
Abstract
The Alzheimer's disease (AD) epidemic is a looming crisis, with an urgent need for new therapies to delay or prevent symptom onset and progression. There is growing awareness that clinical trials must target stage-appropriate pathophysiological mechanisms to effectively develop disease-modifying treatments. Advances in AD biomarker research have demonstrated changes in amyloid-beta (Aβ), brain metabolism and other pathophysiologies prior to the onset of memory loss, with some markers possibly changing one or two decades earlier. These findings suggest that amyloid-based therapies would optimally be targeted at the earliest clinically detectable stage (such as mild cognitive impairment (MCI)) or before. Postmortem data indicate that tau lesions in the locus coeruleus (LC), the primary source of subcortical norepinephrine (NE), may be the first identifiable pathology of AD, and recent data from basic research in animal models of AD indicate that loss of NE incites a neurotoxic proinflammatory condition, reduces Aβ clearance and negatively impacts cognition - recapitulating key aspects of AD. In addition, evidence linking NE deficiency to neuroinflammation in AD also exists. By promoting proinflammatory responses, suppressing anti-inflammatory responses and impairing Aβ degradation and clearance, LC degeneration and NE loss can be considered a triple threat to AD pathogenesis. Remarkably, restoration of NE reverses these effects and slows neurodegeneration in animal models, raising the possibility that treatments which increase NE transmission may have the potential to delay or reverse AD-related pathology. This review describes the evidence supporting a key role for noradrenergic-based therapies to slow or prevent progressive neurodegeneration in AD. Specifically, since MCI coincides with the onset of clinical symptoms and brain atrophy, and LC pathology is already present at this early stage of AD pathogenesis, MCI may offer a critical window of time to initiate novel noradrenergic-based therapies aimed at the secondary wave of events that lead to progressive neurodegeneration. Because of the widespread clinical use of drugs with a NE-based mechanism of action, there are immediate opportunities to repurpose existing medications. For example, NE transport inhibitors and NE-precursor therapies that are used for treatment of neurologic and psychiatric disorders have shown promise in animal models of AD, and are now prime candidates for early-phase clinical trials in humans.
Similar content being viewed by others

The locus coeruleus and norephinephrine
The locus coeruleus (LC) is the major subcortical site for the synthesis of norepinephrine (NE) [1]. The LC preferentially projects to the thalamus, hippocampus, the frontal and entorhinal cortices and, to a minor extent, most other brain regions. Due to its extensive innervation of multiple forebrain regions and the widespread distribution of noradrenergic receptors, the noradrenergic system is involved in many behavioral and physiologic processes. The role of the LC noradrenergic system in cognitive processes, arousal and wakefulness is covered in several extensive reviews [2–6]. In addition to declining with normal aging, altered NE transmission has been reported in major brain disorders in psychiatry (depression, attention deficit disorder, Tourette's, psychosis, post-traumatic stress disorder), neurology (epilepsy, Parkinson's, Alzheimer's disease (AD)) and sleep [7, 8].
Locus coeruleus loss in Alzheimer's disease
Extensive LC degeneration is nearly universal in AD [9–13] and is among the earliest pathologies [11, 14, 15], with LC neuropathology detectable as early as 10 years before neurocognitive signs [16–18]. Alterations in NE have long been known to be linked to cognitive, mood and neuropsychiatric symptoms [6, 19–24]. A number of studies have also demonstrated significant correlations between LC cell death (or decreased cortical NE levels) and severity and duration of dementia in AD [25, 26]. Neurofibrillary changes in the LC occur in prodromal stages of AD (that is, mild cognitive impairment (MCI)), and even in some young, cognitively normal individuals [16–18], preceding amyloid-beta (Aβ) deposition. However, whether the LC represents the initial site of pathology or reflects a nonspecific response to brain insults is still under debate [27].
An additional complication is that compensatory changes in the degenerating noradrenergic system appear to occur in AD; despite decreases in tissue forebrain NE in AD, surviving LC neurons show increased abundance of mRNA for tyrosine hydroxylase, the rate-limiting NE biosynthetic enzyme, sprouting of dendrites and axonal projections [28], and increased cerebrospinal fluid levels of NE are observed in AD patients [29–32]. The knowledge gaps present in these areas highlight the need for additional investigations into the mechanism by which LC loss contributes to AD.
Locus coeruleus and norepinephrine in AD pathogenesis: preclinical studies
The strong correlation between LC degeneration, NE depletion and severity of AD in patients has prompted multiple studies of the contribution of LC dysfunction to AD progression through the use of animal models. The primary tool for studying the effects of LC degeneration and NE depletion in vivo is the neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (dsp-4), which reliably lesions the LC while leaving other aminergic systems intact. Transgenic mice that overexpress human amyloid precursor protein (APP) with familial Alzheimer mutations recapitulate many aspects of AD neuropathology and cognitive deficits, and have been used extensively to study AD. However, most of these mouse lines do not show the frank LC degeneration that occurs in human AD. To determine the functional consequences of LC loss in AD, therefore, several laboratories have used dsp-4 to lesion LC neurons in these transgenic mice.
