA. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature. 1959;183:302–3.
Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet. 1991;87:81–3.
Morris JK, Alberman E, Scott C, Jacobs P. Is the prevalence of Klinefelter syndrome increasing? Eur J Hum Genet EJHG. 2008;16:163–70. https://doi.org/10.1038/sj.ejhg.5201956.
Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet Lond Engl. 2004;364:273–83. https://doi.org/10.1016/S0140-6736(04)16678-6.
Laron Z, Dickerman Z, Zamir R, Galatzer A. Paternity in Klinefelter’s syndrome--a case report. Arch Androl. 1982;8:149–51.
Terzoli G, Lalatta F, Lobbiani A, Simoni G, Colucci G. Fertility in a 47,XXY patient: assessment of biological paternity by deoxyribonucleic acid fingerprinting. Fertil Steril. 1992;58:821–2. https://doi.org/10.1016/S0015-0282(16)55334-5.
Hancock JL, Daker MG. Testicular hypoplasia in a boar with abnormal sex chromosome constitution (39 XXY). J Reprod Fertil. 1981;61:395–7.
Dunn HO, Lein DH, McEntee K. Testicular hypoplasia in a Hereford bull with 61,XXY karyotype: the bovine counterpart of human Klinefelter’s syndrome. Cornell Vet. 1980;70:137–46.
Meyers-Wallen VN. Genetics of sexual differentiation and anomalies in dogs and cats. J Reprod Fertil Suppl. 1993;47:441–52.
Aksglaede L, Juul A. Therapy of endocrine disease: testicular function and fertility in men with Klinefelter syndrome: a review. Eur J Endocrinol. 2013;168:R67–76. https://doi.org/10.1530/EJE-12-0934.
Corona G, Pizzocaro A, Lanfranco F, Garolla A, Pelliccione F, Vignozzi L, et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2017;23:265–75. https://doi.org/10.1093/humupd/dmx008.
Coerdt W, Rehder H, Gausmann I, Johannisson R, Gropp A. Quantitative histology of human fetal testes in chromosomal disease. Pediatr Pathol. 1985;3:245–59.
Aksglaede L, Wikström AM, Rajpert-De Meyts E, Dunkel L, Skakkebaek NE, Juul A. Natural history of seminiferous tubule degeneration in Klinefelter syndrome. Hum Reprod Update. 2006;12:39–48. https://doi.org/10.1093/humupd/dmi039.
Wikström AM, Raivio T, Hadziselimovic F, Wikström S, Tuuri T, Dunkel L. Klinefelter syndrome in adolescence: onset of puberty is associated with accelerated germ cell depletion. J Clin Endocrinol Metab. 2004;89:2263–70. https://doi.org/10.1210/jc.2003-031725.
Van Saen D, Vloeberghs V, Gies I, Mateizel I, Sermon K, De Schepper J, et al. When does germ cell loss and fibrosis occur in patients with Klinefelter syndrome? Hum Reprod Oxf Engl. 2018;33:1009–22. https://doi.org/10.1093/humrep/dey094.
Rives N, Milazzo JP, Perdrix A, Castanet M, Joly-Hélas G, Sibert L, et al. The feasibility of fertility preservation in adolescents with Klinefelter syndrome. Hum Reprod Oxf Engl. 2013;28:1468–79. https://doi.org/10.1093/humrep/det084.
Sandberg AA, Koepf GF, Ishihara T, Hauschka TS. AN XYY HUMAN MALE. Lancet. 1961;278:488–9. https://doi.org/10.1016/S0140-6736(61)92459-X.
Skakkebaek NE, Hultén M, Jacobsen P, Mikkelsen M. Quantification of human seminiferous epithelium. II. Histological studies in eight 47,XYY men. J Reprod Fertil. 1973;32:391–401.
Abdel-Razic MM, Abdel-Hamid IA, ElSobky ES. Nonmosaic 47,XYY syndrome presenting with male infertility: case series. Andrologia. 2012;44:200–4. https://doi.org/10.1111/j.1439-0272.2010.01129.x.
Chandley AC, Fletcher J, Robinson JA. Normal meiosis in two 47,XYY men. Hum Genet. 1976;33:231–40.
Speed RM, Faed MJ, Batstone PJ, Baxby K, Barnetson W. Persistence of two Y chromosomes through meiotic prophase and metaphase I in an XYY man. Hum Genet. 1991;87:416–20.
Gabriel-Robez O, Delobel B, Croquette MF, Rigot JM, Djlelati R, Rumpler Y. Synaptic behaviour of sex chromosome in two XYY men. Ann Genet. 1996;39:129–32.
Mahadevaiah SK, Evans EP, Burgoyne PS. An analysis of meiotic impairment and of sex chromosome associations throughout meiosis in XYY mice. Cytogenet Cell Genet. 2000;89:29–37. https://doi.org/10.1159/000015585.
