
Abstract
Aryl hydrocarbon receptor (AhR) was originally identified as an environmental sensor that responds to pollutants. Subsequent research has revealed that AhR recognizes multiple exogenous and endogenous molecules, including uremic toxins retained in the body due to the decline in renal function. Therefore, AhR is also considered to be a uremic toxin receptor. As a ligand-activated transcriptional factor, the activation of AhR is involved in cell differentiation and senescence, lipid metabolism and fibrogenesis. The accumulation of uremic toxins in the body is hazardous to all tissues and organs. The identification of the endogenous uremic toxin receptor opens the door to investigating the precise role and molecular mechanism of tissue and organ damage induced by uremic toxins. This review focuses on summarizing recent findings on the role of AhR activation induced by uremic toxins in chronic kidney disease, diabetic nephropathy and acute kidney injury. Furthermore, potential clinical approaches to mitigate the effects of uremic toxins are explored herein, such as enhancing uremic toxin clearance through dialysis, reducing uremic toxin production through dietary interventions or microbial manipulation, and manipulating metabolic pathways induced by uremic toxins through controlling AhR signaling. This information may also shed light on the mechanism of uremic toxin-induced injury to other organs, and provide insights into clinical approaches to manipulate the accumulated uremic toxins.
Graphical Abstract

Similar content being viewed by others


Introduction
The kidney is the organ that excretes metabolic waste products, including creatinine, urea and uric acid, from the body. When metabolic waste products cannot be appropriately eliminated by the kidney, the accumulation of uremic toxins and disruption of the body’s internal environmental homeostasis are hazardous to all tissues and organs [1]. In recent years, the identification of endogenous uremic toxin receptors has opened the door to research on the precise role and molecular mechanism of uremic toxins in the tissue and organ, leading to the emergence of valuable insights [2].
Aryl hydrocarbon receptor (AhR) is an important receptor of uremic toxins. AhR was initially identified as an environmental sensor that responds to pollutants, including halogenated aromatic hydrocarbons and polycyclic aromatic hydrocarbons [3]. Growing evidence has suggested that AhR not only is a receptor for xenobiotics but can also be activated by various physiological ligands, such as metabolites derived from the host, gut microbiota or natural plants. Numerous studies have demonstrated that AhR activation is widely involved in cell differentiation, cellular senescence, lipid metabolism, intestinal balance, immune response and fibrogenesis [4,5,6,7]. Recent studies have indicated that AhR activation by the accumulation of uremic toxins may be implicated in various kidney diseases, including chronic kidney disease (CKD), CKD-associated complications, diabetic nephropathy (DN), acute kidney injury (AKI) and systemic lupus erythematosus (SLE) [8, 9]. Reducing uremic toxins by improving their clearance or inhibiting their production benefits clinical treatment outcomes [9]. However, these therapies possess inherent advantages and limitations that may contribute to poor outcomes for patients with kidney diseases. Targeting AhR with agonists or antagonists has shown promising initial efficacy in various kidney disease models [2]. Given the importance of understanding the effects of AhR activation by uremic toxins on kidney diseases and complications, this review summarizes the recent understanding of the mechanisms of uremic toxin-activated AhR signaling pathways and their effects on different renal diseases and also simply discusses current therapeutic strategies for targeting both uremic toxins and AhR activation.
Uremic toxins
During the development of CKD, some metabolic waste products (including uremic toxins) are retained in the circulation and tissues due to a decreased glomerular filtration rate (GFR) and renal structural and physiological dysfunction [1]. Many uremic toxins are products of dietary constituents. For instance, p-cresyl sulfate (PCS) is derived from tyrosine; kynurenine (KYN) and indoxyl sulfate (IS) are derived from tryptophan (Trp); and trimethylamine-N-oxide (TMAO) is derived from dietary fish, red meat and eggs [10].
L-tyrosine can be reversibly converted to phenol by tyrosine phenol-lyase [11]. In addition, L-tyrosine can also be reversibly converted to 4-hydroxyphenylpyruvate by tyrosine transaminase [12], aromatic-amino-acid transaminase [13] or phenylalanine dehydrogenase [14]. 4-Hydroxyphenylpyruvate is the precursor of 4-hydroxyphenylacetate, which is catalyzed by p-hydroxyphenylpyruvate oxidase [12], and can subsequently be decarboxylated to p-cresol by p-hydroxyphenylacetate decarboxylase [15]. These enzymes are present in the gut microbiota. The majority of p-cresol is sulfated into the PCS by aryl sulfotransferases [16], and a small fraction is metabolized to p-cresyl glucuronide by UDP-glucuronyltransferases in the gut mucosa and liver [17, 18] (Fig. 1).

Metabolic pathway of tyrosine into p-cresyl sulfate (PCS). l-tyrosine can be reversibly converted to 4-hydroxyphenylpyruvate by tyrosine transaminase, aromatic-amino-acid transaminase or phenylalanine dehydrogenase. 4-Hydroxyphenylpyruvate is the precursor of 4-hydroxyphenylacetate, which is catalyzed by p-hydroxyphenylpyruvate oxidase, and can subsequently be decarboxylated to p-cresol by p-hydroxyphenylacetate decarboxylase. The majority of p-cresol is sulfated into the PCS by aryl sulfotransferases, and a small fraction is metabolized to p-cresyl glucuronide by UDP-glucuronyltransferases. The processes marked by the yellow box occur in the gut microbiota, and the processes marked by the blue box occur in the gut mucosa and liver of the host
The essential amino acid Trp is mainly degraded by three known metabolic pathways that can be initiated in the host, plant or microbiota: the KYN pathway (90–95% of Trp), serotonin pathway (1–2% of Trp) and indolic pathway (4%-6% of Trp) [19] (Fig. 2). In the KYN pathway, Trp is converted to N-formylkynurenine (NFK) by the rate-limiting enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine-2,3-dioxygenase (IDO-1/2). NFK is converted to KYN by kynurenine formamidase (AFMID). Subsequently, KYN is converted to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO). Then, 3-HK is converted by kynureninase (KYNU) to 3-hydroxyanthralinic acid (3-HAA), which is converted by 3-hydroxyanthranilate 3,4-dioxygenase (HAAO) to quinolinic acid (QA). QA can be converted to NAD+, a key coenzyme in energy metabolism. 3-HK can also be catalyzed by kynurenine amino transferase (KAT) to produce xanthurenic acid (XA). KYN is also converted to anthralinic acid (AA) by KYNU. KAT can catalyze KYN to produce kynurenine quinolinic acid, also known as kynurenic acid (KYNA) [20]. TDO is highly expressed in the liver and brain, and IDO-1/2 is widely expressed in various tissues [2, 21].

Metabolic pathway of tryptophan (Trp). Trp is mainly degraded through three known metabolic pathways that can be initiated by the host, plant or microbiota. These include the KYN pathway (90–95% of Trp), serotonin pathway (1–2% of Trp) and indolic pathway (4–6% of Trp). Several compounds, including indoxyl sulfate (IS), kynurenine (KYN), kynurenic acid (KYNA), and indole-3-acetic acid (IAA), are recognized as uremic toxins and are marked with blue boxes. The compound acting as AhR ligands is marked with an asterisk (*)
In the serotonin pathway, Trp is metabolized by Trp hydroxylase enzyme (TpH), which produces 5-hydroxytryptophan (5-HTP). 5-HTP is further metabolized into 5-hydroxytryptamine (5-HT), also known as serotonin [22].
In the indole pathway, Trp is converted into indole by tryptophanase-positive microbiota. Indole is absorbed in the liver and then oxidized by cytochrome P450 family 2 subfamily E member 1 (CYP2E1) to hydroxyindole, which is converted into IS by sulfotransferases [23]. Some bacterial species use Trp and metabolize it to various indolic derivatives. For example, Lactobacillus spp. metabolize Trp to indole-3-aldehyde (I3A), Bifdobacterium spp. metabolize Trp to indole-3-lactic acid (ILA), and Bacteroides spp. metabolize Trp to indole-3-acetic acid (IAA) [8].
Trp photolysis by ultraviolet or visible light triggers several photochemical products, such as 1-(1H-indol-3-yl)-9H-pyrido[3,4-b]indole [24] and 6-formylindolo[3,2-b]carbazole (FICZ) [25].
Tryptophan-derived phytochemical indole-3-carbinol (I3C), which is produced in cruciferous brassica genus vegetables, including cauliflower, cabbage, and brussels sprouts, can be converted into indolo[3,2-b]carbazole (ICZ) by nonenzymatic condensation reactions in the stomach [26].
Choline is derived from eggs, fish and meat and can be metabolized to trimethylamine (TMA) by the choline-utilizing TMA lyase (CutC/D). L-carnitine is found in red meat and fish and can be metabolized to TMA by the carnitine Rieske-type oxygenase/reductase (CntA/B) [27,28,29]. YeaW and YeaX, the homologs of CntA/B, can also metabolize choline, carnitine and betaine to generate TMA. These effects are dependent on the gut microbiota [30]. TMA produced in the gut is absorbed into the blood and transported to the liver, where flavin monooxygenase 3 (FMO3) catalyzes TMA into TMAO [31]. Apart from dietary precursors of TMAO, most preformed TMAO, which is independent of gut microbes, is found in fish, humans [32] and rats [31, 33] (Fig. 3).

Pathways of trimethylamine-N-oxide (TMAO) production. Foods are enriched in TMAO precursors (choline, carnitine and betaine) or TMAO itself. Choline can be metabolized to trimethylamine (TMA) by the choline-utilizing TMA lyase (CutC/D). L-carnitine can be metabolized to TMA by the carnitine Rieske-type oxygenase/reductase (CntA/B). YeaW and YeaX, the homologs of CntA/B, can also metabolize choline, carnitine and betaine to generate TMA. The above processes occur in the microbiota. TMA produced in the gut is absorbed into the blood and transported to the liver, where flavin monooxygenase 3 (FMO3) catalyzes TMA into TMAO. Dietary TMAO can bypass processing by the gut microbiota before intestinal absorption
The European Uremic Toxin Work Group in 2003 classified uremic toxins into three categories based on dialysis clearance and physicochemical characteristics. The first category comprises small molecule toxins with a molecular weight of less than 500 Da, solubility in water, non-protein binding, and easy elimination through hemodialysis (HD), which include urea, creatinine, creatine, uric acid and xanthine. The second category comprises medium molecule toxins with a molecular weight exceeding 500 Da which are less efficiently cleared by HD; these toxins include β2-microglobulin, interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α. Finally, protein-bound toxins are difficult to eliminate using conventional dialysis techniques, including PCS, IS, KYN, KYNA and IAA [34]. However, a conference on uremic toxins in 2020 challenges this classification as follows: First, the current physiochemical subdivisions based on molecular weight can be considered arbitrary and artificial. Second, the protein-bound degree of these uremic solutes is variable, and the molecular weights of these solutes remain uncertain. Third, the HD in the original classification only applies to conventional HD and not peritoneal or other dialysis. Fourth, the original classification does not consider the compartmental partitioning behavior of solutes within the body. Fifth, some uremic toxins already exist before the initiation of dialysis. Therefore, experts recommended that the definition of uremic toxins should be based on HD strategies, membranes, and removal patterns while adapting to technological advancements [35]. In addition, experts approved a scoring system in 2008 for classifying uremic toxins according to the experimental and clinical evidence of their toxicity. The highest-scoring uremic toxins were PCS, β2-microglobulin, asymmetric dimethyl arginine, KYN, carbamylated compounds, fibroblast growth factor (FGF)-23, IL-6, TNF-α and symmetric dimethyl arginine. The second highest-scoring uremic toxins are advanced glycation end products, IS, uric acid, ghrelin, IAA, parathyroid hormone, phenyl acetic acid, TMAO, retinol binding protein, endothelin, immunoglobulin light chains, IL-1β, IL-8, neuropeptide Y, lipids and lipoproteins [36]. Based on a new classification schema proposed by experts [35], this review further summarized the classification of uremic toxins according to metabolic pathways and dialysis modalities (Table 1).