In general, dsp-4 lesions of the LC exacerbate AD-like neuropathology and cognitive deficits, suggesting that LC degeneration plays a causal role in AD progression. For example, the first study to use this approach showed that dsp-4 lesions of the LC in APP23 mice resulted in increased Aβ deposition, neurodegeneration, neuronal loss, cognitive deficits and microglial activation, and reduced cerebral glucose metabolism [33]. Importantly, the effects of dsp-4 were confined to forebrain areas that received projections directly from the LC, while brain regions that receive noradrenergic innervation from non-LC cell groups were unaffected. APP/presenilin-1 (PS1) mice treated with dsp-4 displayed severe loss of norepinephrine transporter (NET) in the LC and cortex, along with a loss of noradrenergic innervation [34]. Lesioning of the LC induced accelerated amyloid deposition and neuron death with age, and more severe deficits in spatial memory compared with vehicle-treated animals [34]. The mechanism underlying the increased amyloid deposition appears to be related to reduced clearance, as occurs in sporadic AD [35], due to the inhibition of Aβ1-42 (Aβ42) phagocytosis by microglia rather than an influence on APP production or processing [36]. NE has several strong influences on microglial function, and in general suppresses the production of proinflammatory cytokines and promotes the production of anti-inflammatory molecules. Thus, it is not surprising that dsp-4 treatment also exacerbates the neuroinflammatory response in multiple brain regions of APP/PS1 mice [36, 37]. Interestingly, a recent study reported that in addition to increased Aβ deposition, dsp-4 lesions of the LC in APP/PS1 mice also resulted in olfactory deficits, another common and early pathology seen in AD patients [38].
Among the questions raised by these findings, an important issue with therapeutic implications is whether the effects of LC lesions in AD mouse models are due solely to the loss of NE itself, the loss of co-transmitters in LC neurons, collateral damage from the neuro degenerative process itself, or some combination thereof. To help resolve these issues, we recently crossed APP/PS1 mice with dopamine β-hydroxylase knockout (DBH-/-) mice that lack the ability to synthesize NE but have intact LC neurons [39]. While APP/PS1 and DBH-/- single-mutant mice each displayed moderate hippocampal long-term potentiation (LTP) and spatial memory impairments, the two mutations had an additive effect, resulting in double mutants with severely compromised LTP and maze performance. Somewhat surprisingly, the genetic loss of NE had no apparent effect on AD-like neuropathology in the double mutant. Nondegenerative loss of NE produced by Ear2 knockout, which prevents the development of most LC neurons, also exacerbated LTP and memory deficits but had no effect on plaque deposition in APP/PS1 mice. However, dsp-4 worsened neuropathology in the APP/PS1, DBH-/- double mutant. Combined, these results indicate that the LC neuronal loss contributes to distinct aspects of AD; loss of NE itself impairs synaptic plasticity and cognitive performance, while the physical process of LC neuron degeneration exacerbates AD-like neuropathology.
In summary, combining expression of familial AD mutations with LC lesions or NE deficiency appears to more closely recapitulate the neuropathological and cognitive symptoms of AD compared with mutant APP expression alone, and implicates LC loss as a crucial component of AD.
Neuroinflammation is a key mechanism linking loss of locus coeruleus neurons and norepinephrine innervation with AD
Recent studies provide insights into the mechanisms by which LC dysfunction and NE loss facilitate AD pathogenesis. There is growing evidence suggesting that the inflammatory response induced and/or augmented by LC degeneration is a key mechanism contributing to the initiation and progression of AD pathogenesis. Microglia, astrocytes and endothelia are among the major targets of NE, and, under normal conditions, these cells control the delicate balance of the inflammatory response. In general, NE is an anti-inflammatory molecule; acting via β-adrenergic receptors, NE suppresses the expression of multiple proinflammatory genes, including major histocompatibility complex class II, TNFα, inducible nitric oxide synthase and IL-1β, while simultaneously promoting the expression of anti-inflammatory molecules such as NF-κB, inhibitory IκB, heat shock protein-70 and chemokine monocyte chemotactic protein-1 in astrocytes and microglia [7, 40]. That NE deficiency results in undesirable proinflammatory effects is therefore not surprising.
One of the first pieces of evidence connecting LC degeneration and neuroinflammation in an AD model was reported by Heneka and colleagues [41]. Injections of Aβ42 in the cortex of rats induced severe cortical inflammation and the expression of several proinflammatory genes - including inducible nitric oxide synthase/nitric oxide synthase-2, IL-1β and IL-6 - within hours. This neuroinflammation was profoundly exacerbated when LC neurons were lesioned with dsp-4 prior to the cortical injection of Aβ42. In addition, dsp-4 pretreatment increased inducible nitric oxide synthase expression solely in neurons rather than in microglial cells, more accurately replicating the expression pattern seen in AD patients [41]. Augmented forebrain microglial and astroglial activation and proinflammatory gene expression that coincide with the development of other AD-like neuropathologies such as Aβ plaques were also obtained using dsp-4 and the APPV171 and APP/PS1 transgenic mouse models of AD [36]. LC lesions profoundly increased the Aβ plaque load, brain inflammation and spatial memory deficits concurrently in APP23 transgenic mice. In addition, dsp-4 treatment was associated with a switch in microglial cytokine expression from a neuroprotective anti-inflammatory profile to a proinflammatory and neurotoxic profile [33, 36, 42].
Because NE promotes microglia-mediated degradation and phagocytosis of Aβ in cell culture [43], another deleterious effect of LC degeneration on the neuroinflammatory response is the dysfunction of cellular machinery involved in Aβ metabolism and clearance. For example, in V717F APP transgenic mice, dsp-4 lesions of the LC produce a fivefold increase in Aβ plaques that is accompanied by microglial and astroglia activation and decreased expression of the Aβ plaque-degrading enzyme, metallopeptidase neprilysin [42]. Another study showed that NE suppressed Aβ-induced cytokine and chemokine production and increased microglial migration and phagocytosis in cell culture, while dsp-4 lesions prevented the recruitment of microglia to Aβ plaques and impaired Aβ phagocytosis in APP/PS1 transgenic mice [36].