Rodriguez TA, Burgoyne PS. Evidence that sex chromosome asynapsis, rather than excess Y gene dosage, is responsible for the meiotic impairment of XYY mice. Cytogenet Cell Genet. 2000;89:38–43. https://doi.org/10.1159/000015559.
Rives N, Siméon N, Milazzo JP, Barthélémy C, Macé B. Meiotic segregation of sex chromosomes in mosaic and non-mosaic XYY males: case reports and review of the literature. Int J Androl. 2003;26:242–9.
Gurbuz F, Ceylaner S, Erdogan S, Topaloglu AK, Yuksel B. Sertoli cell only syndrome with ambiguous genitalia. J Pediatr Endocrinol Metab JPEM. 2016;29:849–52. https://doi.org/10.1515/jpem-2015-0458.
Jain M, VeeraMohan V, Chaudhary I, Halder A. The Sertoli cell only syndrome and Glaucoma in a sex - determining region Y (SRY) positive XX infertile male. J Clin Diagn Res JCDR. 2013;7:1457–9. https://doi.org/10.7860/JCDR/2013/5186.3169.
Délot EC, Vilain EJ. Nonsyndromic 46,XX Testicular Disorders of Sex Development. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993. Available: http://www.ncbi.nlm.nih.gov/books/NBK1416/.
Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet EJHG. 2015;23:1025–32. https://doi.org/10.1038/ejhg.2014.237.
Tallapaka K, Venugopal V, Dalal A, Aggarwal S. Novel RSPO1 mutation causing 46,XX testicular disorder of sex development with palmoplantar keratoderma: A review of literature and expansion of clinical phenotype. Am J Med Genet A. 2018;176:1006–10. https://doi.org/10.1002/ajmg.a.38646.
Koulischer L, Schoysman R. Studies of the mitotic and meiotic chromosomes in infertile males. J Genet Hum. 1975;23(SUPPL):58–70.
Chandley AC. The chromosomal basis of human infertility. Br Med Bull. 1979;35:181–6.
Retief AE, Van Zyl JA, Menkveld R, Fox MF, Kotzè GM, Brusnickỳ J. Chromosome studies in 496 infertile males with a sperm count below 10 million/ml. Hum Genet. 1984;66:162–4.
Bourrouillou G, Dastugue N, Colombies P. Chromosome studies in 952 infertile males with a sperm count below 10 million/ml. Hum Genet. 1985;71:366–7.
Hens L, Bonduelle M, Liebaers I, Devroey P, Van Steirteghem AC. Chromosome aberrations in 500 couples referred for in-vitro fertilization or related fertility treatment. Hum Reprod Oxf Engl. 1988;3:451–7.
Chandley AC, Seuánez H, Fletcher JM. Meiotic behavior of five human reciprocal translocations. Cytogenet Cell Genet. 1976;17:98–111. https://doi.org/10.1159/000130694.
Johannisson R, Schwinger E, Wolff HH, vom Ende V, Löhrs U. The effect of 13;14 Robertsonian translocations on germ-cell differentiation in infertile males. Cytogenet Cell Genet. 1993;63:151–5. https://doi.org/10.1159/000133524.
Gabriel-Robez O, Ratomponirina C, Rumpler Y, Le Marec B, Luciani JM, Guichaoua MR. Synapsis and synaptic adjustment in an infertile human male heterozygous for a pericentric inversion in chromosome 1. Hum Genet. 1986;72:148–52.
Turner JMA, Mahadevaiah SK, Fernandez-Capetillo O, Nussenzweig A, Xu X, Deng C-X, et al. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat Genet. 2005;37:41–7. https://doi.org/10.1038/ng1484.
Barasc H, Congras A, Mary N, Trouilh L, Marquet V, Ferchaud S, et al. Meiotic pairing and gene expression disturbance in germ cells from an infertile boar with a balanced reciprocal autosome-autosome translocation. Chromosome Res Int J Mol Supramol Evol Asp Chromosome Biol. 2016;24:511–27. https://doi.org/10.1007/s10577-016-9533-9.
Vogt P, Chandley AC, Hargreave TB, Keil R, Ma K, Sharkey A. Microdeletions in interval 6 of the Y chromosome of males with idiopathic sterility point to disruption of AZF, a human spermatogenesis gene. Hum Genet. 1992;89:491–6.
Vogt PH, Edelmann A, Kirsch S, Henegariu O, Hirschmann P, Kiesewetter F, et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5:933–43.
Vogt PH. Azoospermia factor (AZF) in Yq11: towards a molecular understanding of its function for human male fertility and spermatogenesis. Reprod BioMed Online. 2005;10:81–93.
Reijo R, Alagappan RK, Patrizio P, Page DC. Severe oligozoospermia resulting from deletions of azoospermia factor gene on Y chromosome. Lancet Lond Engl. 1996;347:1290–3.