AhR signaling
Compounds produced by Trp metabolism have been demonstrated to be potential AhR ligands, including KYN, KYNA, XA, 3-HK, 3-HAA, QA, tryptamine, IAA, 3-methylindole (skatole), I3A, ILA, indole, IS, I3C, ICZ, FICZ [19], 5-HTP [37], and indole-3-acetaldehyde (IAAld) [8]. IS is a potent ligand of AhR that exhibits 500-fold greater potency in the transcriptional activation of human AhR than mouse AhR [23]. FICZ has structural similarities to ICZ, and both are important endogenous AhR agonists. FICZ binds to the AhR with higher affinity than tetrachlorodibenzo-p-dioxin (TCDD), a well-known potent agonist of AhR [38]. However, the precursor I3C acts as a weak AhR ligand [39]. Under pathological stimuli, AhR is widely expressed in a variety of cells, including epithelial cells [40], vascular smooth muscle cells [41], endothelial cells, immune cells [42], hepatocytes [43], astrocytes and neurons [44].
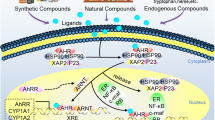
AhR is a member of the bHLH-PAS family and is an evolutionarily conserved transcription factor. Structurally, AhR contains a bHLH domain and two repeats of a PAS domain, known as PAS-A and PAS-B [45,46,47] (Fig. 4). Under physiological conditions, the AhR PAS-B domain is attached to heat shock protein 90 (HSP90) [48]. The AhR bridge motif between PAS-A and PAS-B tightly binds to the HSP90 dimer and is threaded through the lumen of HSP90. HSP90 plays a crucial role in maintaining a high-affinity ligand-binding conformation. The amino acid residues connecting AhR PAS-B to the C-terminal transactivation domain form a long loop that folds back to the AhR PAS-B domain and interface with X-associated protein 2 (XAP2, also known as ARA9 or AIP), potentially interacting with the co-chaperone p23 [45]. These interactions effectively sequester the AhR molecule within the HSP90/XAP2/p23 complex, thereby stabilizing AhR in the cytoplasm [48]. In the presence of ligands, the DE-loop and a group of conserved pocket inner residues within the AhR PAS-B domain are responsible for ligand binding [48]. Activation of AhR involves conformational changes that expose the nuclear localization sequence in its N-terminal region, triggering translocation to the nucleus. In the nucleus, this complex dissociates and releases AhR [49]. Subsequently, AhR binds to the aryl hydrocarbon receptor nuclear translocator (ARNT) through interactions in the bHLH and PAS-A domains [50]. The outcome is the recruitment of transcriptional coactivators, such as histone acetyltransferase steroid receptor coactivator (SRC)-1, SRC-2 and p300, IκB kinase α (IKKα), brahma-related gene 1, and RNA initiation factors, to target promoters to enhance transcriptional activity [51,52,53]. This AhR/ARNT/coactivator complex binds to target genes containing consensus DRE or XRE (referred to as dioxin-response element or xenobiotic-responsive element) sequences (5’-GCGTG-3’) and regulates the transcription of target genes, including Cyp1a1, Cyp1a2 [53], aryl hydrocarbon receptor repressor (AhRR) [54], nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 (Nlrp3) [55], IL-10 [9] and IL-22 [56]. Additionally, AhR regulates the transcription of target genes that do not harbor the canonical XRE recognition site in promoter regions by interacting with additional transcription factors, such as estrogen receptor (ER), krüppel-like factor 6 (KLF6), nuclear factor-κB (NF-κB), and MAF bZIP transcription factor (c-Maf) [57,58,59,60]. Furthermore, AhR can directly regulate the transcription of nuclear factor erythroid 2-related factor 2 (Nrf2) [61].

The structure of mouse AhR. AhR contains a basic helix-loop-helix (bHLH) domain and two repeats of the Per-Arnt-Sim (PAS) domain, known as PAS-A and PAS-B. The bHLH and PAS-A domains of AhR are responsible for ARNT binding, and the PAS-B domain is responsible for ligand binding. The C-terminal acidic domain of AhR interacts with cullin 4B (CUL4B)
The transcriptional activity of AhR cannot explain all the cellular functions attributed to this receptor. Several studies have reported that AhR also functions as an E3 ubiquitin ligase. In the nucleus, AhR, together with damaged-DNA binding protein 1 (DDB1), RING-box protein 1 (Rbx1), transducin-β-like protein 3 (TBL3), ARNT and scaffold protein cullin 4B (CUL4B), forms a novel CUL4B ubiquitin ligase complex, CUL4BAhR. Ligand-activated AhR acts as a substrate-specific adaptor component targeting ER-α and androgen receptor (AR) for ubiquitin-mediated degradation. Furthermore, a study confirmed that the conserved C-terminal acidic domain of AhR interacts with the N-terminal region of CUL4B [62]. The role of AhR E3 ubiquitin ligase was implicated in β-catenin degradation, which occurs independently but cooperatively with the APC-dependent pathway to suppress intestinal carcinogenesis [63]. AhR also targeted peroxisome proliferator-activated receptor γ (PPARγ) for proteasomal degradation to regulate adipocyte differentiation [64]. However, the specific mechanism by which this molecular switch mediates the transcriptional activity or E3 ubiquitin ligase activity of AhR has not been fully elucidated. Several investigations have attempted to address this question, with Luecke-Johansson et al. proposing that ARNT plays a crucial role in determining the dual functions of AhR. Their findings revealed that the absence of ARNT significantly impeded the transcriptional activation of AhR but did not affect its E3 ubiquitin ligase function [65]. Kuocheng Lu et al. also demonstrated that different IS concentrations can modulate ARNT as a molecular switch for AhR. Low-dose IS exposure increased nuclear ARNT expression, facilitating the formation of the IS/AhR/ARNT complex in the nucleus. However, high-dose IS exposure decreased ARNT expression, inhibiting the transcriptional activity of AhR in the nucleus and increasing the function of AhR E3 ligase in the cytoplasm [66]. However, it should be noted that the two aforementioned studies ignored the involvement of ARNT in CUL4BAhR complex formation. The cytoplasmic functions of AhR have been gradually elucidated. Ligand-activated cytoplasmic AhR has been reported to act as a protein adaptor that links SRC to janus kinase 2 (JAK2) and mediates SRC phosphorylation by JAK2, which activates the phosphatidylinositol 3-kinase (PI3K)/AKT, mitogen-activated extracellular signal-regulated kinase (MEK)/extracellular signal-regulated kinase (ERK) [67] and yes-associated protein (YAP)-ERK signaling pathways [68]. Ligand-activated cytoplasmic AhR also protects tissue factor (TF) from ubiquitination and degradation to increase thrombotic risk [41] (Fig. 5).

AhR signaling pathway. Before ligand binding, AhR remains stable in the cytoplasm within the HSP90/XAP2/p23 complex. When exposed to AhR ligands, such as uremic toxins, pollutants or natural plants, AhR changes its conformation, thus exposing the nuclear localization sequence in its N-terminal region and triggering translocation to the nucleus. In the nucleus, AhR is released from this complex and activated. Activated AhR binds to ARNT and some coactivators to regulate the transcription of target genes containing consensus XRE (xenobiotic response element), such as Cyp1a1, Cyp1a2, AhRR, Nlrp3, IL-10 and IL-22. In addition, AhR regulates the transcription of target genes that do not harbor the canonical XRE recognition site in their promoter regions by interacting with additional transcription factors, such as ER, KLF6, NF-κB and c-Maf. Furthermore, AhR directly regulates the transcription of Nrf2. Additionally, AhR, together with DDB1, Rbx1, TBL3, ARNT and CUL4B, assembles into the novel CUL4B ubiquitin ligase complex CUL4BAhR to regulate target proteins for ubiquitin degradation, such as ER-α, AR, β-catenin and PPARγ. Ligand-activated cytoplasmic AhR can act as a protein adaptor that links SRC to JAK2, activating the PI3K/AKT, MEK/ERK and YAP/ERK signaling pathways. AhR can also protect tissue factor (TF) from ubiquitination and degradation
The AhR signaling pathway is regulated at three levels: (i) the production and metabolism of ligands that act as agonists or antagonists [8] and (ii) activity disruption by competitors such as AhRR and hypoxia inducible factor (HIF)-1α. AhRR inhibits AhR signal transduction by binding to XREs and ARNT [69] or recruiting corepressors such as ankyrin repeat and LEM domain containing 2 gene, histone deacetylase (HDAC) 4 and HDAC5, which form a negative feedback loop to prevent the overactivation of AhR [70]. HIF1α inhibits AhR activity by interacting with ARNT (also known as HIF-1β) [71]. (iii) Degradation of AhR. After AhR is separated from DNA, it is exported from the nucleus and subjected to proteasomal degradation [72]. AhR can be phosphorylated in a glycogen synthase kinase-3-dependent manner, leading to lysosomal degradation of the AhR protein [73]. Conversely, AhR can be deubiquitinated by the deubiquitinating enzyme ubiquitin C-terminal hydrolase L3 [74]. These mechanisms ensure the proper balance of AhR biology.
Uremic toxin-activated AhR in kidney diseases
Proximal renal tubular epithelial cells (RTECs), which possess various transporters on the cell membrane, are responsible for the absorption and secretion of substances, including drugs, metabolites and environmental toxins [75]. Most uremic toxins are transported via the solute carrier family members organic anion transporters 1 and 3 (OAT1 and OAT3) [75]. A study revealed elevated levels of plasma uremic toxins such as IS, XA and KYN in OAT1 knockout mice [76]. An imbalance between the production and excretion of uremic toxins can contribute to their accumulation within the body, which disturbs normal physiological functions and energy metabolism [1]. When renal function is impaired, the accumulation of uremic toxins accelerates the progression of kidney diseases by activating the AhR signaling pathway, and this damage occurs not only in the kidney but also in other organs, such as the heart, vessel, liver and muscle (Table 2).
Uremic toxin-activated AhR in CKD
CKD is defined by persistent urine abnormalities and structural or functional impairments suggestive of a loss of functional nephrons [77]. CKD is a major public health problem that affects nearly 9.1% of the global population [78]. As the global population ages and the incidences of diabetes, hypertension and other diseases increase, the incidence of CKD also gradually increases [77]. The majority of patients with CKD are at high risk of cardiovascular disease (CVD) and death. When patients with CKD progress to end-stage renal disease (ESRD), the optimal treatment strategy is renal replacement therapy, such as dialysis or kidney transplantation, which has limited accessibility and is extremely susceptible to cardiovascular mortality [77]. It was estimated that CVD mortality in patients who underwent kidney transplantation is 2.3 times greater than that in the general population [79].
The accumulation of uremic toxins and activation of AhR in CKD
In CKD patients, plasma Trp levels were unchanged, and metabolites of Trp, including KYN, 5-HTP, serotonin and QA, were significantly increased at CKD stage 3 [80]. Plasma KYNA [80], IAA [81] and serum IS [82] levels increased significantly at the CKD stage 4. These uremic toxins increased progressively with increasing CKD stage [80,81,82]. IAA levels decreased substantially after kidney transplantation. Nontransplanted CKD patients with above-median IAA concentrations had a significantly higher risk of overall mortality and cardiovascular events than patients with below-median levels [81]. IS is also an independent risk factor for cardiovascular events in patients with CKD [82, 83]. IS has been shown to be positively correlated with aortic calcification and pulse wave velocity [82]. Patients on HD with high plasma IS concentrations were at a higher risk of developing first heart failure [84]. PCS is another widely studied and protein-bound uremic toxin. Serum PCS levels were increased in CKD patients and associated with CKD progression and all-cause mortality [85]. Similarly, the levels of serum uremic toxins have been observed in animal models of CKD. The serum IS concentrations were significantly higher in adenine diet-fed mice and rats, IS mice given water containing IS, and 5/6 nephrectomized rats than in controls, and the serum KYN levels were elevated in adenine diet-fed mice [86, 87]. Plasma TMAO was markedly increased, and elevated TMAO was associated with a 2.8-fold increase in the risk of 5-year all-cause mortality in CKD patients. High TMAO levels portend poorer prognosis among non-CKD subjects [88]. Taken together, these studies indicate that some uremic toxins are independent predictors of overall mortality, CKD progression and cardiovascular events in CKD patients.
With the accumulation of AhR ligands in serum, upregulated AhR expression and activation are also observed in CKD patients and animals. AhR activity was higher in the sera of 20 ESRD patients on HD than in those of controls (activity range 3.02–7.62 vs. 1.1–2.38) [41]. Similarly, another clinical study involving 116 patients with CKD revealed a significant increase in serum AhR activity. The mRNA levels of AhR target genes Cyp1a1 and AhRR were increased in patient blood cells, suggesting activation of the AhR signaling pathway in CKD patients. In addition, significant increases in serum AhR activity and Cyp1a1 mRNA level in the aorta and heart were detected in both 5/6 nephrectomy-induced CKD mice and mice injected with IS for 5 consecutive days, whereas increased Cyp1a1 mRNA level was not observed in AhR knockout mice [89]. In kidneys of mice with unilateral ureteral obstruction-induced renal fibrosis, an increase in AhR mRNA level was accompanied by significant increases in the expressions of AhR target genes, including Cyp1a1, Cyp1a2 and Cyp1b1, suggesting AhR signaling pathway activation in mouse kidneys [90]. AhR activation was also confirmed in the kidneys of 5/6 nephrectomized rats and patients with idiopathic membranous nephropathy and IgA nephropathy [91].