A few epidemiological studies have investigated interactions between NE and neuroinflammation in AD. A small pilot study in a Spanish population found that a SNP associated with low DBH activity alone had no effect, but significantly increased AD risk in combination with SNPs in the IL-1A or IL-6 genes [44]. This result was partially confirmed and extended in an independent study with a larger sample population and wider patient demographics. This follow-up study reported a significant association between the low-activity variant of DBH alone and AD risk that was mostly attributable to males over the age of 75, and also replicated the interaction between DBH and IL-1A polymorphisms [45]. Interestingly, SNPs that are thought to increase adrenergic signaling have also been linked to a risk for developing AD. Individuals homozygous for the C allele of ADRB1 (the β1-adrenergic receptor) and the the T allele of GNB3 (the G protein β3 subunit gene), which are associated with increased cAMP levels and mitogen-activated protein kinase activation, have an increased risk for AD [46]. A Chinese case-control study found that a β2-adrenergic receptor polymorphism which enhances responsiveness is also associated with the risk of sporadic late-onset AD [47]. These studies highlight the complicated nature of noradrenergic signaling in AD; activation of some receptor subtypes may suppress neuroinflammation and neuropathology, while other receptors may exacerbate aspects of the disease.
Recent biomarker studies in living subjects have also confirmed a proinflammatory state in AD [48–51]. Of note, increased proinflammatory and decreased anti-inflammatory markers account for the majority of changes detectable in a large panel of cerebrospinal fluid analytes in MCI and AD [49, 50]. By promoting proinflammatory responses, suppressing anti-inflammatory responses and impairing Aβ degradation and clearance, LC degeneration and NE loss can therefore be considered a triple threat to AD pathogenesis.
Treatments that increase norepinephrine in AD animal models ameliorate AD-like pathology and cognitive decline
In vitro and animal studies have provided the most compelling evidence that increasing NE could have beneficial effects on both AD neuropathology and cognitive symptoms. In vitro challenge of human acute monocytic leukemia cells (THP-1) with Aβ42 induced cytotoxicity and provoked a neuroinflammatory response that was dose-dependently attenuated by NE [52]. Treatment with cAMP or forskolin, a protein kinase A activator, had similar effects, suggesting that NE's protective effects were regulated, at least in part, via stimulation of β-adrenergic receptors and the corresponding activation of the cAMP/protein kinase A signaling pathway [52]. Another in vitro study in hNT neuronal and primary hippocampal cultures revealed a neuroprotective effect of NE against both Aβ42- and Aβ25-35-induced increases in oxidative stress, mitochondrial dysfunction and cell death [53]. The neuroprotective effects were mediated by activation of β-adrenoceptor/cAMP signaling and also required the brain-derived neurotrophic factor/tropo myosin-related kinase B pathway, although some β-receptor-independent effects of NE persisted [53].
The earliest in vivo animal studies using noradrenergic pharmacotherapies focused on the α2-adrenergic autoreceptor. The α2-antagonists that enhance NE release, such as piperoxane, reversed memory deficits in aged mice as assessed by performance in a step-down inhibitory avoidance response task [54]. Another α2 -antagonist, fluparoxan, prevented age-related decline in the spontaneous alternation task (a test of spatial working memory) in APP/PS1 mice, although it had no effect in other memory tasks such as object recognition or the Morris water maze, and occurred in the absence of obvious concomitant change in pathology [55]. Drugs targeting other NE receptors and transporters have also been tested in animal models of AD. Desipramine, a tricyclic antidepressant that inhibits endogenous NE re-uptake, induced the production of the anti-inflammatory cytokine monocyte chemotactic protein-1 [56]. CL316243, a selective β3-adrenergic receptor agonist, rescued performance in a learning paradigm by chicks given intracranial injections of Aβ42 [57]. Recently, β-adrenoceptor activation of cAMP/protein kinase A signaling was found to reverse the synaptotoxic effects of human Aβ oligomers on LTP and behavior [58].
Compelling evidence in favor of noradrenergic treatments for AD has also been observed using the NE precursor, L-threo-3,4-dihydroxyphenylserine (L-DOPS). For example, L-DOPS restored the balance of the brain inflammatory system, facilitated microglial migration and Aβ phagocytosis, and reversed learning deficits in dsp-4 lesioned APP transgenic mice [36], and also partially rescued spatial memory deficits in the DBH-/-, APP/PS1 double-mutant mice [39]. Treatment of 5xFAD mice, which have robust and early development of AD-like neuropathology, with a combination of L-DOPS and the NET inhibitor, atomoxetine, elevated brain NE levels, increased expression of Aβ clearance enzymes and brain-derived neurotrophic factor, reduced inflammatory changes and Aβ burden, and improved spatial memory [59].
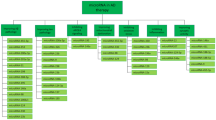
To generate further proof-of-principle for the efficacy of NET inhibitors in AD, we took advantage of norepinephrine transporter knockout mice (NET KO) that lack the NET completely, and have elevated basal extracellular NE levels, similar to what might be observed with chronic NET inhibitor treatment [60]. We crossed the NET KO mice to APP/PS1 transgenic mice that overexpress mutant human APP and PS1 and develop age-dependent Aβ plaques, and examined AD-like neuropathology by western blot assay at 6 months of age and by immunocytochemistry at 1 year of age. As shown in Figure 1a, APP/PS1 mice that carry wildtype copies of NET (NET WT, APP/PS1) contain heavy plaque load in the hippocampus and cortex, as detected by immune-histochemistry using antiserum 2964 against fibrillar Aβ42 [61]. The Aβ levels were much higher in female NET WT, APP/PS1 mice compared with males (Figure 1b), as reported previously for APP/PS1 and other lines of APP transgenic mice (for example, [62]). Remarkably, plaques were almost completely abolished in littermate APP/PS1 mice that lack the NET (NET KO, APP/PS1). Similar results were obtained with western blots of brain homogenates (Figure 1b).