Patrat C, Bienvenu T, Janny L, Faure A-K, Fauque P, Aknin-Seifer I, et al. Clinical data and parenthood of 63 infertile and Y-microdeleted men. Fertil Steril. 2010;93:822–32. https://doi.org/10.1016/j.fertnstert.2008.10.033.
Kaplan E, Shwachman H, Perlmutter AD, Rule A, Khaw K-T, Holsclaw DS. Reproductive failure in males with cystic fibrosis. N Engl J Med. 1968;279:65–9. https://doi.org/10.1056/NEJM196807112790203.
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–904. https://doi.org/10.1016/S0140-6736(09)60327-5.
Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45:1160–7. https://doi.org/10.1038/ng.2745.
Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–4.
Tsui LC. The spectrum of cystic fibrosis mutations. Trends Genet TIG. 1992;8:392–8.
Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. https://doi.org/10.1146/annurev.ge.29.120195.004021.
Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, et al. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998;393:79–82. https://doi.org/10.1038/30006.
Modiano G, Ciminelli BM, Pignatti PF. Cystic fibrosis and lactase persistence: a possible correlation. Eur J Hum Genet EJHG. 2007;15:255–9. https://doi.org/10.1038/sj.ejhg.5201749.
Alfonso-Sánchez MA, Pérez-Miranda AM, García-Obregón S, Peña JA. An evolutionary approach to the high frequency of the Delta F508 CFTR mutation in European populations. Med Hypotheses. 2010;74:989–92. https://doi.org/10.1016/j.mehy.2009.12.018.
Borzan V, Tomašević B, Kurbel S. Hypothesis: possible respiratory advantages for heterozygote carriers of cystic fibrosis linked mutations during dusty climate of last glaciation. J Theor Biol. 2014;363:164–8. https://doi.org/10.1016/j.jtbi.2014.08.015.
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–80. https://doi.org/10.1126/science.2570460.
Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2011;10(Suppl 2):S86–102. https://doi.org/10.1016/S1569-1993(11)60014-3.
Ferlin A, Raicu F, Gatta V, Zuccarello D, Palka G, Foresta C. Male infertility: role of genetic background. Reprod BioMed Online. 2007;14:734–45.
Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–80. https://doi.org/10.1056/NEJM199506013322204.
Yu J, Chen Z, Ni Y, Li Z. CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): a systemic review and meta-analysis. Hum Reprod Oxf Engl. 2012;27:25–35. https://doi.org/10.1093/humrep/der377.
Llabador MA, Pagin A, Lefebvre-Maunoury C, Marcelli F, Leroy-Martin B, Rigot JM, et al. Congenital bilateral absence of the vas deferens: the impact of spermatogenesis quality on intracytoplasmic sperm injection outcomes in 108 men. Andrology. 2015;3:473–80. https://doi.org/10.1111/andr.12019.
Xu WM, Chen J, Chen H, Diao RY, Fok KL, Dong JD, et al. Defective CFTR-dependent CREB activation results in impaired spermatogenesis and azoospermia. PLoS One. 2011;6:e19120. https://doi.org/10.1371/journal.pone.0019120.
Molina LCP, Pinto NA, Rodríguez PT, Romarowski A, Sanchez AV, Visconti PE, et al. Essential role of CFTR in PKA-dependent phosphorylation, alkalinization, and hyperpolarization during human sperm capacitation. J Cell Physiol. 2017;232:1404–14. https://doi.org/10.1002/jcp.25634.
Patat O, Pagin A, Siegfried A, Mitchell V, Chassaing N, Faguer S, et al. Truncating mutations in the adhesion G protein-coupled receptor G2 gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am J Hum Genet. 2016;99:437–42. https://doi.org/10.1016/j.ajhg.2016.06.012.
Yang B, Wang J, Zhang W, Pan H, Li T, Liu B, et al. Pathogenic role of ADGRG2 in CBAVD patients replicated in Chinese population. Andrology. 2017;5:954–7. https://doi.org/10.1111/andr.12407.
Obermann H, Samalecos A, Osterhoff C, Schröder B, Heller R, Kirchhoff C. HE6, a two-subunit heptahelical receptor associated with apical membranes of efferent and epididymal duct epithelia. Mol Reprod Dev. 2003;64:13–26. https://doi.org/10.1002/mrd.10220.
Zhang D-L, Sun Y-J, Ma M-L, Wang Y, Lin H, Li R-R, et al. Gq activity- and β-arrestin-1 scaffolding-mediated ADGRG2/CFTR coupling are required for male fertility. Bagnat M, editor. eLife. 2018;7:e33432. https://doi.org/10.7554/eLife.33432.
Davies B, Baumann C, Kirchhoff C, Ivell R, Nubbemeyer R, Habenicht U-F, et al. Targeted deletion of the epididymal receptor HE6 results in fluid dysregulation and male infertility. Mol Cell Biol. 2004;24:8642–8. https://doi.org/10.1128/MCB.24.19.8642-8648.2004.