The above studies consistently suggest that uremic toxins are accumulated and AhR is activated during the progression of CKD.
The function of uremic toxin-activated AhR in CKD
It was reported that IS and uremic serum induced AhR activation, as validated by nuclear translocation and increased expressions of target genes, including Cyp1a1, Cyp1b1 and AhRR, which were abrogated by AhR antagonists CB7993113 and CH223191 [41]. Approximately 90% of IS circulates in a protein-bound way among HD patients [92]. A study indicated that both albumin-bound and free IS induced dose-dependent AhR activity in vascular smooth muscle cells [41]. Numerous studies have focused on the harmful effects of AhR activation by uremic toxins on cardiovascular dysfunction during CKD. Serum IS levels in ESRD patients were positively correlated with serum AhR activity and vascular smooth muscle cellular TF activity. In primary cultured human aortic vascular smooth muscle cells, the activation of AhR by IS stabilized TF via inhibiting TF ubiquitination and degradation, thus accelerating thrombosis [41]. Similar studies also showed that IS and IAA upregulated TF expression by activating AhR in human umbilical vein endothelial cells (HUVECs) and peripheral blood mononuclear cells. And the effect was suppressed by treatment with AhR siRNA or the AhR inhibitor geldanamycin. Plasma TF levels were significantly higher in CKD patients than in healthy controls, and TF levels were even higher in CKD patients requiring HD than in non-dialysis patients. In addition, plasma TF levels were positively correlated with IS and IAA levels. The procoagulant state induced by increased TF expression and the direct proatherogenic effect of AhR activation accelerated atherogenesis in CKD [42]. However, other researchers observed that IAA-activated AhR promoted TF transcription independently of binding to the TF promoter in HUVECs. In fact, TF upregulation by IAA was regulated by the AhR/p38 mitogen-activated protein kinase (MAPK)/NF-κB pathway, which increased thrombotic risk [93]. Activated AhR by uremic solutes IS and IAA, as well as TCDD and FICZ, promoted neuronal pentraxin 1 transcription in HUVECs, and the mRNA level of neuronal pentraxin 1 was increased in the aortas of adenine-induced CKD mice [94]. Another study showed that IS reduced the fast transient outward potassium current-related proteins and current densities by activating the reactive oxygen (ROS)/MAPK and NF-κB signaling pathways, prolonging action potential duration and QT interval in neonatal rat ventricular myocytes and hearts of CKD rats. This result helps to account for the high prevalence of ventricular arrhythmias related to sudden cardiac death in CKD patients [95].
CKD is well recognized as a distinct contributor to cardiac hypertrophy. A study clarified the relationship between uremic toxins and cardiac hypertrophy. Treatment of cardiomyocytes with uremic serum collected from patients with CKD stage 5 who have accumulated diverse uremic toxins induced mitochondrial oxidative damage. Mitochondrial damage increased VDAC-mediated mitochondrial outer membrane permeabilization, leading to the release of mitochondrial DNA. Mitochondrial DNA activated cyclic GMP-AMP synthase/stimulator of interferon gene/NF-κB pathway and then stimulated ornithine decarboxylase upregulation and putrescine accumulation, which promoted cardiac hypertrophy [96].
CKD imposes a potent and independent risk for peripheral artery disease (PAD). In a study involving a cohort of 1,091,201 patients, those with CKD exhibited a striking threefold increase in the prevalence of PAD compared with the non-CKD patients [97]. A recent study demonstrated that plasma IS levels were elevated by 1.6-fold, KYN levels were raised by 2.2-fold, and KYNA and XA levels were heightened by 1.5-fold in PAD patients with adverse limb events compared to those without adverse limb events. However, there were no significant differences in the levels of Trp, AA, or QA between the 2 groups. Plasma from PAD patients with adverse events activated AhR activity in human dermal microvascular endothelial cells 60% more compared with the group without adverse events. Uremic toxins were found to suppress the Wnt/β-catenin pathway by augmenting AhR-mediated β-catenin ubiquitination and degradation in human dermal microvascular endothelial cells, which was also verified in adenine-induced CKD and IS solute-specific mouse models with hindlimb ischemia. Notably, inhibiting AhR activity with CH223191 normalized postischemic angiogenesis in adenine-induced CKD mice to a non-CKD level [98]. Another study explored the role of AhR activation in the myopathy of PAD and CKD. The expression and activity of AhR in skeletal muscle were greater in PAD patients with CKD than in PAD patients with normal renal function or non-PAD adult controls. Skeletal muscle-specific AhR knockout promoted ischemic muscle perfusion recovery and arteriogenesis and preserved ischemic muscle mass, contractile function, mitochondrial respiratory function and paracrine vasculogenic signaling between myofibers and vascular cells in adenine-induced CKD mice with hindlimb ischemia. These findings indicate that AhR inhibition is a potential therapeutic for PAD patients with CKD [99]. These studies implicate that retained uremic solutes in CKD patients drive PAD progression by disrupting angiogenesis and muscle health in an AhR-dependent manner.
Increasing reports show that uremic toxin-activated AhR creates a vicious cycle between oxidative stress and inflammation, which aggravates the chronic inflammatory environment in CKD. It has been reported that IS induces ROS production [95]. IS-upregulated ROS promoted the expressions of cAMP response element-binding protein and NF-κB, increasing NADPH oxidase (NOX) 4 expression, an enzyme catalyzing the reduction of molecular oxygen to ROS in proximal renal tubules [100]. IS-induced ROS production led to c-Jun N-terminal kinase (JNK) and NF-кB activation independent of AhR regulation in HUVECs. This study also showed that IS-induced AhR activation stimulated the transcriptional activity of activator protein 1 and subsequently upregulated E-selectin expression in HUVECs, which led to the aggravation of leukocyte recruitment to the vascular wall and vascular inflammation. Endothelial cell-specific AhR knockout inhibited leukocyte recruitment [101]. Crosstalk between AhR and NF-κB is also observed in macrophages. During the early stages of IS stimulation, IS-activated AhR was associated with the NF-κB p65 subunit, leading to mutual inhibition of AhR and NF-κB in the cytoplasm. Subsequently, IS-activated AhR translocated into the nucleus and promoted the transcription of suppressor of cytokine signaling 2 (Socs2), a negative modulator of NF-κB, thus inhibiting NF-κB signaling pathway activation. Finally, the mutual inhibition of AhR and NF-κB was diminished, and free activated AhR induced TNF-α expression by binding to the promoter of Tnf-α [102]. Both free and albumin-bound IS triggered proinflammatory macrophage activation and the expression of proinflammatory cytokines, such as IL-1β, TNF-α and monocyte chemotactic protein 1, in 5/6 nephrectomy-induced CKD mice [103]. These findings indicate that AhR may promote inflammation in CKD.
Furthermore, uremic toxins impair the antioxidant capacity of cells against oxidative stress. Glutathione is a marker of oxidative stress and is known as the most potent antioxidant [104]. A study showed that IS, phenyl sulfate, and PCS, but not IAA, at CKD concentrations led to decreases in total glutathione levels, thus rendering tubular epithelial cells vulnerable to oxidative stress [105].
Recently, researchers have realized that CKD patients have a higher risk of developing cognitive impairment and dementia, even in the early stage of CKD [106]. It is noted that the accumulation of uremic toxins may harm cerebral endothelium and cognitive function in CKD [107, 108]. Notably, serum free IS concentrations, but not PCS, were associated with lower cognitive function in patients with HD [109]. The effect of uremic toxins was experimentally explored, and an increase in serum IS concentrations was shown to promote blood–brain barrier disruption associated with cognitive impairment by AhR activation in CKD rats established by an adenine-rich diet or by 5/6 nephrectomy [87]. Similarly, 5/6 nephrectomy-induced CKD mice showed increased IS concentrations in both the blood and brain and AhR activation in the anterior cortex. CKD-induced anxiety, cognitive impairment, astrocyte reactivation in the anterior cingulate cortex, and neuronal activity enhancement in the anterior cingulate cortex and hippocampal CA1 neurons were ameliorated after knocking out neural lineage-specific and astrocyte-specific AhR or treating with AhR antagonist CH223191. Mechanistically, IS-activated AhR downregulated glutamate transporter 1 (GLT1) expression and activity and promoted pro-oxidant NOX1 expression in astrocytes, leading to enhanced neuronal activity and synaptotoxicity in the brain. The study indicates that astrocytic AhR promotes CKD-induced neuron-astrocyte interaction dysfunction and mental disorders [44].
Renal fibrosis is the common ultimate pathological feature of CKD. Uremic toxins are considered to play a determinant pathological role in the progression of renal fibrosis. Peripheral fibroblast activation and tubular injury are the hallmarks of renal fibrosis [110]. In fibroblasts, IS accumulation promoted renal fibroblast activation via an HSP90-dependent pathway [110]. In proximal RTECs, IS and PCS significantly activated the intrarenal renin–angiotensin–aldosterone system by increasing renin, angiotensinogen and angiotensin 1 receptor expressions, and decreasing angiotensin 2 receptor expression. IS and PCS also increased transforming growth factor β1 (TGFβ1) expression and activated the TGFβ/Smad pathway. IS and PCS induced the epithelial-mesenchymal transition (EMT) phenotype by increasing snail family transcriptional repressor expression. EMT was implicated in renal fibrosis [111]. Cellular senescence is a stress-induced cell cycle arrest independent of age. Senescent cells obtain increased secretion of cytokines, chemokines, growth factors, and proteases, which is referred to as the senescence-associated secretory phenotype [112]. Cellular senescence has been found in multiple kidney diseases, especially in CKD. Young CKD patients frequently exhibit characteristics of premature aging, including vascular aging, bone disease, muscle wasting, cognitive dysfunction and frailty. Chronic renal injury induces cellular senescence, and cellular senescence can also accelerate the progression of renal fibrosis [113]. Recent findings have revealed that uremic toxins mediate cellular senescence in CKD. IS and PCS decreased Klotho expression by enhancing DNA methylation of the Klotho gene in RTECs, thus promoting renal fibrosis [114]. IS can also induce the downregulation of Klotho expression and the production of proinflammatory cytokines in macrophages by stimulating M1 polarization. Overexpression of Klotho alleviated kidney fibrosis by inactivating NF-kB signaling and promoting macrophage M2 polarization [115].
An increase in body uremic toxins triggers remote metabolite sensing to mediate toxins and drug clearance. Membrane transporters are generally involved in metabolite sensing and are widely expressed in epithelial barriers. In proximal RTECs, elevated IS levels induced robust increases in the expression and transport activity of OAT1 by activating the AhR/ARNT and EGFR pathways, enhancing IS clearance. EGFR played a pivotal role in ARNT nuclear translocation, suggesting that crosstalk occurs between EGFR and AhR in IS sensing and signaling [40]. Additionally, IS increased the expression and activity of hepatocellular efflux transport protein P-glycoprotein (P-gp) during CKD by activating AhR, thus promoting the clearance of cyclosporine, a P-gp substrate, from the liver [43]. These results indicate that activated AhR promotes the detoxification process by upregulating the expression of membrane transporters in response to the uremic toxin IS during CKD. Unfortunately, increasing the expression of transporters may alter the clearance of drugs and produce secondary effects.
Several studies have observed renal and hepatic changes in systemic AhR knockout rats. AhR knockout rats exhibited urologic pathological changes such as bilateral renal and ureter dilation (hydronephrosis and hydroureter), as well as secondary medullary tubular and uroepithelial degenerative changes. However, AhR knockout mice exhibited impaired liver function, patent hepatic ductus venosus, and persistent hyaloid arteries in the eye [116]. These changes suggest that AhR plays significantly different roles in tissue development and body homeostasis in different species. Activated AhR is predominantly expressed in the proximal and distal tubules and periglomerular regions in animal models of CKD [86]. However, few studies have explored the role of uremic toxin-activated AhR in the renal tubular epithelium during CKD.