Enhancing norepinephrine may be a promising strategy to treat Alzheimer's disease. (a) Plaque deposition in the hippocampus (HP) and cortex (CTX) of 1-year-old APP/PS1 mice (NET WT,APP/PS1) and norepinephrine transporter knockout APP/PS1 mice (NET KO,APP/PS1) as detected by immunohistochemistry using antiserum 2964 against fibrillar Aβ42. (b) Abundance of full-length amyloid precursor protein (APP), the APP C-terminal fragment (CTF), and Aβ in male and female NET WT,APP/PS1 and NET KO,APP/ PS1 mice at 6 months of age as detected by western blot using mAb 6E10 against amino acid residues 1 to 16 of Aβ. Tubulin was used as a loading control. Aβ, amyloid-beta; NET WT, norepinephrine transporter wildtype; PS1, presenilin-1.
These results suggest that attenuating NET activity can reduce Aβ levels, perhaps by increasing phagocytosis or another NE-mediated mechanism described in this review. Interestingly, full-length APP and the C-terminal fragment of APP were also reduced. The reasons for this are not clear, but raise the possibility that a change in APP production or turnover contributes to the decrease in Aβ levels. Consistent with this finding, selective lesion of the ascending noradrenergic bundle with 6-hydroxydopamine in rats increased cortical APP [63]. Combined with the results that atomoxetine + L-DOPS reduces AD-like neuropathology and cognitive deficits in 5xFAD mice [59], these data support the use of NET inhibitors in AD patient populations.
While studies using NE pharmacotherapy in AD models show promise for disease treatment, these studies must be interpreted with caution because the effects of noradrenergic drugs are complicated by multiple adrenergic receptor subtypes with different distributions and signaling capabilities. There are a number of studies that suggest noradrenergic stimulation actually increases certain proinflammatory markers, and that some adrenergic receptor blockade can be therapeutic. Pharmacological activation of β-adrenergic receptors (especially β2-adrenergic) increases mRNA and protein levels for IL-1B and 1L-6 in macrophages, microglia and brain parenchyma [64–66]. Administration of adrenergic receptor antagonists in vivo can protect against the inflammatory response induced by a foot shock [67], peripheral bacterial challenge [68] or ischemia [69, 70]. Nevibolol, a β1-blocker, can also reduce amyloid production in TG2576 mice that have established amyloid and cognitive impairment, although it does not improve cognition [71]. One potential explanation for the dual beneficial and harmful effects of adrenergic receptor stimulation is that the loss of LC neurons coupled with the compensatory sprouting by surviving cells probably creates a situation where NE transmission is compromised in some brain regions, and overactive in others [6, 19–24, 28].
Clinical studies of pharmacotherapies that modulate norepinephrine in AD
Most clinical studies using noradrenergic pharmacotherapy to date have been primarily focused on treating the aggression and other behavioral disturbances that occur in many late-stage AD patients. β-adrenergic receptor antagonists (that is, propranolol) are somewhat effective in the treatment of aggression and agitation, which may be caused by NE overstimulation [72, 73], while antidepressants inhibiting NE reuptake, such as the tricyclic imipramine, have been used to treat depression, which may be caused by NE deficiency [74]. Tantalizing pieces of evidence continue to support the idea of increasing NE to treat cognitive impairment in AD. For example, clonidine - which suppresses NE release by activating the α2-adrenergic autoreceptor - impairs short-term recognition memory in patients [75], suggesting that facilitating NE release may be beneficial. The same group determined that clonidine could also enhance spatial working memory in AD patients [76], however, highlighting the complexity of these processes. Several clinical studies examining hypertension suggest that β-blockers may have therapeutic effects on inflammation and dementia. Dementia incidence and annual rate of cognitive decline tend to be lower in older patients that take β-blockers for hypertension [77–79]. The β1-antagonists nevibolol and metoprolol have been shown to attenuate the release of atherosclerotic inflammatory markers such as soluble intercellular adhesion molecule-1 in humans after 1 year of treatment [80]. Since hypertension itself is a risk factor for AD, however, it is difficult to know whether the benefits of β-blockade are mediated by direct effects on neuroinflammation or are indirect effects mediated by control of hypertension.
Overall, the strong links between LC/NE loss in AD and disease progression in AD animal models combined with human clinical and preclinical data demonstrate the exciting disease-modifying potential of drugs that modulate NE levels. The urgent and essential next step is to translate these discoveries to humans. Although NE pharmacotherapies are widely used in medicine, drugs that regulate NE transmission in the brain could have complicated effects in AD. The integrity of the LC and pharmacological responsiveness in prodromal stages of AD are poorly understood. While preclinical studies suggest potential for NE-enhancing therapies to reduce neuroinflammation and amyloid burden and to ameliorate cognitive impairment, clinical observations in AD patients also suggest the potential to impact noncognitive symptoms of AD including mood, apathy, disinhibition, sleep, agitation and aggression [81, 82].
Several NE pharmacotherapies are already used in clinical practice for a variety of neurological and psychiatric disorders, including attention-deficit disorder, depression and orthostatic hypotension. NET inhibitors such as atomoxetine, a US Food and Drug Administration-approved drug that is a widely prescribed treatment for children and adults with attention-deficit hyperactive disorder, and reboxetine, approved in many countries around the world for depression, have been used safely in older subjects. The NE prodrug L-DOPS crosses the blood-brain barrier and has been used safely in Asia for several decades to treat hypotension. As mentioned above, treatment of 5xFAD transgenic mice (which accumulate amyloid burden at early ages) with a combination of L-DOPS and atomoxetine elevated brain NE levels, increased expression of Aβ clearance enzymes and brain-derived neurotrophic factor, reduced inflammatory changes and Aβ burden, and improved spatial memory [59].
