
Abstract
UV–vis and photoluminescence spectra of the hydrothermally synthesized crystalline lithium metasilicate (Li2SiO3) and lithium disilicate (Li2Si2O5) nanomaterials are studied. The intensity of the bands in the emission spectra increases with increasing reaction time in both compounds. The electronic band structure along with density of states calculated by the density functional theory (DFT) method indicates that Li2SiO3 and Li2Si2O5 have an indirect energy band gap of 4.575 and 4.776 eV respectively. The optical properties, including the dielectric, absorption, reflectivity, and energy loss spectra of the compounds, are calculated by DFT method and analyzed based on the electronic structures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Silicates are the most abundant and most complicated class of minerals on earth that have tremendous technological applications in fields such as catalysis, microelectronics, biomedicine, photonics, and traditional glass and ceramic industries [1]. In particular, the crystalline lithium silicates are present as important phases in silicate glass ceramics [2] and are of research interest because of their technological applications in areas such as CO2 captures [3–12], lithium battery cathode materials [13], fast ion conductors [14], optical waveguides [15], and tritium breeding materials [16, 17].
Synthesis of lithium silicates has been achieved using different methods, such as solid state reaction, precipitation, sol–gel method, extrusion-spherodisation process, rotating/melting procedures, combustion, electrochemical method, and recently via hydrothermal method. However, most of the time, a mixture of Li2SiO3, Li2Si2O5, Li4SiO4, and SiO2 were obtained [13, 15, 18–21]. On the other hand, the synthesis of nanocrystalline ceramic materials imposes a challenge on the traditional solid state synthesis methods which fail to offer a sufficiently narrow size distribution and desired homogeneity at the nanometer level [22]. However, the hydrothermal synthesis method has an advantage for the production of highly crystalline and pure nanoparticles [23].
Moreover, despite of some significant experimental achievements, our knowledge on the electronic structure and optical properties of the crystalline lithium silicates is still rather limited. The electronic structure of the lithium metasilicate (Li2SiO3) and lithium disilicates (Li2Si2O5) are previously calculated [1]. However, the predicted band gaps are wider than even those experimentally measured for the related nanocrystals described in this research work. Moreover, the optical properties of these materials are not calculated.
Recently, we have reported the synthesis of highly crystalline and pure lithium metasilicate and lithium disilicate nanomaterials through a mild condition via hydrothermal method [24]. Herein, we will report the powder X-ray diffraction (PXRD) and scanning electron microscopy (SEM) analysis results in more details. In addition, the UV–vis and photoluminescence spectra of the obtained materials will be discussed. Moreover, we will present the electronic and optical properties of the synthesized materials through the density functional theory calculations.
Methods
The synthesis procedures are reported previously [24]. Phase identifications were performed on a powder X-ray diffractometer Siemens D5000 (Siemens AG, Munich, Germany) using Cu-Kα radiation. The morphology of the obtained materials was examined with a Philips XL30 scanning electron microscope (North Billerica, MA, USA). Absorption and photoluminescence spectra were recorded on a Jena Analytik Specord 40 (AnalytikJena UK, Wembley, UK) and a PerkinElmer LF-5 spectrometer (PerkinElmer, Waltham, MA, USA) respectively.