Uremic toxin-activated AhR in DN
DN is defined as kidney damage due to diabetes and has become the predominant contributing factor to CKD. DN occurs in approximately 40% of people with type 2 diabetes (T2D) and type 1 diabetes (T1D) [117]. DN mainly manifests as hyperfiltration, urinary protein, and progressive decline in renal function [118].
The accumulation of uremic toxins and activation of AhR in DN
Notably, compared with nondiabetic patients, the plasma of diabetic patients had lower Trp levels and significantly higher Trp metabolite levels such as 5-HTP, 5-hydroxyindoleacetic acid, KYNA, 3-HK, and XA [119]. Serum IS levels were fourfold higher in streptozotocin (STZ)-induced DN mice compared with controls [120].
AhR expression is increased in DN patients [91]. An increase in AhR activity is also observed in DN patients. Serum AhR activity was increased in the microalbuminuria, macroalbuminuria and ESRD patients compared with normoalbuminuria subjects, and the ESRD group showed higher AhR activity compared with the microalbuminuria and macroalbuminuria groups. Moreover, the serum AhR activity was negatively correlated with eGFR and positively correlated with serum creatinine levels. These findings suggested that serum AhR activity is a significant independent risk factor for DN [121].
The function of uremic toxin-activated AhR in DN
One study confirmed the role and mechanism of AhR activation in DN. STZ-induced diabetic mouse kidneys exhibited elevation in glomerular mesangial cell (MC) activation, macrophage infiltration, extracellular matrix protein deposition, cyclooxygenase (COX-2)/prostaglandin E2 production, lipid peroxidation, oxidative stress, NOX activity and N-ɛ-carboxymethyl lysine formation, which was attenuated by AhR knockout or AhR inhibitor α-NF. N-ɛ-carboxymethyl lysine triggered the transportation of AhR to the nucleus, where it bound to the promoters of Cox-2, fibronectin and collagen IV to produce extracellular matrix proteins in MCs and RTECs. These results suggest that activated AhR plays an important role in MC activation, macrophage infiltration, and extracellular matrix protein accumulation in DN [122]. In addition, treatment with Tangshen Formula, a traditional Chinese herbal medicine, for 12 weeks, significantly attenuated inflammation, renal histologic injury and urinary albumin excretion by inhibiting the upregulation of AhR expression in DN rats [120].
Uremic toxin-activated AhR in AKI
AKI is defined as the sudden loss of kidney function. Slow deterioration of kidney function or persistent kidney dysfunction in AKI is associated with irreversible loss of renal cells and nephrons, potentially leading to CKD [123]. The incidence of AKI is growing by 10% annually, and AKI affects up to 20% of hospitalized patients, with up to 50% of intensive care unit-admitted patients [124]. The main features of AKI induced by ischemia reperfusion (IR), drugs, and sepsis are apoptosis, oxidative stress, inflammation, mitochondrial dysfunction, and abnormalities within the renal vascular system [125, 126].
The accumulation of uremic toxins and activation of AhR in AKI
Clinical studies have shown that serum IS levels are significantly upregulated in AKI patients [127]. IR-induced AKI mice exhibited elevated plasma IS concentrations but no significant change in KYN. Renal AhR activity was increased in IR-induced AKI mice [86].
The function of uremic toxin-activated AhR in AKI
Studies showed that renal AhR expression was decreased in IR mice, along with impaired renal function, increased secretion of inflammatory factors and increased apoptosis. Treatment with the AhR agonist FICZ attenuated renal inflammation, pathological injury and apoptosis by inhibiting the NF-κB and JNK signaling pathways [128]. A similar study also revealed that IR-induced AKI mice treated with the AhR agonist leflunomide exhibited less apoptosis and necrosis and higher mitochondrial membrane potential than AKI mice. Leflunomide affected the infiltration of immune cells and stem cells in injured kidneys by increasing regulatory T cells, IL-10-positive cells and stem cell subsets (e.g., mesenchymal and hematopoietic stem cells and endothelial progenitor cells) and reducing IL-17- and IL-23-expressing cells [129]. In addition, activated AhR can relieve cisplatin-induced AKI. Elevated miR-125b transcription induced by Nrf2 inhibited the translation of AhRR mRNA, which increased the transcriptional activity of AhR. Activated AhR promoted the expression of mouse double minute 2 (MDM2), leading to the inhibition of p53 activity. The decrease in p53 promoted tubular cell survival against cisplatin toxicity and protected the kidney from cisplatin-induced acute injury [130]. AhR knockout mice were more susceptible to malaria and developed high plasma heme levels and AKI during malaria, suggesting that AhR limits renal damage during malaria [131]. These studies indicate that AhR may represent a novel renoprotective mechanism for AKI. However, the role of AhR in AKI remains controversial. Several studies have shown the pro-injury effect of AhR in AKI. AhR expression was increased in RTECs after cisplatin treatment. Knockdown of AhR by siRNA inhibited the IS-induced increase in ROS levels in cisplatin-treated RTECs, indicating that the IS/AhR/ROS axis contributes to oxidative stress. ROS elevation may result in apoptosis and renal damage in cisplatin-induced AKI [132]. A similar study showed that AhR was increased in cisplatin-induced AKI mice kidneys and RTECs. AhR inhibition by BAY2416964 and tubular conditional deletion of AhR both alleviated cisplatin-induced kidney dysfunction and tubular injury by inhibiting cellular senescence. Mechanistically, AhR upregulated the expression of methyltransferase enhancer of zeste homolog 2 (EZH2), and EZH2 conversely enhanced AhR expression via weakening H3K27me3 transcriptional inhibition on the AhR promoter [133]. This finding suggests that increased AhR is implicated in cisplatin-associated cellular senescence, and inhibition of AhR is a promising therapeutic strategy against AKI. Interestingly, a study showed that AhR was activated under anoxia or reoxygenation in primary proximal RTECs. The AhR inhibitor CH223191 did not affect cellular senescence under anoxia or reoxygenation [134].
Role of AhR in other kidney-related diseases
Renal damage is one of the typical clinical manifestations of SLE. A study showed that AhR was significantly increased in B cells of SLE patients with renal injury compared to SLE patients without renal injury, indicating that AhR may be a potential marker for predicting SLE with renal damage [135].
Obstructive sleep apnea (OSA) is a highly prevalent sleep-related breathing disorder. The main hallmark of OSA is chronic intermittent hypoxia (CIH), which contributes to systemic hypertension (HTN). The CIH-induced HTN rat kidney cortex and medulla showed higher expression and activation of AhR. In CIH-induced HTN rats, administration of AhR antagonist CH223191 (5 mg/kg/day, gavage, daily) for 14 days prevented the increase in systolic blood pressure by 53 ± 12% and diastolic blood pressure by 44 ± 16%. These findings suggest that renal AhR activation promotes the progression of HTN induced by CIH [136].
Therapeutic strategy
The accumulation of uremic toxins contributes to multiple organ injuries by activating AhR. Two principal therapeutic options are available to alleviate uremic toxin-induced injury: reducing the levels of uremic toxins and developing pharmacologic approaches to target AhR to mitigate their toxic effects [10] (Table 3).
Reducing uremic toxins
Reducing circulating uremic toxins is a viable strategy for preventing or alleviating kidney diseases. Inhibiting the production and/or enhancing the clearance of uremic toxins are two rational and effective approaches [10].
Blood purification
Conventional HD is the main technique for reducing high concentrations of uremic toxins in the blood. HD transports solutes across a semipermeable membrane through diffusion and mainly applies to remove water-soluble small molecular-weight uremic toxins. Middle molecular-weight molecules and protein-bound uremic toxins are poorly removed [137]. Hemodiafiltration (HDF) transports solutes through diffusion and convection and effectively removes small and middle molecular-weight uremic toxins. However, HDF leads to loss of potential albumin and nutrients during treatment and the consequent need for reinfusion [137]. Adsorption-based hemoperfusion can remove middle and large molecular-weight and protein-bound uremic toxins [137]. Absorbents for hemoperfusion are usually made of polymeric resins, activated carbon, carbon nanotubes and zeolites [138]. Graphene oxide is an exceptional material because of its outstanding mechanical properties, modifiable surface functionalization and controllable interlayer distance [138]. However, the direct use of graphene oxide as an adsorbent in hemoperfusion can contribute to hemolysis and decrease blood cell and platelet levels, which may harm patients [138]. In contrast, cellulose acetate (CA) is an adsorbent material with good water and solute permeabilities and excellent hemocompatibility [138].
Improvements in the materials and production processes to increase the removal effect of uremic toxins may improve their applicability in extracorporeal purification systems. Abhishek Tyagi et al. developed a CA-functionalized graphene oxide composite material for hemoperfusion, which cleared creatinine from 83.23 to 54.87 μmol/l and uric acid from 93.4 to 54.14 μmol/l, thus restoring to normal levels within 30 min [138]. The water-dispersal adsorbent poly-β-cyclodextrin added into the dialysate can remove 96% PCS in the plasma via adsorbent once-through mode [139]. Adding poly-β-cyclodextrin cross-linked with epichlorohydrin for two hours to the dialysate can result in a twice increase in the ability to remove IS [140]. Cationic metal–organic frameworks, utilizing tetrakis ethene as a ligand skeleton, pyridyl units as functional groups, and nickel/silver nitrate as metal nodes, could almost completely remove PCS within 3 hours through anion exchange with high adsorption capacities and good adsorption kinetics [141]. In the future, optimizing sorbent materials with technical characteristics to enhance dialysis efficiency is a crucial research direction.
Gastrointestinal dialysis
Oral administration of cathartic compounds is a well-known method for promoting the excretion of uremic toxins and excess fluids [10]. The carbon adsorbent AST-120 has received the most attention due to its ability to absorb uremic toxin precursors in the intestinal tract and then excrete the precursors in feces, thereby reducing the absorption of uremic toxins into the blood [10]. The oral adsorbent AST-120 prevented renal accumulation of IS and PCS in adenine-induced CKD mice. However, AST-120 did not improve renal function and attenuate tubular injury and renal fibrosis in adenine-induced CKD mice [142]. Administration of AST-120 significantly decreased serum IS levels in mice with 5/6 nephrectomy-induced CKD and arteriovenous fistula. AST-120 attenuated neointima formation by inhibiting the expressions of matrix metalloproteinase (MMP)-2, MMP-9, TNF-α, and TGFβ1 in neointima tissue [143]. The therapeutic efficacy of AST-120 in CKD patients is also controversial. A multicenter, randomized, controlled trial showed that AST-120 can slow the deterioration of renal function as evidenced by inhibition of the decrease in eGFR, but it did not significantly slow disease progression in patients with moderate to severe CKD during 1 year [144]. A systematic review and meta-analysis including eight studies also demonstrated that AST-120 can effectively reduce IS levels, but controversy remained regarding slowing CKD progression and all-cause mortality [145]. So the clinical use of AST-120 for the treatment of CKD needs to be carefully considered.
Nutritional therapy
Nutritional therapy has been recommended for the management of patients with CKD for more than a century [146]. A diet rich in animal proteins increases populations of proteolytic bacteria that ferment dietary protein and generate uremic toxins, such as PCS, IS and TMAO. A low protein diet exhibits favorable effects on CKD progression due to the reduction of these substrates [147]. One study involving 29 healthy subjects and 20 wild-type friend leukemia virus mice revealed the influence of dietary protein intake on the mammalian metabolome. Human results showed that plasma and urinary IS levels were significantly lower, as were urinary indoxyl glucuronide, KYNA and QA, in the low protein diet group (target of 9% of total energy intake derived from protein intake) compared to the high protein diet group (target of > 25% of total energy intake derived from protein intake). The mouse results showed that the plasma p-cresyl glucuronide, phenyl sulfate and phenylacetic acid levels were decreased in the control diet (21% crude protein) compared to the high protein diet (45% crude protein). These results indicate that a low protein diet is a feasible approach for lowering uremic toxin levels in CKD patients [148]. A meta-analysis of 16 controlled trials of dietary protein restriction in CKD patients revealed that low protein intake (< 0.8 g/kg per day) or very low intake (< 0.4 g/kg per day) for 6–36 months preserved kidney function, slowed the progression to ESRD and reduced the rate of all-cause death compared to a high protein diet (> 0.8 g/kg/day) [149]. However, a large Modification of Diet in Kidney Disease (MDRD) Study revealed that a very low protein diet (0.28 g/kg/day) did not delay CKD progression and even increased the risk of death between 6 and 12 years of follow-up [150].