In clinical studies, atomoxetine has also been shown to improve working memory, response inhibition and other executive functions in patients with attention-deficit hyperactivity disorder [83–86]. Several small studies have examined atomoxetine treatment in older patients with neurodegenerative disease to assess safety, tolerability and symptomatic effects. Marsh and colleagues studied 12 patients with Parkinson's disease with doses up to 100 mg daily (mean tolerated dose 89.6 mg), with excellent safety, tolerability and improved executive function [82]. Weintraub and colleagues found that 80 mg once daily was well tolerated by Parkinson's disease subjects as a treatment for depression; only four of 29 patients withdrew because of adverse effects [87]. Although atomoxetine was ineffective for the treatment of depression in the study, atomoxetine was associated with improvement of global cognition. A 6-month phase II trial in mild to moderate AD tested up to 80 mg atomoxetine once daily in 47 subjects [88]. Although atomoxetine was well tolerated (only five subjects withdrew because of adverse effects), there were no significant improvements in cognitive function, global clinical impression or neuropsychiatric symptoms. However, this study was not powered for clinical efficacy and, more importantly, did not investigate the potential anti-inflammatory neuroprotective role of NE pharmacotherapy. Moreover, since patients with mild to moderate AD already have extensive neurodegeneration, most investigators now realize the best chance for neuroprotection will come from earlier intervention.
Logical next steps would therefore be to test NE pharmacotherapies for their potential anti-inflammatory and other neuroprotective mechanisms in phase II trials with individuals with preclinical or early clinical (that is, MCI) stages of AD. For example, it would be important to evaluate the effect of NE-based treatments such as atomoxetine and L-DOPS on biomarkers of AD pathology and inflammation [49, 50, 89, 90]. A potential target would be cerebrospinal fluid inflammatory markers, which have been used successfully as surrogate markers of drug response in multiple sclerosis [91, 92] and are among novel biomarkers that distinguish MCI and AD from other neurodegenerative diseases and correlate with both baseline cognitive impairment and subsequent cognitive decline [50].
In sum, there is a growing body of evidence linking LC neurodegeneration and altered NE neurotransmission to the pathogenesis of AD, in addition to the long-established links with cognitive and behavioral symptoms. Preclinical studies demonstrate that restoration of NE function has great potential to slow neurodegeneration by enhancing anti-inflammatory and suppressing proinflammatory responses, facilitating amyloid clearance and via other protective mechanisms. However, the complexities of NE signaling and multiplicity of effects of adrenergic receptor subtypes, together with the limitations of animal studies, underscore the importance of translating these studies to humans. The availability of clinically approved drugs that enhance central nor adrenergic function provides a timely opportunity to repurpose their use to determine their potential as a novel disease-modifying therapeutic strategy.
Abbreviations
- Aβ:
-
amyloid-beta
- AD:
-
Alzheimer's disease
- APP:
-
amyloid precursor protein
- DBH:
-
dopamine β-hydroxylase
- DBH-/-:
-
dopamine β-hydroxylase knockout
- dsp-4:
-
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine
- IL:
-
interleukin
- LC:
-
locus coeruleus
- L-DOPS:
-
L-threo-3:4-dihydroxyphenylserine
- LTP:
-
long-term potentiation
- MCI:
-
mild cognitive impairment
- NE:
-
norepinephrine
- NET:
-
norepinephrine transporter
- NET KO:
-
norepinephrine transporter knockout
- NET WT:
-
norepinephrine transporter wildtype
- NF:
-
nuclear factor
- PS1:
-
presenilin-1
- SNP:
-
single nucleotide polymorphism
- TNF:
-
tumor necrosis factor.
References
Freedman R, Foote SL, Bloom FE: Histochemical characterization of a neocortical projection of the nucleus locus coeruleus in the squirrel monkey. J Comp Neurol. 1975, 164: 209-231.
Berridge CW, Schmeichel BE, Espana RA: Noradrenergic modulation of wakefulness/arousal. Sleep Med Rev. 2012, 16: 187-197.
Berridge CW, Waterhouse BD: The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003, 42: 33-84.
Ramos BP, Arnsten AF: Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther. 2007, 113: 523-536.
Robbins TW, Arnsten AF: The neuropsychopharmacology of fronto-executive function: monoaminergic modulation. Annu Rev Neurosci. 2009, 32: 267-287.
Sara SJ: The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009, 10: 211-223.
Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E: Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002, 41: 357-365.
Szot P: Common factors among Alzheimer's disease, Parkinson's disease, and epilepsy: possible role of the noradrenergic nervous system. Epilepsia. 2012, 53 (Suppl 1): 61-66.
Bondareff W, Mountjoy CQ, Roth M, Rossor MN, Iversen LL, Reynolds GP, Hauser DL: Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis Assoc Disord. 1987, 1: 256-262.
Chan-Palay V, Asan E: Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. 1989, 287: 373-392.
Haglund M, Sjobeck M, Englund E: Locus ceruleus degeneration is ubiquitous in Alzheimer's disease: possible implications for diagnosis and treatment. Neuropathology. 2006, 26: 528-532.
Lyness SA, Zarow C, Chui HC: Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging. 2003, 24: 1-23.
Zarow C, Lyness SA, Mortimer JA, Chui HC: Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003, 60: 337-341.
Forno L: Pathology of Parkinsonism. J Neurosurg. 1966, 2: 266-271.
Mann DM, Lincoln J, Yates PO, Stamp JE, Toper S: Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry. 1980, 136: 533-541.
Braak H, Del Tredici K: Alzheimer's pathogenesis: is there neuron-to-neuron propagation?. Acta Neuropathol. 2011, 121: 589-595.