Computational details
The electronic band structures along with the density of states (DOS) of the compounds are calculated by density functional theory (DFT) using one of the three non-local gradient-corrected exchange-correlation functionals (generalized gradient approximation-Perdew-Burke-Ernzerhof parametrization, GGA-PBE). Calculations were performed with the CASTEP code [25, 26], which uses a plane wave basis set for the valence electrons and norm-conserving pseudopotential [27] for the core electrons. The number of plane waves included in the basis was determined by a cutoff energy Ec of 500.0 eV. The summation over the Brillouin zone was carried out with a k-point sampling using a Monkhorst-Pack grid [28] with parameters of 5 × 5 × 5 and 4 × 5 × 2 for Li2SiO3 and Li2Si2O5, respectively. Pseudoatomic calculations were performed for Li-2 s2, Si-3s23p2, O-2s22p4. The parameters used in the calculations and convergence criteria were set by the default values of the CASTEP code, e.g., reciprocal space pseudo-potentials representations, eigen-energy convergence tolerance of 1 × 10-6 eV, Gaussian smearing scheme with the smearing width of 0.1 eV, and Fermi energy convergence tolerance of 1 × 10-7 eV.
Results and discussion
PXRD analysis
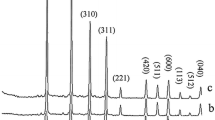
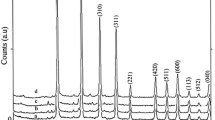
Figure 1 represents the PXRD patterns of the obtained Li2SiO3 nanomaterials after reaction times of 48, 72, and 96 h. The PXRD measurements confirm that when the Li/Si molar ratio in the reaction mixture is 1:2, a pure phase of the orthorhombic Li2SiO3 (space group of Cmc 21[29–35]) is formed. In contrast, as shown in Figure 2, with the Li/Si molar ratio of 1:3 in the reaction mixture, a mixture of meta-stable Li2Si2O5 (space group of Pbcn[36, 37]) and Li2SiO3 is obtained after 48 h. By increasing the reaction time to 72, 96, or 120 h, a pure highly crystalline phase of meta-stable Li2Si2O5 is obtained. A stable form of this compound crystallizes in the space group of Ccc 2 [38]. However, most papers refer to a monoclinic cell [19, 39–43] despite noticing a discrepancy in diffraction peak intensities between the experiment and calculation. The monoclinic cell has a different symmetry but the same size as the Ccc 2 stable form (β = 90°) [44]. Particle sizes that were measured via Debye-Sherrer equation are as follows: Lithium metasilicate particle sizes are 26.12, 26.82, and 24.58 nm for 48, 72, and 96 h reaction times, respectively. Lithium disilicate particle sizes are 20.696, 22.50, and 23.86 nm for 72, 96, and 120 h reaction times, respectively. Also, interplanar spacing in the crystalline material are calculated via Bragg’s law (n λ = 2dhkl sin θ)). Thus compared to those of the nanoparticles of pure lithium silicates, with increasing the reaction time, the diffraction lines in the powder XRD patterns of the nanoparticles of lithium metasilicates shift to higher 2θ values (Δ 2θ = 26.84(48 h) - 26.88(96 h) = 0.04° and Δd = 3.3177 Å (48 h) - 3.3128 Å (96 h) = 0.0049 Å; and with increasing the reaction time, the diffraction lines in the powder XRD patterns of the nanoparticles of lithium disilicates shift to lower 2θ values Δ2θ = 30.66(72 h) - 30.59(120 h) = 0.07°, Δd = 2.9190 Å (120 h) - 2.9125 Å (72 h) = 0.065 Å. According to above measurements, particle sizes measured with Debye-Sherrer equation are in good agreement with interplanar spacing in the crystalline material measured via Bragg’s law.