Due to the uncertain efficacy and potential increased risk of protein malnutrition in protein restriction regimens, the use of a low (0.6–0.8 g/kg per day) or very low (0.3–0.4 g/kg per day) protein diet is partly limited [146]. Some studies have focused on compensating for missing essential amino acids by supplementing transamination-based ketoanalogues (KA) in a low or very low protein diet. In a randomized controlled trial, 207 patients with CKD stage 4 + were allocated to a low protein diet (0.6 g/kg per day) or KA-supplemented vegetarian very low-protein diet (0.3 g/kg vegetable proteins and 0.125 g/kg KA per day). Patients on a KA-supplemented vegetarian very low-protein diet had a lower risk of reaching the composite end point (> 50% eGFR reduction or dialysis initiation) than those on a conventional low protein diet after 18 months of follow-up. A KA-supplemented vegetarian very low-protein diet also improved calcium-phosphorus metabolism and increased serum bicarbonate levels, which alleviated uremic symptoms and deferred dialysis initiation [151].
Previous dietary trials often focused on restricting total protein intake. Actually, the types of protein intake are more important, which produces a vegetarian diet [147, 152]. Vegetable proteins may induce renal changes comparable to a low protein diet and prevent the proteinuric and vasodilatory effects of meat [152]. A randomized study involving 113 healthy volunteers who were given red meat, white meat or non-meat protein (all meals prepared with 25% calories from protein) reported that chronic dietary red meat increased systemic TMAO levels by enhancing dietary precursors, increasing gut microbial TMA/TMAO production from carnitine, and reducing renal TMAO excretion [153]. The study on the oral ingestion of deuterium-labeled L-carnitine showed that vegetarians/vegans had significantly lower TMAO levels than omnivores because vegetarians/vegans had decreased gut microbiota catabolism [29]. Fiber consumption can slow CKD progression by improving the intestinal microbiota composition and reducing toxic metabolites [154]. A high-fiber diet also induced the production of beneficial metabolites, such as short-chain fatty acids (SCFAs) produced by butyrate-producing bacteria. SCFAs not only provide energy for the intestinal flora and allow the incorporation of amino acids from the colon into bacterial proteins and excretion instead of fermentation into uremic solutes, but also benefit the maintenance of intestinal epithelial functionality and integrity [154]. A prospective monocentric study using a seven-day diet record in 58 HD patients reported that IS and PCS concentrations were negatively correlated with fiber intake and positively correlated with the protein/fiber index in anuric HD patients [155].
A low protein diet, KA-supplemented diet and vegetarian diet exhibit potential benefits in reducing uremic toxin levels and slowing CKD progression. However, the benefits of these diets are often counteracted by poor patient compliance. Therefore, patient-tailored diets that reduce uremic toxins should be established for the management of CKD [10].
Targeting microbiota
The microbial diversity and abundance of gut bacterial species are altered in patients with CKD or AKI [156]. For example, in kidney transplant recipients, the abundances of pathogenic bacteria, including Ruminococcacea and E. coli, were increased, whereas the abundances of protective bacteria, such as Alistipes senegalensis and Bacteroidales sp., were reduced. The metabolites of the microbiota were also significantly altered, such as a decrease in SCFAs in kidney transplant recipients [157]. Some therapies, such as dietary control as described above, and administration of probiotics, prebiotics or synbiotics, have been potential options to target the microbiome for ameliorating kidney injury and uremic toxins production [158].
A study showed that supplementation of Faecalibacterium prausnitzii to 5/6 nephrectomy surgery-induced CKD mice reduced plasma PCS and TMAO levels but not IS levels, and ameliorated renal dysfunction and inflammation [159]. Lactobacillus paracasei X11 has been shown to possess excellent uric acid-lowering activity and oral administration of Lactobacillus paracasei X11 reduced serum uric acid and renal inflammation in hyperuricemic mice [160]. However, supplementation of CKD patients undergoing HD with well-known Bifidobacteria, Lactobacilli and Streptococci failed to reduce uremic toxins [161].
Prebiotics, nondigestible foods stimulating the growth of beneficial bacteria in the colon, include fructo-oligosaccharides, galactose-oligosaccharides, xylose-oligosaccharides, inulin, resistant starch, pectin, other fiber components, and milk oligosaccharides [162]. A randomized trial enrolling 59 patients with CKD stage 3–5 revealed that β-glucan prebiotic intervention decreased plasma IS, PCS, and p-cresyl glucuronide levels [163]. The study in individuals at high risk of CVD showed that β-glucan increased Bifidobacterium and Lactobacillus, increasing the production of SCFAs [164].
In practice, synbiotics are combinations of probiotics and prebiotics. Nondialysis adult participants with CKD stage 4 or 5 were recruited for a crossover trial of synbiotic therapy (combination of high molecular-weight inulin, fructo-oligosaccharides and galacto-oligosaccharides with nine different strains across the Lactobacillus, Bifidobacterium, and Streptococcus genera) over 6 weeks. The results showed that synbiotic therapy reduced serum PCS but not IS and altered the intestinal microbiome [165].
Regarding the mechanism of probiotics, prebiotics or synbiotics, several meta-analysis studies have shown that supplementation with probiotics, prebiotics, and synbiotics in CKD patients could decrease inflammation, improve the oxidative imbalance between pro-oxidant factors and anti-oxidant enzymes, and ameliorate the lipid profile [166]. Synbiotics enhance the integrity of the intestinal epithelial barrier and the growth of protective bacteria, inhibit the growth of pathogenic bacteria, improve the host immune system and increase the production of the beneficial metabolites SCFAs to suppress the production of uremic toxins [166, 167]. It should be noted that elevation of plasma uremic toxin levels is primarily attributed to decreased excretion due to a decline in kidney function rather than an increase in generation because culture of fecal samples from CKD patients showed no difference in PCS, indole and IAA production [168]. These results prompt a serious reconsideration of microbial manipulation as a therapeutic strategy to reduce the burden of uremic toxins.
Developing natural AhR agonists and antagonists
Natural products are abundant and crucial sources for drug discovery. Numerous studies have revealed and considered natural AhR agonists and antagonists as alternative therapies for improving CKD and inhibiting renal fibrosis [91]. The level of 1-aminopyrene, a polycyclic aromatic hydrocarbon metabolite, was increased in the remnant kidneys of 5/6 nephrectomized rats. Treatment of RTECs with 1-aminopyrene activated the AhR, suggesting that 1-aminopyrene is an agonist of AhR [91]. Three flavonoids 5',7',3',4',5'-pentahydroxy flavanone, barleriside A and rhoifolin screened and identified from Semen Plantagini showed strong interactions with rat AhR and strong antagonistic effects on AhR activity, suggesting that they are potent AhR antagonists. Three flavonoids alleviated 1-aminopyrene-induced upregulation of profibrotic protein expression in RTECs. Dietary 5',7',3',4',5'‐pentahydroxy flavanone and barleriside A alleviated the decline in renal function and renal fibrosis in 5/6 nephrectomized rats by inhibiting AhR activation [91]. Vitamin B12 and folic acid (FA) were reported as natural antagonists of AhR. Vitamin B12 or FA deficiency in mice induced an increase in AhR transcriptional activity in the liver and accumulation of erythroid progenitors in bone marrow in an AhR-dependent manner. Treatment with vitamin B12 or FA rescued mice from TCDD- or FICZ-induced anemia and thrombocytopenia [169]. Baicalein, an important flavonoid compound isolated from the roots of Scutellaria baicalensis Georgi [170], was able to bind to AhR as predicted by molecular docking models, and induced AhR activation, indicating that baicalein is an AhR agonist [171]. Administration of baicalein (200 mg/kg) significantly decreased serum uric acid and urea nitrogen levels to attenuate hyperuricemia and renal injury [170]. The renoprotective effect of baicalein was also observed in mice with aristolochic acid nephropathy through AhR-dependent CYP1A1/2 induction in the liver [172].
Considering the double-edged sword effects of AhR in kidney diseases, the selection of AhR agonists or antagonists should be cautious and confirmed in experimental and clinical studies. However, AhR is still an intriguing and valuable therapeutic target for kidney diseases because of its important effect on renal injury and associated complications and response to uremic toxins.
Conclusions
As the receptor for multiple uremic toxins, AhR is elevated and activated following the accumulation of uremic toxins in the body. Accumulation of uremic toxins affects all organs and tissues, so revealing the roles of AhR activation is attracting more and more research attention. This review systematically generalizes and summarizes various functions and signaling pathways of uremic toxin-activated AhR in current nephropathy studies. Uremic toxin-activated AhR exerts detrimental biological effects on the development of CKD, CKD-associated cognitive impairment, anxiety, obstructive sleep apnea, ischemic myopathy and CVD, and DN. Uremic toxin-activated AhR increases drug and toxins clearance in CKD. In contrast, uremic toxin-activated AhR in AKI are controversial because of both protective and detrimental effects (Fig. 6). Therefore, the strategies of renal protection targeting AhR and related mechanisms, such as reducing uremic toxins or modulating AhR activation, are on the way to investigations. Uremic toxins are influenced not only by renal excretion but also by dietary intake processed in the intestinal microbiota and biotransformed in the liver, all of which can vary between individuals and may be considered targets for intervention. Although targeting uremic toxins and the AhR pathway are promising approaches, further elucidation of AhR regulation and investigations into the effects of specific agonists/antagonists are required to develop optimal therapies for human kidney disease treatment.

The impact of uremic toxin-activated AhR on kidney diseases and complications. Activation of AhR by uremic toxins has been implicated in various organs attributed to renal damage. In the brain, AhR activation promotes blood–brain barrier disruption, cognitive impairment, anxiety and neuron–astrocyte interaction dysfunction. AhR activation promotes the progression of systemic hypertension in obstructive sleep apnea. In drug metabolism, AhR activation facilitates drug clearance in the liver. Similarly, AhR activation in skeletal muscle exacerbates ischemic myopathy. The cardiovascular system is also impacted, with AhR activation in vessels contributing to thrombosis, peripheral artery disease, vascular inflammation and systemic hypertension. In the kidney, AhR activation promotes toxin clearance, chronic kidney disease and diabetic nephropathy and mediates acute kidney injury. AhR may be a potential marker for predicting systemic lupus erythematosus with renal damage. This figure was created with BioRendercom
Availability of data and materials
Not applicable.