Braak H, Del Tredici K: The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011, 121: 171-181.
Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM: Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007, 28: 327-335.
Amaral DG, Foss JA: Locus coeruleus lesions and learning. Science. 1975, 188: 377-378.
Amaral DG, Sinnamon HM: The locus coeruleus: neurobiology of a central noradrenergic nucleus. Progr Neurobiol. 1977, 9: 147-196.
Forstl H, Burns A, Levy R, Cairns N: Neuropathological correlates of psychotic phenomena in confirmed Alzheimer's disease. Br J Psychiatry. 1994, 165: 53-59.
Forstl H, Levy R, Burns A, Luthert P, Cairns N: Disproportionate loss of noradrenergic and cholinergic neurons as cause of depression in Alzheimer's disease-a hypothesis. Pharmacopsychiatry. 1994, 27: 11-15.
Ressler KJ, Nemeroff CB: Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol Psychiatry. 1999, 46: 1219-1233.
Weinshenker D: Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008, 5: 342-345.
German DC, Manaye KF, White CL, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM: Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992, 32: 667-676.
Yates CM, Simpson J, Gordon A, Maloney AF, Allison Y, Ritchie IM, Urquhart A: Catecholamines and cholinergic enzymes in pre-senile and senile Alzheimer-type dementia and Down's syndrome. Brain Res. 1983, 280: 119-126.
Attems J, Thal DR, Jellinger KA: The relationship between subcortical tau pathology and Alzheimer's disease. Biochem Soc Trans. 2012, 40: 711-715.
Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA: Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J Neurosci. 2006, 26: 467-478.
Elrod R, Peskind ER, DiGiacomo L, Brodkin KI, Veith RC, Raskind MA: Effects of Alzheimer's disease severity on cerebrospinal fluid norepinephrine concentration. Am J Psychiatry. 1997, 154: 25-30.
Peskind ER, Wingerson D, Murray S, Pascualy M, Dobie DJ, Le Corre P, Le Verge R, Veith RC, Raskind MA: Effects of Alzheimer's disease and normal aging on cerebrospinal fluid norepinephrine responses to yohimbine and clonidine. Arch Gen Psychiatry. 1995, 52: 774-782.
Raskind MA, Peskind ER: Neurobiologic bases of noncognitive behavioral problems in Alzheimer disease. Alzheimer Dis Assoc Disord. 1994, 8 (Suppl 3): 54-60.
Raskind MA, Peskind ER, Halter JB, Jimerson DC: Norepinephrine and MHPG levels in CSF and plasma in Alzheimer's disease. Arch Gen Psychiatry. 1984, 41: 343-346.
Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M: Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006, 26: 1343-1354.
Jardanhazi-Kurutz D, Kummer MP, Terwel D, Vogel K, Dyrks T, Thiele A, Heneka MT: Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem Int. 2010, 57: 375-382.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ: Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010, 330: 1774-
Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP: Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA. 2010, 107: 6058-6063.
Jardanhazi-Kurutz D, Kumm er MP, Terwel D, Vogel K, Thiele A, Heneka MT: Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience. 2011, 176: 396-407.
Rey NL, Jardanhazi-Kurutz D, Terwel D, Kummer MP, Jourdan F, Didier A, Heneka MT: Locus coeruleus degeneration exacerbates olfactory deficits in APP/PS1 transgenic mice. Neurobiol Aging. 2012, 33 (426): e1-e11.
Hammerschmidt T, Kummer M P, Terwel D, Martinez A, Gorji A, Pape HC, Rommelfanger KS, Schroeder JP, Stoll M, Schultze J, Weinshenker D, Heneka MT: Selective loss of noradrenaline exacerbates early cognitive dysfunction and synaptic deficits in APP/PS1 mice. Biol Psychiatry. 2013, 73: 454-463.
Marien MR, Colpaert FC, Rosenquist AC: Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev. 2004, 45: 38-78.
Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL: Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. 2002, 22: 2434-2442.
Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, Feinstein DL: Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007, 28: 1206-1214.
Kong Y, Ruan L, Qian L, Liu X, Le Y: Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci. 2010, 30: 11848-11857.
Mateo I, Infante J, Rodríguez E, Berciano J, Combarros O, Llorca J: Interaction between dopamine beta-hydroxylase and interleukin genes increases Alzheimer's disease risk. J Neurol Neurosurg Psychiatry. 2006, 77: 278-279.
Combarros O, Warden DR, Hammond N, Cortina-Borja M, Belbin O, Lehmann MG, Wilcock GK, Brown K, Kehoe PG, Barber R, Coto E, Alvarez V, Deloukas P, Gwilliam R, Heun R, Kolsch H, Mateo I, Oulhaj A, Arias-Vásquez A, Schuur M, Aulchenko YS, Ikram MA, Breteler MM, van Duijn CM, Morgan K, Smith AD, Lehmann DJ: The dopamine beta-hydroxylase-1021C/T polymorphism is associated with the risk of Alzheimer's disease in the Epistasis Project. BMC Med Genet. 2010, 11: 162-
Bullido MJ, Ramos MC, Ruiz-Gómez A, Tutor AS, Sastre I, Frank A, Coria F, Gil P, Mayor F, Valdivieso F: Polymorphism in genes involved in adrenergic signaling associated with Alzheimer's. Neurobiol Aging. 2004, 25: 853-859.
Yu JT, Tan L, Ou JR, Zhu JX, Liu K, Song JH, Sun YP: Polymorphisms at the beta2-adrenergic receptor gene influence Alzheimer's disease susceptibility. Brain Res. 2008, 1210: 216-222.
Galasko D, Montine TJ: Biomarkers of oxidative damage and inflammation in Alzheimer's disease. Biomark Med. 2010, 4: 27-36.
Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, McCluskey L, Elman L, Karlawish J, Hurtig HI, Siderowf A, Lee VM, Soares H, Trojanowski JQ: Biomarker discovery for Alzheimer's disease, frontotemporal lobar degeneration, and Parkinson's disease. Acta Neuropathol. 2010, 120: 385-399.
Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, Pickering E, Kuhn M, Chen Y, McCluskey L, Elman L, Karlawish J, Hurtig HI, Siderowf A, Lee VM, Soares H, Trojanowski JQ: Novel CSF biomarkers for Alzheimer's disease and mild cognitive impairment. Acta Neuropathol. 2010, 119: 669-678.
Wyss-Coray T: Inflammation in Alzheimer disease: driving force, bystander or beneficial response?. Nat Med. 2006, 12: 1005-1015.
Yang JH, Lee EO, Kim SE, Suh YH, Chong YH: Norepinephrine differentially modulates the innate inflammatory response provoked by amyloid-beta peptide via action at beta-adrenoceptors and activation of cAMP/PKA pathway in human THP-1 macrophages. Exp Neurol. 2012, 236: 199-206.
Counts SE, Mufson EJ: Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem. 2010, 113: 649-660.
Zornetzer SF: Catecholamine system involvement in age-related memory dysfunction. Ann N Y Acad Sci. 1985, 444: 242-254.
Scullion GA, Kendall DA, Marsden CA, Sunter D, Pardon MC: Chronic treatment with the alpha2-adrenoceptor antagonist fluparoxan prevents age-related deficits in spatial working memory in APPxPS1 transgenic mice without altering beta-amyloid plaque load or astrocytosis. Neuropharmacology. 2011, 60: 223-234.
Madrigal JL, Garcia-Bueno B, Hinojosa AE, Polak P, Feinstein DL, Leza JC: Regulation of MCP-1 production in brain by stress and noradrenaline-modulating drugs. J Neurochem. 2010, 113: 543-551.
Gibbs ME, Maksel D, Gibbs Z, Hou X, Summers RJ, Small DH: Memory loss caused by beta-amyloid protein is rescued by a beta(3)-adrenoceptor agonist. Neurobiol Aging. 2010, 31: 614-624.
Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ: Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron. 2013, 77: 929-941.
Kalinin S, Polak PE, Lin SX, Sakharkar AJ, Pandey SC, Feinstein DL: The noradrenaline precursor L-DOPS reduces pathology in a mouse model of Alzheimer's disease. Neurobiol Aging. 2012, 33: 1651-1663.
Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang YM, Caron MG: Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci. 2000, 3: 465-471.
Wahle T, Thal DR, Sastre M, Rentmeister A, Bogdanovic N, Famulok M, Heneka MT, Walter J: GGA1 is expressed in the human brain and affects the generation of amyloid beta-peptide. J Neurosci. 2006, 26: 12838-12846.
Wang J, Tanila H, Puloivali J, Dadish I, van Groen T: Gender differences in the amount and deposition of amyloidbeta in APPswe double transgenic mice. Neurobiol Dis. 2003, 14: 318-327.
Wallace W, Ahlers ST, Gotlib J, Bragin V, Sugar J, Gluck R, Shea PA, Davis KL, Haroutunian V: Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation. Proc Natl Acad Sci USA. 1993, 90: 8712-8716.
Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM: Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007, 19: 251-260.
Tomozawa Y, Yabuuchi K, Inoue T, Satoh M: Participation of cAMP and cAMP-dependent protein kinase in beta-adrenoceptor-mediated interleukin-1 beta mRNA induction in cultured microglia. Neurosci Res. 1995, 22: 399-409.
Maruta E, Yabuuchi K, Nishiyori A, Takami S, Minami M, Satoh M: Beta2- adrenoceptors on the glial cells mediate the induction of interleukin-1beta mRNA in the rat brain. Brain Res Mol Brain Res. 1997, 49: 291-294.
Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M: Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005, 135: 1295-1307.
Johnson JD, Cortez V, Kennedy SL, Foley TE, Hanson H, Fleshner M: Role of central beta-adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav Immun. 2008, 22: 1078-1086.
Savitz SI, Erhardt JA, Anthony JV, Gupta G, Li X, Barone FC, Rosenbaum DM: The novel beta-blocker, carvedilol, provides neuroprotection in transient focal stroke. J Cereb Blood Flow Metab. 2000, 20: 1197-1204.
Heeba GH, Mahmoud ME, El Hanafy AA: Anti-inflammatory potential of curcumin and quercetin in rats: role of oxidative stress, heme oxygenase-1 and TNF-alpha. Toxicol Ind Health. 2012, doi: 10.1177/0748233712462444,
Wang J, Wright HM, Vempati P, Li H, Wangsa J, Dzhuan A, Habbu K, Knable LA, Ho L, Pasinetti GM: Investigation of nebivolol as a novel therapeutic agent for the treatment of Alzheimer's disease. J Alzheimer's Dis. 2013, 33: 1147-1156.
Greendyke RM, Kanter DR, Schuster DB, Verstreate S, Wootton J: Propranolol treatment of assaultive patients with organic brain disease. A double-blind crossover, placebo-controlled study. J Nerv Ment Dis. 1986, 174: 290-294.
Shankle WR, Nielson KA, Cotman CW: Low-dose propranolol reduces aggression and agitation resembling that associated with orbitofrontal dysfunction in elderly demented patients. Alzheimer Dis Assoc Disord. 1995, 9: 233-237.
Reifler BV, Teri L, Raskind M, Veith R, Barnes R, White E, McLean P: Double-blind trial of imipramine in Alzheimer's disease patients with and without depression. Am J Psychiatry. 1989, 146: 45-49.