Clulow J, Jones RC, Hansen LA, Man SY. Fluid and electrolyte reabsorption in the ductuli efferentes testis. J Reprod Fertil Suppl. 1998;53:1–14.
Khan MJ, Pollock N, Jiang H, Castro C, Nazli R, Ahmed J, et al. X-linked ADGRG2 mutation and obstructive azoospermia in a large Pakistani family. Sci Rep. 2018;8:16280. https://doi.org/10.1038/s41598-018-34262-5.
Hihnala S, Kujala M, Toppari J, Kere J, Holmberg C, Höglund P. Expression of SLC26A3, CFTR and NHE3 in the human male reproductive tract: role in male subfertility caused by congenital chloride diarrhoea. Mol Hum Reprod. 2006;12:107–11. https://doi.org/10.1093/molehr/gal009.
Zhou Q, Clarke L, Nie R, Carnes K, Lai LW, Lien YH, et al. Estrogen action and male fertility: roles of the sodium/hydrogen exchanger-3 and fluid reabsorption in reproductive tract function. Proc Natl Acad Sci U S A. 2001;98:14132–7. https://doi.org/10.1073/pnas.241245898.
Pholpramool C, Borwornpinyo S, Dinudom A. Role of Na+ /H+ exchanger 3 in the acidification of the male reproductive tract and male fertility. Clin Exp Pharmacol Physiol. 2011;38:403–9. https://doi.org/10.1111/j.1440-1681.2011.05525.x.
Wang Y-Y, Lin Y-H, Wu Y-N, Chen Y-L, Lin Y-C, Cheng C-Y, et al. Loss of SLC9A3 decreases CFTR protein and causes obstructed azoospermia in mice. PLoS Genet. 2017;13. https://doi.org/10.1371/journal.pgen.1006715.
Wu Y-N, Lin Y-H, Chiang H-S. SLC9A3 is a novel pathogenic gene in Taiwanese males with congenital bilateral absence of the vas deferens. Eur Urol Suppl. 2018;17:e1092. https://doi.org/10.1016/S1569-9056(18)31593-8.
Lee C-H, Wu C-C, Wu Y-N, Chiang H-S. Gene copy number variations in Asian patients with congenital bilateral absence of the vas deferens. Hum Reprod Oxf Engl. 2009;24:748–55. https://doi.org/10.1093/humrep/den413.
Kuo Y-M, Duncan JL, Westaway SK, Yang H, Nune G, Xu EY, et al. Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia. Hum Mol Genet. 2005;14:49–57. https://doi.org/10.1093/hmg/ddi005.
Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14:1197–213. https://doi.org/10.1038/nm.f.1895.
Yatsenko AN, Georgiadis AP, Röpke A, Berman AJ, Jaffe T, Olszewska M, et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med. 2015;372:2097–107. https://doi.org/10.1056/NEJMoa1406192.
Sha Y, Zheng L, Ji Z, Mei L, Ding L, Lin S, et al. A novel TEX11 mutation induces azoospermia: a case report of infertile brothers and literature review. BMC Med Genet. 2018;19. https://doi.org/10.1186/s12881-018-0570-4.
He W-B, Tu C-F, Liu Q, Meng L-L, Yuan S-M, Luo A-X, et al. DMC1 mutation that causes human non-obstructive azoospermia and premature ovarian insufficiency identified by whole-exome sequencing. J Med Genet. 2018;55:198–204. https://doi.org/10.1136/jmedgenet-2017-104992.
Gershoni M, Hauser R, Yogev L, Lehavi O, Azem F, Yavetz H, et al. A familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genes. Genet Med Off J Am Coll Med Genet. 2017;19:998–1006. https://doi.org/10.1038/gim.2016.225.
McNally FJ. Mechanisms of spindle positioning. J Cell Biol. 2013;200:131–40. https://doi.org/10.1083/jcb.201210007.
Li Y, Yagi H, Onuoha EO, Damerla RR, Francis R, Furutani Y, et al. DNAH6 and its interactions with PCD genes in Heterotaxy and primary ciliary dyskinesia. PLoS Genet. 2016;12:e1005821. https://doi.org/10.1371/journal.pgen.1005821.
Okutman O, Muller J, Skory V, Garnier JM, Gaucherot A, Baert Y, et al. A no-stop mutation in MAGEB4 is a possible cause of rare X-linked azoospermia and oligozoospermia in a consanguineous Turkish family. J Assist Reprod Genet. 2017;34:683–94. https://doi.org/10.1007/s10815-017-0900-z.
Osterlund C, Töhönen V, Forslund KO, Nordqvist K. Mage-b4, a novel melanoma antigen (MAGE) gene specifically expressed during germ cell differentiation. Cancer Res. 2000;60:1054–61.