Abbreviations
- AhR:
-
Aryl hydrocarbon receptor
- CKD:
-
Chronic kidney disease
- DN:
-
Diabetic nephropathy
- AKI:
-
Acute kidney injury
- SLE:
-
Systemic lupus erythematosus
- GFR:
-
Glomerular filtration rate
- eGFR:
-
Estimated glomerular filtration rate
- PCS:
-
P-cresyl sulfate
- KYN:
-
Kynurenine
- IS:
-
Indoxyl sulfate
- Trp:
-
Tryptophan
- NFK:
-
N-formylkynurenine
- TDO:
-
Tryptophan 2,3-dioxygenase
- IDO:
-
Indoleamine-2,3-dioxygenase
- AFMID:
-
Kynurenine formamidase
- 3-HK:
-
3-Hydroxykynurenin
- KMO:
-
Kynurenine 3-monooxygenase
- KYNU:
-
Kynureninase
- 3-HAA:
-
3-Hydroxyanthralinic acid
- HAAO:
-
3-Hydroxyanthranilate 3,4-dioxygenase
- QA:
-
Quinolinic acid
- KAT:
-
Kynurenine amino transferase
- XA:
-
Xanthurenic acid
- AA:
-
Anthralinic acid
- KYNA:
-
Kynurenic acid
- TpH:
-
Tryptophan hydroxylase enzyme
- 5-HTTP:
-
5-Hydroxytryptophan
- 5-HT:
-
5-Hydroxytryptamine
- CYP2E1:
-
Cytochrome P450 family 2 subfamily E member 1
- I3A:
-
Indole-3-aldehyde
- ILA:
-
Indole-3-lactic acid
- IAA:
-
Indole-3-acetic acid
- UV:
-
Ultraviolet
- FICZ:
-
6-Formylindolo[3,2-b]carbazole
- I3C:
-
Indole-3-carbinol
- ICZ:
-
Indolo[3,2-b]carbazole
- IL:
-
Interleukin
- TNF:
-
Tumor necrosis factor
- FGF:
-
Fibroblast growth factor
- IAAld:
-
Indole-3-acetaldehyde
- TCDD:
-
Tetrachlorodibenzo-p-dioxin
- XAP2:
-
X-associated protein 2
- HSP90:
-
Heat shock protein 90
- ARNT:
-
Aryl hydrocarbon receptor nuclear translocator
- SRC:
-
Steroid receptor coactivator
- IKKα:
-
IκB kinase α
- DRE:
-
Dioxin-response element
- XRE:
-
Xenobiotic-responsive element
- AhRR:
-
Aryl hydrocarbon receptor repressor
- Nlrp3:
-
Nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3
- ER:
-
Estrogen receptor
- KLF6:
-
Krüppel-like factor 6
- NF-κB:
-
Nuclear factor-κB
- c-Maf:
-
MAF bZIP transcription factor
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- DDB1:
-
Damaged-DNA binding protein 1
- Rbx1:
-
RING-box protein 1
- TBL3:
-
Transducin-β-like protein 3
- CUL4B:
-
Cullin 4B
- AR:
-
Androgen receptor
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- HIF:
-
Hypoxia inducible factor
- HDAC:
-
Histone deacetylase
- OAT:
-
Organic anion transporter
- CVD:
-
Cardiovascular disease
- ESRD:
-
End-stage renal disease
- TMAO:
-
Trimethylamine-N-oxide
- FMO3:
-
Flavin monooxygenase 3
- RTEC:
-
Renal tubular epithelial cell
- TF:
-
Tissue factor
- HUVEC:
-
Human umbilical vein endothelial cell
- MAPK:
-
Mitogen-activated protein kinase
- PAD:
-
Peripheral artery disease
- NOX:
-
NADPH oxidase
- Socs2:
-
Suppressor of cytokine signaling 2
- GLT1:
-
Glutamate transporter 1
- TGFβ1:
-
Transforming growth factor β1
- EMT:
-
Epithelial-mesenchymal transition
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal-regulated kinase
- YAP:
-
Yes-associated protein
- JAK2:
-
Janus kinase 2
- PI3K:
-
Phosphatidylinositol 3-kinase
- MEK:
-
Mitogen-activated extracellular signal-regulated kinase
- miR:
-
MicroRNA
- P-gp:
-
P-glycoprotein
- T2D:
-
Type 2 diabetes
- T1D:
-
Type 1 diabetes
- STZ:
-
Streptozotocin
- COX-2:
-
Cyclooxygenase
- IR:
-
Ischemia reperfusion
- MDM2:
-
Mouse double minute 2
- EZH2:
-
Enhancer of zeste homolog 2
- OSA:
-
Obstructive sleep apnea
- CIH:
-
Chronic intermittent hypoxia
- HTN:
-
Hypertension
- HD:
-
Hemodialysis
- HDF:
-
Hemodiafiltration
- CA:
-
Cellulose acetate
- MMP:
-
Matrix metalloproteinase
- SCFAs:
-
Short-chain fatty acids
- FA:
-
Folic acid
- KA:
-
Ketoanalogues
- JNK:
-
c-Jun N-terminal kinase
References
Deltombe O, Van Biesen W, Glorieux G, Massy Z, Dhondt A, Eloot S. Exploring protein binding of uremic toxins in patients with different stages of chronic kidney disease and during hemodialysis. Toxins (Basel). 2015;7(10):3933–46.
Zhao H, Chen L, Yang T, Feng YL, Vaziri ND, Liu BL, et al. Aryl hydrocarbon receptor activation mediates kidney disease and renal cell carcinoma. J Transl Med. 2019;17(1):302.
Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS proteins in cancer. Nat Rev Cancer. 2013;13(12):827–41.
Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity. 2018;48(1):19–33.
Salminen A. Aryl hydrocarbon receptor (AhR) reveals evidence of antagonistic pleiotropy in the regulation of the aging process. Cell Mol Life Sci. 2022;79(9):489.
Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. 2020;11(1):4457.
Sayed TS, Maayah ZH, Zeidan HA, Agouni A, Korashy HM. Insight into the physiological and pathological roles of the aryl hydrocarbon receptor pathway in glucose homeostasis, insulin resistance, and diabetes development. Cell Mol Biol Lett. 2022;27(1):103.
Liu JR, Miao H, Deng DQ, Vaziri ND, Li P, Zhao YY. Gut microbiota-derived tryptophan metabolism mediates renal fibrosis by aryl hydrocarbon receptor signaling activation. Cell Mol Life Sci. 2021;78(3):909–22.
Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva MT, et al. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol. 2018;19(6):571–82.
Ravid JD, Kamel MH, Chitalia VC. Uraemic solutes as therapeutic targets in CKD-associated cardiovascular disease. Nat Rev Nephrol. 2021;17(6):402–16.
Xu S, Zhang Y, Li Y, Xia X, Zhou J, Shi G. Production of L-tyrosine using tyrosine phenol-lyase by whole cell biotransformation approach. Enzyme Microb Technol. 2019;131: 109430.
Blakley ER. The catabolism of L-tyrosine by an Arthrobacter sp. Can J Microbiol. 1977;23(9):1128–39.
Powell JT, Morrison JF. The purification and properties of the aspartate aminotransferase and aromatic-amino-acid aminotransferase from Escherichia coli. Eur J Biochem. 1978;87(2):391–400.
Seah SY, Britton KL, Rice DW, Asano Y, Engel PC. Single amino acid substitution in Bacillus sphaericus phenylalanine dehydrogenase dramatically increases its discrimination between phenylalanine and tyrosine substrates. Biochemistry. 2002;41(38):11390–7.
Selmer T, Andrei PI. p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol. Eur J Biochem. 2001;268(5):1363–72.
Brix LA, Barnett AC, Duggleby RG, Leggett B, McManus ME. Analysis of the substrate specificity of human sulfotransferases SULT1A1 and SULT1A3: site-directed mutagenesis and kinetic studies. Biochemistry. 1999;38(32):10474–9.
Burchell B, Coughtrie MW. UDP-glucuronosyltransferases. Pharmacol Ther. 1989;43(2):261–89.
Gryp T, Vanholder R, Vaneechoutte M, Glorieux G. p-Cresyl sulfate. Toxins (Basel). 2017;9(2):52.
Wyatt M, Greathouse KL. Targeting dietary and microbial tryptophan-indole metabolism as therapeutic approaches to colon cancer. Nutrients. 2021;13(4):1189.
Cervenka I, Agudelo LZ, Ruas JL. Kynurenines: tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 2017;357(6349).
Keskin DB, Marshall B, Munn D, Mellor AL, Gearhart DA. Decreased protein nitration in macrophages that overexpress indoleamine 2,3-dioxygenase. Cell Mol Biol Lett. 2007;12(1):82–102.
Sugiyama Y, Mori Y, Nara M, Kotani Y, Nagai E, Kawada H, et al. Gut bacterial aromatic amine production: aromatic amino acid decarboxylase and its effects on peripheral serotonin production. Gut Microbes. 2022;14(1):2128605.
Schroeder JC, Dinatale BC, Murray IA, Flaveny CA, Liu Q, Laurenzana EM, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 2010;49(2):393–400.
Diani-Moore S, Ma Y, Labitzke E, Tao H, David Warren J, Anderson J, et al. Discovery and biological characterization of 1-(1H-indol-3-yl)-9H-pyrido[3,4-b]indole as an aryl hydrocarbon receptor activator generated by photoactivation of tryptophan by sunlight. Chem Biol Interact. 2011;193(2):119–28.
Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci. 2005;85(2):935–43.
Centofanti F, Buono A, Verboni M, Tomino C, Lucarini S, Duranti A, et al. Synthetic methodologies and therapeutic potential of indole-3-carbinol (I3C) and its derivatives. Pharmaceuticals (Basel). 2023;16(2):240.
Zhen J, Zhou Z, He M, Han HX, Lv EH, Wen PB, et al. The gut microbial metabolite trimethylamine N-oxide and cardiovascular diseases. Front Endocrinol (Lausanne). 2023;14:1085041.
Wiedeman AM, Barr SI, Green TJ, Xu Z, Innis SM, Kitts DD. Dietary choline intake: current state of knowledge across the life cycle. Nutrients. 2018;10(10):1513.
Koeth RA, Lam-Galvez BR, Kirsop J, Wang Z, Levison BS, Gu X, et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest. 2019;129(1):373–87.
Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, et al. gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014;20(5):799–812.
Cho CE, Caudill MA. Trimethylamine-N-oxide: friend, foe, or simply caught in the cross-fire? Trends Endocrinol Metab. 2017;28(2):121–30.
Cho CE, Taesuwan S, Malysheva OV, Bender E, Tulchinsky NF, Yan J, et al. Trimethylamine-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: a randomized controlled trial. Mol Nutr Food Res. 2017;61(1).
Bjorndal B, Ramsvik MS, Lindquist C, Nordrehaug JE, Bruheim I, Svardal A, et al. A phospholipid-protein complex from antarctic krill reduced plasma homocysteine levels and increased plasma trimethylamine-oxide (TMAO) and carnitine levels in male wistar rats. Mar Drugs. 2015;13(9):5706–21.
Vanholder R, De Smet R, Glorieux G, Argiles A, Baurmeister U, Brunet P, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63(5):1934–43.
Rosner MH, Reis T, Husain-Syed F, Vanholder R, Hutchison C, Stenvinkel P, et al. Classification of uremic toxins and their role in kidney failure. Clin J Am Soc Nephrol. 2021;16(12):1918–28.
Vanholder R, Pletinck A, Schepers E, Glorieux G. Biochemical and clinical impact of organic uremic retention solutes: a comprehensive update. Toxins (Basel). 2018;10(1):33.
Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22(3):358–69.
Wei YD, Helleberg H, Rannug U, Rannug A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem Biol Interact. 1998;110(1–2):39–55.
Fernandez-Gallego N, Sanchez-Madrid F, Cibrian D. Role of AHR ligands in skin homeostasis and cutaneous inflammation. Cells. 2021;10(11):3176.
Jansen J, Jansen K, Neven E, Poesen R, Othman A, van Mil A, et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc Natl Acad Sci U S A. 2019;116(32):16105–10.
Shivanna S, Kolandaivelu K, Shashar M, Belghasim M, Al-Rabadi L, Balcells M, et al. The aryl hydrocarbon receptor is a critical regulator of tissue factor stability and an antithrombotic target in uremia. J Am Soc Nephrol. 2016;27(1):189–201.
Gondouin B, Cerini C, Dou L, Sallee M, Duval-Sabatier A, Pletinck A, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013;84(4):733–44.
Santana Machado T, Poitevin S, Paul P, McKay N, Jourde-Chiche N, Legris T, et al. Indoxyl sulfate upregulates liver P-glycoprotein expression and activity through aryl hydrocarbon receptor signaling. J Am Soc Nephrol. 2018;29(3):906–18.
Huang YJ, Hung CC, Hsu PC, Lee PY, Tsai YA, Hsin YC, et al. Astrocytic aryl hydrocarbon receptor mediates chronic kidney disease-associated mental disorders involving GLT1 hypofunction and neuronal activity enhancement in the mouse brain. Glia. 2023;71(4):1057–80.
Gruszczyk J, Grandvuillemin L, Lai-Kee-Him J, Paloni M, Savva CG, Germain P, et al. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat Commun. 2022;13(1):7010.
Wu D, Potluri N, Kim Y, Rastinejad F. Structure and dimerization properties of the aryl hydrocarbon receptor PAS-A domain. Mol Cell Biol. 2013;33(21):4346–56.
Seok SH, Lee W, Jiang L, Molugu K, Zheng A, Li Y, et al. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc Natl Acad Sci U S A. 2017;114(21):5431–6.
Wen Z, Zhang Y, Zhang B, Hang Y, Xu L, Chen Y, et al. Cryo-EM structure of the cytosolic AhR complex. Structure. 2023;31(3):295–308.
Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–40.
Schulte KW, Green E, Wilz A, Platten M, Daumke O. Structural basis for aryl hydrocarbon receptor-mediated gene activation. Structure. 2017;25(7):1025–33.
Wu L, Whitlock JP Jr. Mechanism of dioxin action: Ah receptor-mediated increase in promoter accessibility in vivo. Proc Natl Acad Sci U S A. 1992;89(11):4811–5.
Kurita H, Schnekenburger M, Ovesen JL, Xia Y, Puga A. The Ah receptor recruits IKKalpha to its target binding motifs to phosphorylate serine-10 in histone H3 required for transcriptional activation. Toxicol Sci. 2014;139(1):121–32.
Taylor RT, Wang F, Hsu EL, Hankinson O. Roles of coactivator proteins in dioxin induction of CYP1A1 and CYP1B1 in human breast cancer cells. Toxicol Sci. 2009;107(1):1–8.
Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999;13(1):20–5.
Huai W, Zhao R, Song H, Zhao J, Zhang L, Zhang L, et al. Aryl hydrocarbon receptor negatively regulates NLRP3 inflammasome activity by inhibiting NLRP3 transcription. Nat Commun. 2014;5:4738.
Lowe MM, Mold JE, Kanwar B, Huang Y, Louie A, Pollastri MP, et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE. 2014;9(2): e87877.
Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423(6939):545–50.
Wilson SR, Joshi AD, Elferink CJ. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J Pharmacol Exp Ther. 2013;345(3):419–29.
Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000;19(48):5498–506.
Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11(9):854–61.
Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem. 2005;280(21):20340–8.
Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446(7135):562–6.
Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A. 2009;106(32):13481–6.
Dou H, Duan Y, Zhang X, Yu Q, Di Q, Song Y, et al. Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARgamma for proteasomal degradation. J Biol Chem. 2019;294(48):18504–15.
Luecke-Johansson S, Gralla M, Rundqvist H, Ho JC, Johnson RS, Gradin K, et al. A molecular mechanism to switch the aryl hydrocarbon receptor from a transcription factor to an E3 ubiquitin ligase. Mol Cell Biol. 2017;37(13).
Liu WC, Shyu JF, Lim PS, Fang TC, Lu CL, Zheng CM, et al. Concentration and duration of indoxyl sulfate exposure affects osteoclastogenesis by regulating NFATc1 via aryl hydrocarbon receptor. Int J Mol Sci. 2020;21(10).
Ye M, Zhang Y, Gao H, Xu Y, Jing P, Wu J, et al. Activation of the aryl hydrocarbon receptor leads to resistance to EGFR TKIs in non-small cell lung cancer by activating src-mediated bypass signaling. Clin Cancer Res. 2018;24(5):1227–39.
Zhang D, Ning J, Ramprasath T, Yu C, Zheng X, Song P, et al. Kynurenine promotes neonatal heart regeneration by stimulating cardiomyocyte proliferation and cardiac angiogenesis. Nat Commun. 2022;13(1):6371.
Sakurai S, Shimizu T, Ohto U. The crystal structure of the AhRR-ARNT heterodimer reveals the structural basis of the repression of AhR-mediated transcription. J Biol Chem. 2017;292(43):17609–16.
Oshima M, Mimura J, Yamamoto M, Fujii-Kuriyama Y. Molecular mechanism of transcriptional repression of AhR repressor involving ANKRA2, HDAC4, and HDAC5. Biochem Biophys Res Commun. 2007;364(2):276–82.
Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 2015;21(6):638–46.
Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem. 1999;274(40):28708–15.
Yang Y, Chan WK. Glycogen synthase kinase 3 beta regulates the human aryl hydrocarbon receptor cellular content and activity. Int J Mol Sci. 2021;22(11):6097.
Ouyang L, Yan B, Liu Y, Mao C, Wang M, Liu N, et al. The deubiquitylase UCHL3 maintains cancer stem-like properties by stabilizing the aryl hydrocarbon receptor. Signal Transduct Target Ther. 2020;5(1):78.
Nigam SK, Wu W, Bush KT, Hoenig MP, Blantz RC, Bhatnagar V. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin J Am Soc Nephrol. 2015;10(11):2039–49.
Wikoff WR, Nagle MA, Kouznetsova VL, Tsigelny IF, Nigam SK. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res. 2011;10(6):2842–51.
Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, et al. Chronic kidney disease. Nat Rev Dis Primers. 2017;3:17088.
Collaboration GBDCKD. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395(10225):709–33.
Kiberd B, Keough-Ryan T, Panek R. Cardiovascular disease reduction in the outpatient kidney transplant clinic. Am J Transplant. 2003;3(11):1393–9.
Schefold JC, Zeden JP, Fotopoulou C, von Haehling S, Pschowski R, Hasper D, et al. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: a possible link between chronic inflammation and uraemic symptoms. Nephrol Dial Transplant. 2009;24(6):1901–8.
Liabeuf S, Laville SM, Glorieux G, Cheddani L, Brazier F, Titeca Beauport D, et al. Difference in profiles of the gut-derived tryptophan metabolite indole acetic acid between transplanted and non-transplanted patients with chronic kidney disease. Int J Mol Sci. 2020;21(6):2031.
Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4(10):1551–8.
Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: the emerging role of protein-bound uremic toxins. Circ Res. 2012;111(11):1470–83.
Cao XS, Chen J, Zou JZ, Zhong YH, Teng J, Ji J, et al. Association of indoxyl sulfate with heart failure among patients on hemodialysis. Clin J Am Soc Nephrol. 2015;10(1):111–9.
Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, et al. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. 2011;26(3):938–47.
Walker JA, Richards S, Belghasem ME, Arinze N, Yoo SB, Tashjian JY, et al. Temporal and tissue-specific activation of aryl hydrocarbon receptor in discrete mouse models of kidney disease. Kidney Int. 2020;97(3):538–50.
Bobot M, Thomas L, Moyon A, Fernandez S, McKay N, Balasse L, et al. Uremic toxic blood-brain barrier disruption mediated by AhR activation leads to cognitive impairment during experimental renal dysfunction. J Am Soc Nephrol. 2020;31(7):1509–21.
Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448–55.
Dou L, Poitevin S, Sallee M, Addi T, Gondouin B, McKay N, et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018;93(4):986–99.
Wang M, Hu HH, Chen YY, Chen L, Wu XQ, Zhao YY. Novel poricoic acids attenuate renal fibrosis through regulating redox signalling and aryl hydrocarbon receptor activation. Phytomedicine. 2020;79: 153323.
Miao H, Cao G, Wu XQ, Chen YY, Chen DQ, Chen L, et al. Identification of endogenous 1-aminopyrene as a novel mediator of progressive chronic kidney disease via aryl hydrocarbon receptor activation. Br J Pharmacol. 2020;177(15):3415–35.
Niwa T, Takeda N, Tatematsu A, Maeda K. Accumulation of indoxyl sulfate, an inhibitor of drug-binding, in uremic serum as demonstrated by internal-surface reversed-phase liquid chromatography. Clin Chem. 1988;34(11):2264–7.
Addi T, Poitevin S, McKay N, El Mecherfi KE, Kheroua O, Jourde-Chiche N, et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch Toxicol. 2019;93(1):121–36.
Vial R, Poitevin S, McKay N, Burtey S, Cerini C. Tryptophan metabolites regulate neuropentraxin 1 expression in endothelial cells. Int J Mol Sci. 2022;23(4):2369.
Yang J, Li H, Zhang C, Zhou Y. Indoxyl sulfate reduces Itof by activating ROS/MAPK and NF-kappaB signaling pathways. JCI Insight. 2022;7(3).
Han W, Du C, Zhu Y, Ran L, Wang Y, Xiong J, et al. Targeting myocardial mitochondria-STING-polyamine axis prevents cardiac hypertrophy in chronic kidney disease. JACC Basic Transl Sci. 2022;7(8):820–40.
Foley RN, Murray AM, Li S, Herzog CA, McBean AM, Eggers PW, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol. 2005;16(2):489–95.
Arinze NV, Yin W, Lotfollahzadeh S, Napoleon MA, Richards S, Walker JA, et al. Tryptophan metabolites suppress the Wnt pathway and promote adverse limb events in chronic kidney disease. J Clin Invest. 2022;132(1).
Balestrieri N, Palzkill V, Pass C, Tan J, Salyers ZR, Moparthy C, et al. Activation of the aryl hydrocarbon receptor in muscle exacerbates ischemic pathology in chronic kidney disease. Circ Res. 2023;133(2):158–76.
Shimizu H, Saito S, Higashiyama Y, Nishijima F, Niwa T. CREB, NF-kappaB, and NADPH oxidase coordinately upregulate indoxyl sulfate-induced angiotensinogen expression in proximal tubular cells. Am J Physiol Cell Physiol. 2013;304(7):C685–92.
Ito S, Osaka M, Edamatsu T, Itoh Y, Yoshida M. Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J Atheroscler Thromb. 2016;23(8):960–75.
Kim HY, Yoo TH, Cho JY, Kim HC, Lee WW. Indoxyl sulfate-induced TNF-alpha is regulated by crosstalk between the aryl hydrocarbon receptor, NF-kappaB, and SOCS2 in human macrophages. FASEB J. 2019;33(10):10844–58.
Nakano T, Katsuki S, Chen M, Decano JL, Halu A, Lee LH, et al. Uremic toxin indoxyl sulfate promotes proinflammatory macrophage activation via the interplay of OATP2B1 and Dll4-notch signaling. Circulation. 2019;139(1):78–96.
Lu CL, Zheng CM, Lu KC, Liao MT, Wu KL, Ma MC. Indoxyl-sulfate-induced redox imbalance in chronic kidney disease. Antioxidants (Basel). 2021;10(6):936.
Edamatsu T, Fujieda A, Itoh Y. Phenyl sulfate, indoxyl sulfate and p-cresyl sulfate decrease glutathione level to render cells vulnerable to oxidative stress in renal tubular cells. PLoS ONE. 2018;13(2): e0193342.
Bugnicourt JM, Godefroy O, Chillon JM, Choukroun G, Massy ZA. Cognitive disorders and dementia in CKD: the neglected kidney-brain axis. J Am Soc Nephrol. 2013;24(3):353–63.
Stinghen AE, Chillon JM, Massy ZA, Boullier A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bEnd.3). Toxins (Basel). 2014;6(6):1742–60.
Seifter JL, Samuels MA. Uremic encephalopathy and other brain disorders associated with renal failure. Semin Neurol. 2011;31(2):139–43.
Lin YT, Wu PH, Liang SS, Mubanga M, Yang YH, Hsu YL, et al. Protein-bound uremic toxins are associated with cognitive function among patients undergoing maintenance hemodialysis. Sci Rep. 2019;9(1):20388.
Milanesi S, Garibaldi S, Saio M, Ghigliotti G, Picciotto D, Ameri P, et al. Indoxyl Sulfate induces renal fibroblast activation through a targetable heat shock protein 90-dependent pathway. Oxid Med Cell Longev. 2019;2019:2050183.
Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE. 2012;7(3): e34026.
Docherty MH, O’Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol. 2019;30(5):726–36.
Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal ageing and disease. Nat Rev Nephrol. 2017;13(2):77–89.
Sun CY, Chang SC, Wu MS. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012;81(7):640–50.
Lv J, Chen J, Wang M, Yan F. Klotho alleviates indoxyl sulfate-induced heart failure and kidney damage by promoting M2 macrophage polarization. Aging (Albany NY). 2020;12(10):9139–50.
Harrill JA, Hukkanen RR, Lawson M, Martin G, Gilger B, Soldatow V, et al. Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicol Appl Pharmacol. 2013;272(2):503–18.
Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28(1):164–76.
Xiong X, Liu C, Shen M, Yang Q, Zhao Q, Li X, et al. Circular RNA expression profile in transgenic diabetic mouse kidneys. Cell Mol Biol Lett. 2021;26(1):25.
Matsuoka K, Kato K, Takao T, Ogawa M, Ishii Y, Shimizu F, et al. Concentrations of various tryptophan metabolites are higher in patients with diabetes mellitus than in healthy aged male adults. Diabetol Int. 2017;8(1):69–75.
Zhao T, Zhang H, Yin X, Zhao H, Ma L, Yan M, et al. Tangshen formula modulates gut Microbiota and reduces gut-derived toxins in diabetic nephropathy rats. Biomed Pharmacother. 2020;129: 110325.
Kim JT, Kim SS, Jun DW, Hwang YH, Park WH, Pak YK, et al. Serum arylhydrocarbon receptor transactivating activity is elevated in type 2 diabetic patients with diabetic nephropathy. J Diabetes Investig. 2013;4(5):483–91.
Lee WJ, Liu SH, Chiang CK, Lin SY, Liang KW, Chen CH, et al. Aryl hydrocarbon receptor deficiency attenuates oxidative stress-related mesangial cell activation and macrophage infiltration and extracellular matrix accumulation in diabetic nephropathy. Antioxid Redox Signal. 2016;24(4):217–31.
Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52.
Neyra JA, Chawla LS. Acute kidney disease to chronic kidney disease. Crit Care Clin. 2021;37(2):453–74.
Yang W, Li X, He L, Zhu S, Lai S, Zhang X, et al. Empagliflozin improves renal ischemia-reperfusion injury by reducing inflammation and enhancing mitochondrial fusion through AMPK-OPA1 pathway promotion. Cell Mol Biol Lett. 2023;28(1):42.