PXRD patterns of the synthesized Li 2 SiO 3 nanomaterials after different times at 180°C. (a) 48, (b) 72, and (c) 96 h.

PXRD patterns of the synthesized Li 2 Si 2 O 5 nanomaterials after different times at 180 °C. (a) 48, (b) 72, (c) 96, and (d) 120 h.
Microstructure analysis
The SEM images of the synthesized Li2SiO3 nanomaterials are given in Figure 3. With the reaction time of 48 h, ununiform sheet-like nanoparticles of Li2SiO3 are obtained (Figure 3a). The thicknesses, widths, and lengths of the resultant sheets are approximately 100 nm, 600 nm and 2 μm, respectively. With increasing the reaction time to 72 h, the morphology of the obtained materials has been changed to the very compact sheets with heterogeneous morphology (Figure 3b). This is while with the reaction time of 96 h, uniform flower-like nanoparticles are obtained (Figure 3c).

The SEM images of the synthesized Li 2 SiO 3 nanomaterials obtained after different times at 180 °C. (a) 48, (b) 72, and (c) 96 h.
Figure 4 represents the SEM images of the synthesized Li2Si2O5 nanomaterials. After 48 h, the morphology of the obtained material is sponge-like, consisting of sheet-like and flower-like nanoparticles (Figure 4a). With increasing the reaction time to 72, 96, and 120 h, the morphology of the obtained materials has been changed to the rectangular sheets and high homogeny in the size is achieved. According to Figure 3 with image magnification of 15,000, it is clear that with increasing reaction time, the grain size has been decreased. Moreover, according to Figure 4, with image magnification of 15,000, it is clear that with increasing the reaction time the grain size has been increased. So Figures 3 and 4 are in agreement with the calculated particle size and interplanar spacing in the crystalline material measured in PXRD analysis section.

The SEM images of the synthesized Li 2 Si 2 O 5 nanomaterials obtained after different times at 180 °C. (a) 48, (b) 72, (c) 96, and (d) 120 h.
Spectroscopic studies
The electronic absorption spectra and also the emission spectra of the synthesized Li2SiO3 and Li2Si2O5 nanomaterials are given in Figures 5 and 6, respectively. An intense absorption band at 276, 275, and 275 nm is observed in the electronic absorption spectra of the Li2SiO3 nanomaterials obtained after 48, 72, and 96 h at 180°C respectively. A similar intense absorption band is observed at 272, 274, and 277 nm in the electronic absorption spectra of the Li2Si2O5 nanomaterials obtained after 48, 72, and 96 h at 180°C, respectively.

Electronic absorption spectra of Li 2 SiO 3 and Li 2 Si 2 O 5 obtained after 96 h at 180 °C. The electronic absorption spectra of the synthesized Li2SiO3 (a) and Li2Si2O5 (b) nanomaterials obtained after 96 h at 180°C.

Emission spectra of Li 2 SiO 3 and Li 2 Si 2 O 5 obtained after 96 h at 180 °C. The emission spectra of the synthesized Li2SiO3 (a) and Li2Si2O5 (b) nanomaterials obtained after 96 h at 180°C.
In the excitation spectrum of the synthesized Li2SiO3 and Li2Si2O5 nanomaterials, a band is observed with maxima at 360 and 250 nm, respectively. Accordingly, in the emission spectrum of the synthesized Li2SiO3 nanomaterials, an intense peak appears at 410.03 nm. In comparison, an intense peak at 291.45 nm is observed in the emission spectrum of the synthesized Li2Si2O5 nanomaterials. With increasing in the reaction time, no shift is observed in the emission spectrum of the obtained Li2SiO3 and Li2Si2O5 nanomaterials. However, increasing band intensities in the emission spectra of both compounds are observed with increasing reaction time.
Structural optimization
The crystal structure and locations of the atoms of the Li2SiO3[45] and Li2Si2O5[36] determined from X-ray diffraction data are used as a starting point for total energy minimization. The optimized unit cells of the Li2SiO3 and Li2Si2O5 are shown in Figures 3 and 4, respectively. Optimization (relaxation) of the atomic positions and crystal cell parameters was performed before the main calculations of the electronic characteristics, total electronic energy, band energy dispersion, density of electronic states, and optical properties.
Electronic structures
The calculated band structure of the compounds along high symmetry points of the first Brillouin zone is plotted in Figure 7, where the labeled k points are present as G (0.000, 0.000, 0.000), Z (0.000, 0.000, 0.500), T (-0.500, 0.500, 0.500), Y (-0.500, 0.500, 0.000), S (0.000, 0.500, 0.000), and R (0.000, 0.500, 0.500) for Li2SiO3; and G (0.000, 0.000, 0.000), Z (0.000, 0.000, 0.500), T (-0.500, 0.000, 0.500), Y (-0.500, 0.000, 0.000), S (-0.500, 0.500, 0.000), X (0.000, 0.500, 0.000), U (0.000, 0.500, 0.500), and R (-0.500, 0.500, 0.500) for Li2Si2O5. It is found that the top of the valence bands (VBs) has a small dispersion, whereas the bottom of the conduction bands (CBs) has a big dispersion for both Li2SiO3 and Li2Si2O5. The lowest energy (4.575 eV) of the conduction bands (CBs) of Li2SiO3 is localized at the G point, and the highest energy (0.00 eV) of VBs is localized at the Z point. In the case of the Li2Si2O5, the lowest energy (4.776 eV) of the conduction bands (CBs) is localized at the G point, and the highest energy (0.00 eV) of VBs is localized at the X point.