Tenenbaum-Rakover Y, Weinberg-Shukron A, Renbaum P, Lobel O, Eideh H, Gulsuner S, et al. Minichromosome maintenance complex component 8 (MCM8) gene mutations result in primary gonadal failure. J Med Genet. 2015;52:391–9. https://doi.org/10.1136/jmedgenet-2014-102921.
Lee KY, Im J-S, Shibata E, Park J, Handa N, Kowalczykowski SC, et al. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat Commun. 2015;6:7744. https://doi.org/10.1038/ncomms8744.
Park J, Long DT, Lee KY, Abbas T, Shibata E, Negishi M, et al. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol Cell Biol. 2013;33:1632–44. https://doi.org/10.1128/MCB.01503-12.
Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, Ranisavljevic N, et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell. 2012;47:523–34. https://doi.org/10.1016/j.molcel.2012.05.048.
Luo M, Yang F, Leu NA, Landaiche J, Handel MA, Benavente R, et al. MEIOB exhibits single-stranded DNA-binding and exonuclease activities and is essential for meiotic recombination. Nat Commun. 2013;4:2788. https://doi.org/10.1038/ncomms3788.
Souquet B, Abby E, Hervé R, Finsterbusch F, Tourpin S, Le Bouffant R, et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013;9:e1003784. https://doi.org/10.1371/journal.pgen.1003784.
Ben Khelifa M, Ghieh F, Boudjenah R, Hue C, Fauvert D, Dard R, et al. A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family. Hum Reprod Oxf Engl. 2018;33:1034–7. https://doi.org/10.1093/humrep/dey073.
Libby BJ, De La Fuente R, O’Brien MJ, Wigglesworth K, Cobb J, Inselman A, et al. The mouse meiotic mutation mei1 disrupts chromosome synapsis with sexually dimorphic consequences for meiotic progression. Dev Biol. 2002;242:174–87. https://doi.org/10.1006/dbio.2001.0535.
Sato H, Miyamoto T, Yogev L, Namiki M, Koh E, Hayashi H, et al. Polymorphic alleles of the human MEI1 gene are associated with human azoospermia by meiotic arrest. J Hum Genet. 2006;51:533–40. https://doi.org/10.1007/s10038-006-0394-5.
Ramasamy R, Bakırcıoğlu ME, Cengiz C, Karaca E, Scovell J, Jhangiani SN, et al. Whole-exome sequencing identifies novel homozygous mutation in NPAS2 in family with nonobstructive azoospermia. Fertil Steril. 2015;104:286–91. https://doi.org/10.1016/j.fertnstert.2015.04.001.
Zangen D, Kaufman Y, Zeligson S, Perlberg S, Fridman H, Kanaan M, et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am J Hum Genet. 2011;89:572–9. https://doi.org/10.1016/j.ajhg.2011.09.006.
Al-Agha AE, Ahmed IA, Nuebel E, Moriwaki M, Moore B, Peacock KA, et al. Primary ovarian insufficiency and azoospermia in carriers of a homozygous PSMC3IP stop gain mutation. J Clin Endocrinol Metab. 2018;103:555–63. https://doi.org/10.1210/jc.2017-01966.
Kherraf Z-E, Christou-Kent M, Karaouzene T, Amiri-Yekta A, Martinez G, Vargas AS, et al. SPINK2 deficiency causes infertility by inducing sperm defects in heterozygotes and azoospermia in homozygotes. EMBO Mol Med. 2017;9:1132–49. https://doi.org/10.15252/emmm.201607461.
Nakamura S, Kobori Y, Ueda Y, Tanaka Y, Ishikawa H, Yoshida A, et al. STX2 is a causative gene for nonobstructive azoospermia. Hum Mutat. 2018;39:830–3. https://doi.org/10.1002/humu.23423.
Fujiwara Y, Ogonuki N, Inoue K, Ogura A, Handel MA, Noguchi J, et al. t-SNARE Syntaxin2 (STX2) is implicated in intracellular transport of sulfoglycolipids during meiotic prophase in mouse spermatogenesis. Biol Reprod. 2013;88:141. https://doi.org/10.1095/biolreprod.112.107110.
Maor-Sagie E, Cinnamon Y, Yaacov B, Shaag A, Goldsmidt H, Zenvirt S, et al. Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J Assist Reprod Genet. 2015;32:887–91. https://doi.org/10.1007/s10815-015-0445-y.
Bolcun-Filas E, Hall E, Speed R, Taggart M, Grey C, de Massy B, et al. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009;5:e1000393. https://doi.org/10.1371/journal.pgen.1000393.
Ayhan Ö, Balkan M, Guven A, Hazan R, Atar M, Tok A, et al. Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J Med Genet. 2014;51:239–44. https://doi.org/10.1136/jmedgenet-2013-102102.