Zhao S, Wu W, Liao J, Zhang X, Shen M, Li X, et al. Molecular mechanisms underlying the renal protective effects of coenzyme Q10 in acute kidney injury. Cell Mol Biol Lett. 2022;27(1):57.
Wang W, Hao G, Pan Y, Ma S, Yang T, Shi P, et al. Serum indoxyl sulfate is associated with mortality in hospital-acquired acute kidney injury: a prospective cohort study. BMC Nephrol. 2019;20(1):57.
Tao S, Guo F, Ren Q, Liu J, Wei T, Li L, et al. Activation of aryl hydrocarbon receptor by 6-formylindolo[3,2-b]carbazole alleviated acute kidney injury by repressing inflammation and apoptosis. J Cell Mol Med. 2021;25(2):1035–47.
Baban B, Liu JY, Mozaffari MS. Aryl hydrocarbon receptor agonist, leflunomide, protects the ischemic-reperfused kidney: role of Tregs and stem cells. Am J Physiol Regul Integr Comp Physiol. 2012;303(11):R1136–46.
Joo MS, Lee CG, Koo JH, Kim SG. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013;4(10): e899.
Lissner MM, Cumnock K, Davis NM, Vilches-Moure JG, Basak P, Navarrete DJ, et al. Metabolic profiling during malaria reveals the role of the aryl hydrocarbon receptor in regulating kidney injury. Elife. 2020;9.
Yabuuchi N, Hou H, Gunda N, Narita Y, Jono H, Saito H. Suppressed hepatic production of indoxyl sulfate attenuates cisplatin-induced acute kidney injury in sulfotransferase 1a1-deficient mice. Int J Mol Sci. 2021;22(4).
Wen L, Ren Q, Guo F, Du X, Yang H, Fu P, et al. Tubular aryl hydratocarbon receptor upregulates EZH2 to promote cellular senescence in cisplatin-induced acute kidney injury. Cell Death Dis. 2023;14(1):18.
Eleftheriadis T, Pissas G, Filippidis G, Liakopoulos V, Stefanidis I. The role of indoleamine 2,3-Dioxygenase in renal tubular epithelial cells senescence under anoxia or reoxygenation. Biomolecules. 2021;11(10).
Ting L, Mingjun S, Yuanyan C, Jingyu Y, Jiang L, Miao X, et al. Association between AhR in B cells and systemic lupus erythematosus with renal damage. Int Immunopharmacol. 2022;113(Pt A): 109381.
Coelho NR, Tomkiewicz C, Correia MJ, Goncalves-Dias C, Barouki R, Pereira SA, et al. First evidence of aryl hydrocarbon receptor as a druggable target in hypertension induced by chronic intermittent hypoxia. Pharmacol Res. 2020;159: 104869.
Magnani S, Atti M. Uremic toxins and blood purification: a review of current evidence and future perspectives. Toxins (Basel). 2021;13(4).
Tyagi A, Ng YW, Tamtaji M, Abidi IH, Li J, Rehman F, et al. Elimination of uremic toxins by functionalized graphene-based composite beads for direct hemoperfusion. ACS Appl Mater Interfaces. 2021;13(5):5955–65.
Li J, Han L, Xie J, Liu S, Jia L. Multi-sites polycyclodextrin adsorbents for removal of protein-bound uremic toxins combining with hemodialysis. Carbohydr Polym. 2020;247: 116665.
Li J, Han L, Liu S, He S, Cao Y, Xie J, et al. Removal of indoxyl sulfate by water-soluble poly-cyclodextrins in dialysis. Colloids Surf B Biointerfaces. 2018;164:406–13.
Zhang M, Li L, Lei L, Kang K, Xiao C. Effectively decontaminating protein-bound uremic toxins in human serum albumin using cationic metal-organic frameworks. ACS Appl Mater Interfaces. 2022;14(50):55354–64.
Sato E, Saigusa D, Mishima E, Uchida T, Miura D, Morikawa-Ichinose T, et al. Impact of the oral adsorbent AST-120 on organ-specific accumulation of uremic toxins: LC-MS/MS and MS imaging techniques. Toxins (Basel). 2017;10(1):19.
Shih YC, Wu CC, Wang SC, Liou JY, Huang PH, Tarng DC. Oral charcoal adsorbents attenuate neointima formation of arteriovenous fistulas. Toxins (Basel). 2020;12(4):237.
Akizawa T, Asano Y, Morita S, Wakita T, Onishi Y, Fukuhara S, et al. Effect of a carbonaceous oral adsorbent on the progression of CKD: a multicenter, randomized, controlled trial. Am J Kidney Dis. 2009;54(3):459–67.
Chen YC, Wu MY, Hu PJ, Chen TT, Shen WC, Chang WC, et al. Effects and safety of an oral adsorbent on chronic kidney disease progression: a systematic review and meta-analysis. J Clin Med. 2019;8(10):1718.
Apetrii M, Timofte D, Voroneanu L, Covic A. Nutrition in chronic kidney disease-the role of proteins and specific diets. Nutrients. 2021;13(3):956.
Carrero JJ, Gonzalez-Ortiz A, Avesani CM, Bakker SJL, Bellizzi V, Chauveau P, et al. Plant-based diets to manage the risks and complications of chronic kidney disease. Nat Rev Nephrol. 2020;16(9):525–42.
Poesen R, Mutsaers HA, Windey K, van den Broek PH, Verweij V, Augustijns P, et al. The influence of dietary protein intake on mammalian tryptophan and phenolic metabolites. PLoS ONE. 2015;10(10): e0140820.
Rhee CM, Ahmadi SF, Kovesdy CP, Kalantar-Zadeh K. Low-protein diet for conservative management of chronic kidney disease: a systematic review and meta-analysis of controlled trials. J Cachexia Sarcopenia Muscle. 2018;9(2):235–45.
Menon V, Kopple JD, Wang X, Beck GJ, Collins AJ, Kusek JW, et al. Effect of a very low-protein diet on outcomes: long-term follow-up of the Modification of Diet in Renal Disease (MDRD) study. Am J Kidney Dis. 2009;53(2):208–17.
Garneata L, Stancu A, Dragomir D, Stefan G, Mircescu G. Ketoanalogue-supplemented vegetarian very low-protein diet and CKD progression. J Am Soc Nephrol. 2016;27(7):2164–76.
Kontessis P, Jones S, Dodds R, Trevisan R, Nosadini R, Fioretto P, et al. Renal, metabolic and hormonal responses to ingestion of animal and vegetable proteins. Kidney Int. 1990;38(1):136–44.
Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur Heart J. 2019;40(7):583–94.
Cigarran Guldris S, Latorre Catala JA, Sanjurjo Amado A, Menendez Granados N, Pineiro Varela E. Fibre intake in chronic kidney disease: what fibre should we recommend? Nutrients. 2022;14(20):4419.
Ebersolt M, Santana Machado T, Mallmann C, Mc-Kay N, Dou L, Bouchouareb D, et al. Protein/fiber index modulates uremic toxin concentrations in hemodialysis patients. Toxins (Basel). 2022;14(9):589.
Saranya GR, Viswanathan P. Gut microbiota dysbiosis in AKI to CKD transition. Biomed Pharmacother. 2023;161: 114447.
Chadban SJ, Singer J, Coates PT. That sinking gut feeling: is transplant-induced dysbiosis contributing to allograft outcomes? Kidney Int. 2023;103(3):454–7.
Mafra D, Borges NA, Lindholm B, Shiels PG, Evenepoel P, Stenvinkel P. Food as medicine: targeting the uraemic phenotype in chronic kidney disease. Nat Rev Nephrol. 2021;17(3):153–71.
Li HB, Xu ML, Xu XD, Tang YY, Jiang HL, Li L, et al. Faecalibacterium prausnitzii attenuates CKD via butyrate-renal GPR43 axis. Circ Res. 2022;131(9):e120–34.
Cao J, Liu Q, Hao H, Bu Y, Tian X, Wang T, et al. Lactobacillus paracasei X11 ameliorates hyperuricemia and modulates gut microbiota in mice. Front Immunol. 2022;13: 940228.
Borges NA, Carmo FL, Stockler-Pinto MB, de Brito JS, Dolenga CJ, Ferreira DC, et al. Probiotic supplementation in chronic kidney disease: a double-blind, randomized. Placebo-controlled Trial J Ren Nutr. 2018;28(1):28–36.
Hutkins RW, Krumbeck JA, Bindels LB, Cani PD, Fahey G Jr, Goh YJ, et al. Prebiotics: why definitions matter. Curr Opin Biotechnol. 2016;37:1–7.
Ebrahim Z, Proost S, Tito RY, Raes J, Glorieux G, Moosa MR, et al. The effect of ss-glucan prebiotic on kidney function, uremic toxins and gut microbiome in stage 3 to 5 chronic kidney disease (CKD) predialysis participants: a randomized controlled trial. Nutrients. 2022;14(4).
Connolly ML, Tzounis X, Tuohy KM, Lovegrove JA. Hypocholesterolemic and prebiotic effects of a whole-grain oat-based granola breakfast cereal in a cardio-metabolic “at risk” population. Front Microbiol. 2016;7:1675.
Rossi M, Johnson DW, Morrison M, Pascoe EM, Coombes JS, Forbes JM, et al. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): a randomized trial. Clin J Am Soc Nephrol. 2016;11(2):223–31.
Zheng HJ, Guo J, Wang Q, Wang L, Wang Y, Zhang F, et al. Probiotics, prebiotics, and synbiotics for the improvement of metabolic profiles in patients with chronic kidney disease: a systematic review and meta-analysis of randomized controlled trials. Crit Rev Food Sci Nutr. 2021;61(4):577–98.
Yu Z, Zhao J, Qin Y, Wang Y, Zhang Y, Sun S. Probiotics, prebiotics, and synbiotics improve uremic, inflammatory, and gastrointestinal symptoms in end-stage renal disease with dialysis: a network meta-analysis of randomized controlled trials. Front Nutr. 2022;9: 850425.
Gryp T, De Paepe K, Vanholder R, Kerckhof FM, Van Biesen W, Van de Wiele T, et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020;97(6):1230–42.
Kim DJ, Venkataraman A, Jain PC, Wiesler EP, DeBlasio M, Klein J, et al. Vitamin B12 and folic acid alleviate symptoms of nutritional deficiency by antagonizing aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2020;117(27):15837–45.
Chen Y, Zhao Z, Li Y, Yang Y, Li L, Jiang Y, et al. Baicalein alleviates hyperuricemia by promoting uric acid excretion and inhibiting xanthine oxidase. Phytomedicine. 2021;80: 153374.
Li YY, Wang XJ, Su YL, Wang Q, Huang SW, Pan ZF, et al. Baicalein ameliorates ulcerative colitis by improving intestinal epithelial barrier via AhR/IL-22 pathway in ILC3s. Acta Pharmacol Sin. 2022;43(6):1495–507.
Wang K, Feng C, Li C, Yao J, Xie X, Gong L, et al. Baicalin protects mice from aristolochic acid I-induced kidney injury by induction of CYP1A through the aromatic hydrocarbon receptor. Int J Mol Sci. 2015;16(7):16454–68.
Sekula M, Janawa G, Stankiewicz E, Stepien E. Endothelial microparticle formation in moderate concentrations of homocysteine and methionine in vitro. Cell Mol Biol Lett. 2011;16(1):69–78.
Delgado-Andrade C. Carboxymethyl-lysine: thirty years of investigation in the field of AGE formation. Food Funct. 2016;7(1):46–57.
Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65(1):8–24.
So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120(6):1791–9.
Jaisson S, Pietrement C, Gillery P. Carbamylation-derived products: bioactive compounds and potential biomarkers in chronic renal failure and atherosclerosis. Clin Chem. 2011;57(11):1499–505.
Jia W, Guo A, Zhang R, Shi L. Mechanism of natural antioxidants regulating advanced glycosylation end products of Maillard reaction. Food Chem. 2023;404(Pt A): 134541.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82170696, 81873609, 82300834, 82370695) and the Science and Technology Commission of Shanghai Municipality (Grant Nos. 18411960900).
Author information
Authors and Affiliations
Contributions
HX wrote the paper. NY proofread the paper. LL and CY reviewed and modified the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, H., Yang, N., Yu, C. et al. Uremic toxins mediate kidney diseases: the role of aryl hydrocarbon receptor. Cell Mol Biol Lett 29, 38 (2024). https://doi.org/10.1186/s11658-024-00550-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-024-00550-4