Calculated band structures of Li 2 SiO 3 (top) Li 2 Si 2 O 5 (bottom).
To our knowledge, the optical band gap of the bulk Li2SiO3 and Li2Si2O5 has not been measured. It is well known that both local-density approximation and GGA density functional theory calculations systematically underestimate the band gap of insulators and semiconductors [1]. On the other hand, nanomaterials, compared to the corresponding bulk materials, have wider band gap and therefore show a blue shift in the electronic absorption and photoluminescence spectra [46, 47]. In the orthogonalized linear combination of atomic orbital calculations, the band gap of Li2SiO3 and Li2Si2O5 was found to be 7.26 and 7.45 eV respectively [48]. Also, a band gap of 5.7 eV [1] and 5.36 eV [49] for Li2SiO3 and 5.5 eV [1] for Li2Si2O5 is predicted by DFT calculations using the GGA within Perdew and Wang (PW91) scheme. However, according to our calculations, the values of the calculated band gap for Li2SiO3 and Li2Si2O5 are 4.575 and 4.776 eV respectively, which are comparable with the experimental values (4.49 and 4.56 eV obtained for Li2SiO3 and Li2Si2O5 nanomaterials obtained after 96 h at 180°C) measured from the electronic absorption spectrum of the synthesized nanomaterials.
The total density of states and partial densities of states for Li2SiO3 and Li2Si2O5 are shown in Figures 8 and 9 respectively. The VBs at -19.42 to -15.00 eV for Li2SiO3 and at -19.61 to -15.00 eV for Li2Si2O5 have significant contributions from O-2 s states; however, small contributions from Si-3 s, 3p and Li-2 s, O-2p states still can be observed at these energy intervals.

Total and partial densities of states for Li 2 SiO 3 . The position of the Fermi level is set at 0.0 eV.

Total and partial densities of states for Li 2 Si 2 O 5 . The position of the Fermi level is set at 0.0 eV.
The most complex VBs are from -8.07 eV in Li2SiO3 and -8.84 eV in Li2Si2O5 to the Fermi level (0.0 eV). According to the partial density of states, it is confirmed that the valence bands at these energy intervals are essentially formed by O-2p for both compounds, along with small admixture Li-2 s, while the contributions from Si-3 s, 3p states in Li2Si2O5 are significant and cannot be neglected. Such characteristic indicates that covalent bonds could be formed among O-2p and Si-3p, 3 s states in Li2Si2O5. However, in the case of Li2SiO3, these contributions are weaker. The valence bands at these energy ranges can be further divided into two parts. Such a splitting characteristic of valence bands reflects different bonding behaviors. The first parts located at -8.84 to -5.19 eV (for Li2SiO3) and -8.07 to -4.62 eV (for Li2Si2O5) are due to the bonding between Si-3 s, 3p, Li-2 s orbits and O-2p orbits, while the second part from -5.19 to -4.62 eV for Li2SiO3 and Li2Si2O5, respectively, to the Fermi level (0.0 eV) indicates the small interaction between Si-3p, Li-2 s orbits and O-2p orbits. Analyzing the PDOS also suggests ionic interactions between Si-2 s, 2p orbits and O-2 s, 2p orbits.
The conduction bands between 4.23 and 14.61 eV for Li2SiO3 come from Si-3 s, 3p states, Li-2 s states, and O-3 s, 3p states. In comparison, the bands between 4.23 and 10.00 eV for Li2Si2O5 come primarily from Si-3p states, with small contribution from Si-3 s states, Li-2 s states, and O-2 s, 2p states. The hybridization between Si-3 s, 3p orbits and O-2 s, 2p orbits at upper valence bands is the important structural character of the two compounds.
Optical properties
The optical properties can be gained from the complex dielectric function [50, 51]:
This is mainly connected with the electronic structures and characterizes the linear response of the material to an electromagnetic radiation, and therefore governs the propagation behavior of radiation in a medium. The imaginary part of the dielectric function ε2(ω) represents the optical absorption in the crystal, which can be calculated from the electronic structure through the joint density of states and the momentum matrix elements between the occupied and the unoccupied wave functions within the selection rules and is given
where e is the electronic charge, and and are the conduction band and valence band wavefunctions at k, respectively.
The real part ε1(ω) is evaluated from the imaginary part ε2(ω) by the Kramers-Kronig transformation. The other optical constants such as the refractive index n(ω), extinction coefficient k(ω), optical reflectivity R(ω) absorption coefficient α(ω), energy loss spectrum L(ω), and the complex conductivity function σ(ω) can be computed from the complex dielectric function ε1(ω), through the following relations [49, 50]:
The dielectric functions of Li2SiO3 and Li2Si2O5 were calculated based on the electronic structure. The ε1(ω) and ε2(ω) as a function of the photon energy are shown in Figure 10 for Li2SiO3 and Li2Si2O5.