Yang X, Zhang H, Jiang Y, Zhang H, Hu X, Zhu D, et al. Association study between MTRR, TAF4B, PIWIL1 variants and non-obstructive azoospermia in northeast Chinese Han population. Clin Lab. 2018;64:1731–8. https://doi.org/10.7754/Clin.Lab.2018.180525.
Falender AE, Freiman RN, Geles KG, Lo KC, Hwang K, Lamb DJ, et al. Maintenance of spermatogenesis requires TAF4b, a gonad-specific subunit of TFIID. Genes Dev. 2005;19:794–803. https://doi.org/10.1101/gad.1290105.
Tan Y-Q, Tu C, Meng L, Yuan S, Sjaarda C, Luo A, et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med Off J Am Coll Med Genet. 2017. https://doi.org/10.1038/gim.2017.130.
Tanaka T, Hosokawa M, Vagin VV, Reuter M, Hayashi E, Mochizuki AL, et al. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc Natl Acad Sci U S A. 2011;108:10579–84. https://doi.org/10.1073/pnas.1015447108.
Shoji M, Tanaka T, Hosokawa M, Reuter M, Stark A, Kato Y, et al. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev Cell. 2009;17:775–87. https://doi.org/10.1016/j.devcel.2009.10.012.
Arafat M, Har-Vardi I, Harlev A, Levitas E, Zeadna A, Abofoul-Azab M, et al. Mutation in TDRD9 causes non-obstructive azoospermia in infertile men. J Med Genet. 2017;54:633–9. https://doi.org/10.1136/jmedgenet-2017-104514.
Boroujeni PB, Sabbaghian M, Totonchi M, Sodeifi N, Sarkardeh H, Samadian A, et al. Expression analysis of genes encoding TEX11, TEX12, TEX14 and TEX15 in testis tissues of men with non-obstructive azoospermia. JBRA Assist Reprod. 2018;22:185–92. https://doi.org/10.5935/1518-0557.20180030.
Wu M-H, Rajkovic A, Burns KH, Yan W, Lin Y-N, Matzuk MM. Sequence and expression of testis-expressed gene 14 (Tex14): a gene encoding a protein kinase preferentially expressed during spermatogenesis. Gene Expr Patterns GEP. 2003;3:231–6.
Greenbaum MP, Yan W, Wu M-H, Lin Y-N, Agno JE, Sharma M, et al. TEX14 is essential for intercellular bridges and fertility in male mice. Proc Natl Acad Sci U S A. 2006;103:4982–7. https://doi.org/10.1073/pnas.0505123103.
Sironen A, Uimari P, Venhoranta H, Andersson M, Vilkki J. An exonic insertion within Tex14 gene causes spermatogenic arrest in pigs. BMC Genomics. 2011;12:591. https://doi.org/10.1186/1471-2164-12-591.
Colombo R, Pontoglio A, Bini M. Two novel TEX15 mutations in a family with nonobstructive azoospermia. Gynecol Obstet Investig. 2017;82:283–6. https://doi.org/10.1159/000468934.
Okutman O, Muller J, Baert Y, Serdarogullari M, Gultomruk M, Piton A, et al. Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum Mol Genet. 2015;24:5581–8. https://doi.org/10.1093/hmg/ddv290.
Yang F, Eckardt S, Leu NA, McLaughlin KJ, Wang PJ. Mouse TEX15 is essential for DNA double-strand break repair and chromosomal synapsis during male meiosis. J Cell Biol. 2008;180:673–9. https://doi.org/10.1083/jcb.200709057.
Ruan J, He X-J, Du W-D, Chen G, Zhou Y, Xu S, et al. Genetic variants in TEX15 gene conferred susceptibility to spermatogenic failure in the Chinese Han population. Reprod Sci Thousand Oaks Calif. 2012;19:1190–6. https://doi.org/10.1177/1933719112446076.
Plaseski T, Noveski P, Popeska Z, Efremov GD, Plaseska-Karanfilska D. Association study of single-nucleotide polymorphisms in FASLG, JMJDIA, LOC203413, TEX15, BRDT, OR2W3, INSR, and TAS2R38 genes with male infertility. J Androl. 2012;33:675–83. https://doi.org/10.2164/jandrol.111.013995.
Yang Y, Guo J, Dai L, Zhu Y, Hu H, Tan L, et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet. 2018;55:628–36. https://doi.org/10.1136/jmedgenet-2017-105145.
Zhang Y-X, Li H-Y, He W-B, Tu C, Du J, Li W, et al. XRCC2 mutation causes premature ovarian insufficiency as well as non-obstructive azoospermia in humans. Clin Genet. 2018. https://doi.org/10.1111/cge.13475.
Gu A-H, Liang J, Lu N-X, Wu B, Xia Y-K, Lu C-C, et al. Association of XRCC1 gene polymorphisms with idiopathic azoospermia in a Chinese population. Asian J Androl. 2007;9:781–6. https://doi.org/10.1111/j.1745-7262.2007.00325.x.