Riekkinen M, Laakso MP, Jakala P: Clonidine impairs sustained attention and memory in Alzheimer's disease. Neuroscience. 1999, 92: 975-982.
Riekkinen P, Riekkinen M: THA improves word priming and clonidine enhances fluency and working memory in Alzheimer's disease. Neuropsychopharmacology. 1999, 20: 357-364.
Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC: Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch Neurol. 2006, 63: 686-692.
Rosenberg PB, Mielke MM, Tschanz J, Cook L, Corcoran C, Hayden KM, Norton M, Rabins PV, Green RC, Welsh-Bohmer KA, Breitner JC, Munger R, Lyketsos CG: Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am J Geriatr Psychiatry. 2008, 16: 883-892.
Hajjar I, Catoe H, Sixta S, Boland R, Johnson D, Hirth V, Wieland D, Eleazer P: Cross-sectional and longitudinal association between antihypertensive medications and cognitive impairment in an elderly population. J Gerontol A Biol Sci Med Sci. 2005, 60: 67-73.
Serg M, Kampus P, Kals J, Zagura M, Zilmer M, Zilmer K, Kullisaar T, Eha J: Nebivolol and metoprolol: long-term effects on inflammation and oxidative stress in essential hypertension. Scand J Clin Lab Invest. 2012, 72: 427-432.
Koppel J, Goldberg TE, Gordon ML, Huey E, Davie s P, Keehlisen L, Huet S, Christen E, Greenwald BS: Relationships between behavioral syndromes and cognitive domains in Alzheimer disease: the impact of mood and psychosis. Am J Geriatr Psychiatry. 2012, 20: 994-1000.
Marsh L, Biglan K, Gerstenhaber M, Williams JR: Atomoxetine for the treatment of executive dysfunction in Parkinson's disease: a pilot open-label study. Mov Disord. 2009, 24: 277-282.
Brown TE, Holdnack J, Saylor K, Adler L, Spencer T, Williams DW, Padival AK, Schuh K, Trzepacz PT, Kelsey D: Effect of atomoxetine on executive function impairments in adults with ADHD. J Atten Disord. 2011, 15: 130-138.
Faraone SV, Biederman J, Spencer T, Michelson D, Adler L, Reimherr F, Glatt SJ: Efficacy of atomoxetine in adult attention-deficit/hyperactivity disorder: a drug-placebo response curve analysis. Behav Brain Func. 2005, 1: 16-
Faraone SV, Biederman J, Spencer T, Michelson D, Adler L, Reimherr F, Seidman L: Atomoxetine and stroop task performance in adult attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2005, 15: 664-670.
Gau SS, Shang CY: Improvement of executive functions in boys with attention deficit hyperactivity disorder: an open-label follow-up study with once-daily atomoxetine. Int J Neuropsychopharmacol. 2010, 13: 243-256.
Weintraub D, Mavandadi S, Mamikonyan E, Siderowf AD, Duda JE, Hurtig HI, Colcher A, Horn SS, Nazem S, Ten Have TR, Stern MB: Atomoxetine for depression and other neuropsychiatric symptoms in Parkinson disease. Neurology. 2010, 75: 448-455.
Mohs RC, Shiovitz TM, Tariot PN, Porsteinsson A P, Baker KD, Feldman PD: Atomoxetine augmentation of cholinesterase inhibitor therapy in patients with Alzheimer disease: 6-month, randomized, double-blind, placebo-controlled, parallel-trial study. Am J Geriatr Psychiatry. 2009, 17: 752-759.
Craig-Schapiro R, Kuhn M, Xiong C, Pickering EH, Liu J, Misko TP, Perrin RJ, Bales KR, Soares H, Fagan AM, Holtzman DM: Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer's disease diagnosis and prognosis. PloS One. 2011, 6: e18850-
Wang Y, Hancock AM, Bradner J, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Edwards KL, Snapinn KW, Goldstein DS, Shi M, Zhang J: Complement 3 and factor h in human cerebrospinal fluid in Parkinson's disease, Alzheimer's disease, and multiple-system atrophy. Am J Pathol. 2011, 178: 1509-1516.
Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, Cross AH: Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol. 2010, 67: 707-714.
Mellergard J, Edstrom M, Vrethem M, Ernerudh J, Dahle C: Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler. 2010, 16: 208-217.
Acknowledgements
TC is supported by the Alzheimer's Disease Research Center (ADRC; 5P50 AG025688, PI AIL). The ADRC supported writing of the manuscript. DW received funding from the Alzheimer's Drud Discovery Foundation and the ADRC for design, collection, analysis and interpretation of data. The ADRC supported writing of the manuscript. AIL has funding from the Alzheimer's Drug Discovery Foundation and philanthropic funds for the phase II clinical trial with atomoxetine. Funding from the Alzheimer's Drug Discovery Foundation, the ADRC and philanthropy supported the writing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
TC, BK, MPK, TH, MTH, DW and AIL declare that they have no competing interests. WTH received one compensated meal from Eli Lilly as part of the Alzheimer's Association International Conference (under $100). WTH has patents pending on cerebrospinal fluid biomarkers for frontotemporal lobar degeneration and plasma biomarkers for AD. Some markers in these panels overlap with cerebrospinal fluid biomarkers to be measured in the atomoxetine trial.
Authors' contributions
All authors contributed to the writing and editing of the manuscript. MTH and MPK generated the data presented in Figure 1.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Chalermpalanupap, T., Kinkead, B., Hu, W.T. et al. Targeting norepinephrine in mild cognitive impairment and Alzheimer's disease. Alz Res Therapy 5, 21 (2013). https://doi.org/10.1186/alzrt175
Published:
DOI: https://doi.org/10.1186/alzrt175