Dielectric functions of Li 2 SiO 3 (top) Li 2 Si 2 O 5 (bottom).
The imaginary part of ε(ω) in Li2SiO3 has three intense bands located at 9.02, 11.11, and 14.35 eV. The first peak corresponds mainly to the transition from O-2p states (VBs) to the empty Li-2 s and Si-3 s states (CBs) above the Fermi level. The second and third peaks are mainly due to the transitions from O-2p states (VBs) to the Si-3p and Li-2 s states (CBs) above the Fermi level. In contrast, Li2Si2O5 has a prominent absorption peak, located at the photon energies of 9 eV and two weaker bands located at 11.74 and 15 eV. The main peak at the 9 eV is due to strong interband transitions between the O-2p states (VBs) and Si-3p empty states (CBs). It is noted that a peak in ε2(ω) does not correspond to a single interband transition since many direct or indirect transitions may be found in the band structure with an energy corresponding to the same peak [52]. The peak amplitudes of Li2SiO3 are larger than those of the Li2Si2O5 crystals, due to the fact that the band structures for the two compounds are not similar.
For the real part ε1(ω) of the dielectric function ε(ω), the most important quantity is the 0 frequency limit ε1(0), which is the electronic part of the static dielectric constant and depends strongly on the band gap. A smaller energy gap yields a larger ε1(0) value. This could be explained on the basis of the Penn model [52]:
The energy gap (Eg) could be determined from this expression by using the values of ε1(0) and the plasma energy hωp. The calculated and experimental Eg and also the calculated static dielectric constants ε1(0) of Li2SiO3 and Li2Si2O5 are listed in Table 1.
The calculated results on the absorption, reflectivity, and energy loss spectra by norm-conserving pseudo-potentials were shown in Figures 11, 12, 13. According to the absorption spectra, the absorption edges are located at 9.11, 11.85, and 14.70 eV for lithium metasilicate and at 8.2, 11.60, and 15 eV for lithium disilicate. The absorption coefficients decrease rapidly in the low-energy region, which is the representative character of the semiconductors and insulators.

Calculated absorption spectra of Li 2 SiO 3 (top) and Li 2 Si 2 O 5 (bottom).

Calculated reflectivity of Li 2 SiO 3 (top) and Li 2 Si 2 O 5 (bottom).