Zheng L, Wang X, Zhou D, Zhang J, Huo Y, Tian H. Association between XRCC1 single-nucleotide polymorphisms and infertility with idiopathic azoospermia in northern Chinese Han males. Reprod BioMed Online. 2012;25:402–7. https://doi.org/10.1016/j.rbmo.2012.06.014.
Jahantigh D, Hosseinzadeh Colagar A. XRCC5 VNTR, XRCC6 -61C>G, and XRCC7 6721G>T gene polymorphisms associated with male infertility risk: evidences from case-control and in silico studies. Int J Endocrinol. 2017;2017:4795076. https://doi.org/10.1155/2017/4795076.
Yan W, Si Y, Slaymaker S, Li J, Zheng H, Young DL, et al. Zmynd15 encodes a histone deacetylase-dependent transcriptional repressor essential for spermiogenesis and male fertility. J Biol Chem. 2010;285:31418–26. https://doi.org/10.1074/jbc.M110.116418.
Mouka A, Izard V, Tachdjian G, Brisset S, Yates F, Mayeur A, et al. Induced pluripotent stem cell generation from a man carrying a complex chromosomal rearrangement as a genetic model for infertility studies. Sci Rep. 2017;7:39760. https://doi.org/10.1038/srep39760.
Yang Y, Luo YY, Wu S, Tang YD, Rao XD, Xiong L, et al. Association between C677T and A1298C polymorphisms of the MTHFR gene and risk of male infertility: a meta-analysis. Genet Mol Res GMR. 2016;15. https://doi.org/10.4238/gmr.15027631.
Holliday R. The inheritance of epigenetic defects. Science. 1987;238:163–70.
Dada R, Kumar M, Jesudasan R, Fernández JL, Gosálvez J, Agarwal A. Epigenetics and its role in male infertility. J Assist Reprod Genet. 2012;29:213–23. https://doi.org/10.1007/s10815-012-9715-0.
Filipponi D, Feil R. Perturbation of genomic imprinting in oligozoospermia. Epigenetics. 2009;4:27–30.
Kobayashi H, Sato A, Otsu E, Hiura H, Tomatsu C, Utsunomiya T, et al. Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum Mol Genet. 2007;16:2542–51. https://doi.org/10.1093/hmg/ddm187.
Hammoud SS, Purwar J, Pflueger C, Cairns BR, Carrell DT. Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril. 2010;94:1728–33. https://doi.org/10.1016/j.fertnstert.2009.09.010.
De Mateo S, Sassone-Corsi P. Regulation of spermatogenesis by small non-coding RNAs: role of the germ granule. Semin Cell Dev Biol. 2014;29:84–92. https://doi.org/10.1016/j.semcdb.2014.04.021.
Hayashi K, Chuva de Sousa Lopes SM, Kaneda M, Tang F, Hajkova P, Lao K, et al. MicroRNA biogenesis is required for mouse primordial germ cell development and spermatogenesis. PLoS One. 2008;3:e1738. https://doi.org/10.1371/journal.pone.0001738.
Maatouk DM, Loveland KL, McManus MT, Moore K, Harfe BD. Dicer1 is required for differentiation of the mouse male germline. Biol Reprod. 2008;79:696–703. https://doi.org/10.1095/biolreprod.108.067827.
Eelaminejad Z, Favaedi R, Sodeifi N, Sadighi Gilani MA, Shahhoseini M. Deficient expression of JMJD1A histone demethylase in patients with round spermatid maturation arrest. Reprod BioMed Online. 2017;34:82–9. https://doi.org/10.1016/j.rbmo.2016.09.005.
Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature. 2007;450:119–23. https://doi.org/10.1038/nature06236.
Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662–71. https://doi.org/10.1016/j.tcb.2010.08.011.
Liu Z, Zhou S, Liao L, Chen X, Meistrich M, Xu J. Jmjd1a demethylase-regulated histone modification is essential for cAMP-response element modulator-regulated gene expression and spermatogenesis. J Biol Chem. 2010;285:2758–70. https://doi.org/10.1074/jbc.M109.066845.
Faure AK, Pivot-Pajot C, Kerjean A, Hazzouri M, Pelletier R, Péoc’h M, et al. Misregulation of histone acetylation in Sertoli cell-only syndrome and testicular cancer. Mol Hum Reprod. 2003;9:757–63.
Sonnack V, Failing K, Bergmann M, Steger K. Expression of hyperacetylated histone H4 during normal and impaired human spermatogenesis. Andrologia. 2002;34:384–90.
Khazamipour N, Noruzinia M, Fatehmanesh P, Keyhanee M, Pujol P. MTHFR promoter hypermethylation in testicular biopsies of patients with non-obstructive azoospermia: the role of epigenetics in male infertility. Hum Reprod Oxf Engl. 2009;24:2361–4. https://doi.org/10.1093/humrep/dep194.
Ferfouri F, Boitrelle F, Ghout I, Albert M, Molina Gomes D, Wainer R, et al. A genome-wide DNA methylation study in azoospermia. Andrology. 2013;1:815–21. https://doi.org/10.1111/j.2047-2927.2013.00117.x.
Cheng Y-S, Wee S-K, Lin T-Y, Lin Y-M. MAEL promoter hypermethylation is associated with de-repression of LINE-1 in human hypospermatogenesis. Hum Reprod Oxf Engl. 2017;32:2373–81. https://doi.org/10.1093/humrep/dex329.
Minor A, Chow V, Ma S. Aberrant DNA methylation at imprinted genes in testicular sperm retrieved from men with obstructive azoospermia and undergoing vasectomy reversal. Reproduction. 2011;141:749–57. https://doi.org/10.1530/REP-11-0008.
Ramasamy R, Ridgeway A, Lipshultz LI, Lamb DJ. Integrative DNA methylation and gene expression analysis identifies discoidin domain receptor 1 association with idiopathic nonobstructive azoospermia. Fertil Steril. 2014;102:968–973.e3. https://doi.org/10.1016/j.fertnstert.2014.06.028.
Li Z, Chen S, Yang Y, Zhuang X, Tzeng C-M. Novel biomarker ZCCHC13 revealed by integrating DNA methylation and mRNA expression data in non-obstructive azoospermia. Cell Death Dis. 2018;4. https://doi.org/10.1038/s41420-018-0033-x.
Marques PI, Fernandes S, Carvalho F, Barros A, Sousa M, Marques CJ. DNA methylation imprinting errors in spermatogenic cells from maturation arrest azoospermic patients. Andrology. 2017;5:451–9. https://doi.org/10.1111/andr.12329.
Marques CJ, Francisco T, Sousa S, Carvalho F, Barros A, Sousa M. Methylation defects of imprinted genes in human testicular spermatozoa. Fertil Steril. 2010;94:585–94. https://doi.org/10.1016/j.fertnstert.2009.02.051.
Lian J, Zhang X, Tian H, Liang N, Wang Y, Liang C, et al. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol RBE. 2009;7:13. https://doi.org/10.1186/1477-7827-7-13.
Wang C, Yang C, Chen X, Yao B, Yang C, Zhu C, et al. Altered profile of seminal plasma microRNAs in the molecular diagnosis of male infertility. Clin Chem. 2011;57:1722–31. https://doi.org/10.1373/clinchem.2011.169714.
Wu W, Qin Y, Li Z, Dong J, Dai J, Lu C, et al. Genome-wide microRNA expression profiling in idiopathic non-obstructive azoospermia: significant up-regulation of miR-141, miR-429 and miR-7-1-3p. Hum Reprod Oxf Engl. 2013;28:1827–36. https://doi.org/10.1093/humrep/det099.
Abu-Halima M, Hammadeh M, Backes C, Fischer U, Leidinger P, Lubbad AM, et al. Panel of five microRNAs as potential biomarkers for the diagnosis and assessment of male infertility. Fertil Steril. 2014;102:989–997.e1. https://doi.org/10.1016/j.fertnstert.2014.07.001.
Dabaja AA, Mielnik A, Robinson BD, Wosnitzer MS, Schlegel PN, Paduch DA. Possible germ cell-Sertoli cell interactions are critical for establishing appropriate expression levels for the Sertoli cell-specific MicroRNA, miR-202-5p, in human testis. Basic Clin Androl. 2015;25:2. https://doi.org/10.1186/s12610-015-0018-z.
Yao C, Yuan Q, Niu M, Fu H, Zhou F, Zhang W, et al. Distinct expression profiles and novel targets of microRNAs in human spermatogonia, pachytene spermatocytes, and round spermatids between OA patients and NOA patients. Mol Ther - Nucleic Acids. 2017;9:182–94. https://doi.org/10.1016/j.omtn.2017.09.007.
Fang N, Cao C, Wen Y, Wang X, Yuan S, Huang X. MicroRNA profile comparison of testicular tissues derived from successful and unsuccessful microdissection testicular sperm extraction retrieval in non-obstructive azoospermia patients. Reprod Fertil Dev. 2018. https://doi.org/10.1071/RD17423.
Acknowledgments
Not applicable
Funding
There are no funding sources to report.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
FG and FV: wrote the paper. BM-P and VM revised the paper. FG, BM-P, VM and FV designed the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ghieh, F., Mitchell, V., Mandon-Pepin, B. et al. Genetic defects in human azoospermia. Basic Clin. Androl. 29, 4 (2019). https://doi.org/10.1186/s12610-019-0086-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12610-019-0086-6