Calculated energy loss function for Li 2 SiO 3 (top) and Li 2 Si 2 O 5 (bottom).
The calculated reflectivity for lithium metasilicate at 0 to 5 eV is lower than 10% and a maximum value of roughly 35.0% is calculated at about 17.53 eV. In comparison, the reflectivity for lithium disilicate at 0 to 5 eV is calculated to be lower than 2%. The calculated reflectivity spectrum of lithium disilicate shows a maximum value of about 15% at 9.9 eV. According to the absorption and reflectivity spectra, it is concluded that lithium metasilicate and lithium disilicate are transmitting for frequencies of <4.00 eV.
The energy loss spectrum describes the energy loss of a fast electron traversing in the material [53]. The main peak is generally defined as the bulk plasma frequency [54]. At energies smaller than 5.0 eV, no distinct peak is calculated due to the fact that ε2(ω) is still large at these energy values. The main peaks of energy loss spectra, as shown in Figure 13, are calculated at about 12.82 and 15.55 eV for lithium disilicate and 19.5 eV for lithium metasilicate. Such calculations may stimulate the experimental investigations.
Conclusions
This study describes the hydrothermal synthesis of highly crystalline and pure lithium metasilicate and lithium disilicate nanoparticles. The PXRD patterns indicate that the pure lithium metasilicate and lithium disilicate crystallized well under hydrothermal condition. SEM images show the reaction time effect on the morphology and homogeneity of the synthesized materials. The intensity of the bands in the emission spectra increases with increasing reaction time in both lithium metasilicate and lithium disilicate.
The electronic band structure along with DOS calculated by the DFT method indicates that Li2SiO3 and Li2Si2O5 have indirect energy band gaps of about 4.575 and 4.776 eV, respectively. The hybridized interactions between Si-3 s, 3p orbits and O-2p orbits are revealed as the important structural characteristics of the two compounds, which leads to large band gaps.
The optical properties, including the dielectric function, absorption coefficient, reflectivity and energy loss spectra, also have been calculated by DFT methods. According to the calculated absorption and reflectivity spectra, Li2SiO3 and Li2Si2O5 are theoretically transmitting for frequencies of <4.00 eV. Therefore, Li2SiO3 and Li2Si2O5 are the excellent visible and IR transparent materials, which have been experimentally proved. Furthermore, for both compounds, the imaginary part ε2(ω) of the dielectric function ε(ω) has been discussed in detail according to the band structure. It is found that the peak intensities in Li2SiO3 are obviously enhanced compared to that in Li2Si2O5.
Authors’ information
SK got his B.S. degree from the University of Birjand in the field of applied chemistry in 2007. He got his M.Sc. degree from the University of Tabriz in the field of inorganic chemistry in August 2010. He is now a Ph.D. student in faculty of chemistry at University of Semnan, Iran in the field of inorganic chemistry. AA got his B.S. and M.Sc. degree from the University of Tabriz, Iran in the field of chemistry in 1972 and 1974, respectively. He got his Ph.D. degree from the University of Paris, France in the field of inorganic chemistry in 1978. He is now a professor in inorganic chemistry at University of Tabriz, Iran. S.W. joo got his B.S. and M.S. degree from seoul national university in Feb. 1982 and 1984. He got his Ph.D. degree from the University of Michigan, Ann Arbor. From 1995 up to present he is the professor in the school of mechanical engineering, yeungnam University. MD got her B.S. and M.Sc. degree from the University of Tabriz, Iran in the field of chemistry and inorganic chemistry in 2004 and 2006, respectively. She got her Ph.D. degree from the University of Tabriz, Iran in the field of inorganic-solid state chemistry in 2010. She is now post doctorate student and associate professor in the research group of Prof. Rostami at School of Engineering Emerging Technologies, University of Tabriz, Iran and in the department of inorganic chemistry in University of Tabriz, Iran. AB got his B.S. and M.Sc. degree from the University of Tabriz, Iran and from the University of Urmia in the field of chemistry and inorganic chemistry in 2004 and 2006, respectively. He got his Ph.D. degree from University of Tabriz, Iran in the field of inorganic chemistry in 2010.
References
Du J, Corrales LR: Characterization of the structural and electronic properties of crystalline lithium silicates. J. Phys. Chem. 2006, 110: 22346.
Beall GH: Design and Properties of Glass-Ceramics. Annu. Rev. Mater. Sci. 1992, 22: 91. 10.1146/annurev.ms.22.080192.000515
Vincent CA: Lithium batteries: a 50-year perspective, 1959–2009. Solid State Ion. 2000, 134: 159. 10.1016/S0167-2738(00)00723-2
Lu CH, Cheng LW: Reaction mechanism and kinetics analysis of lithium nickel oxide during solid-state reaction. J. Mater. Chem. 2000, 10: 1403. 10.1039/a909130k
Broussely M, Perton F, Biensan P, Bodet JM, Labat J, Lecerf A, Delmas C, Rougier A, Peres JP: Li x NiO 2 , a promising cathode for rechargeable lithium batteries. J. Power. Source. 1995, 54: 109. 10.1016/0378-7753(94)02049-9
Subramanian V, Chen CL, Chou HS, Fey GTK: Microwave-assisted solid-state synthesis of LiCoO 2 and its electrochemical properties as a cathode material for lithium batteries. J. Mater. Chem. 2001, 11: 3348. 10.1039/b105008g
Yang X, Tang W, Kanoh H, Ooi K: Synthesis of lithium manganese oxide in different lithium-containing fluxes. J. Mater. Chem. 1999, 9: 2683. 10.1039/a903495a
Kudo H, Okuno K, Ohira S: Tritium release behavior of ceramic breeder candidates for fusion reactors. J. Nucl. Mater. 1988, 155: 524.
Pfeiffer H, Bosch P, Bulbulian S: Sol–gel synthesis of Li 2 ZrSi 6 O 15 powders. J. Mater. Chem. 2000, 10: 1255. 10.1039/a908714a
Pfeiffer H, Bosch P: Thermal stability and high-temperature carbon dioxide sorption on hexa-lithium zirconate (Li 6 Zr 2 O 7 ). Chem. Mater. 2005, 17: 1704. 10.1021/cm047897+
Mosqueda HA, Vazquez C, Bosch P, Pfeiffer H: Nanoscale domain control in multiferroic BiFeO 3 thin films. Chem. Mater. 2006, 18: 2307. 10.1021/cm060122b
Pfeiffer H, Vazquez C, Lara VH, Bosch P: Thermal behavior and CO 2 absorption of Li 2-x Na x ZrO 3 solid solutions. Chem. Mater. 2007, 19: 922. 10.1021/cm0623965
Vinod MP, Bahnemann D: Materials for all-solid-state thin-film rechargeable lithium batteries by sol–gel processing. J. Solid State Electrochem. 2002, 7: 498.
Zhang B, Easteal AJ: Effect of HNO3 on crystalline phase evolution in lithium silicate powders prepared by sol–gel processes. J. Mat. Sci. 2008, 43: 5139. 10.1007/s10853-008-2736-5
Cruz D, Bulbulian S, Lima E, Pfeiffer H: Kinetic analysis of the thermal stability of lithium silicates (Li 4 SiO 4 and Li 2 SiO 3 ). J. Solid State Chem. 2006, 179: 909. 10.1016/j.jssc.2005.12.020
Nakazawa T, Yokoyama K, Noda K: Ab initio MO study on hydrogen release from surface of lithium silicate. J. Nucl. Mat. 1998, 258: 571.
Munakata K, Yokoyama Y: Ab initio study of electron state in Li4Sio4 crystal. J. Nucl. Sci. Technol. 2001, 38: 915. 10.1080/18811248.2001.9715117
Taddia M, Modesti P, Albertazzi A: Determination of macro-constituents in lithium zirconate for tritium-breeding applications. J. Nucl. Mat. 2005, 336: 173. 10.1016/j.jnucmat.2004.09.011
Pfeiffer H, Bosch P, Bulbulian S: Synthesis of lithium silicates. J. Nucl. Mat. 1998, 257: 309. 10.1016/S0022-3115(98)00449-8
van der Laan JG, Kawamura H, Roux N, Yamaki D: Ceramic breeder research and development: progress and focus. J. Nucl. Mat. 2000, 283: 99.
Cruz D, Bulbulian S: Synthesis of Li4SiO4 by a modified combustion method. J. Am. Ceram. Soc. 2005, 88: 1720. 10.1111/j.1551-2916.2005.00262.x
Khomane RB, Sharma BK, Saha S, Kulkarni BD: Reverse microemulsion mediated sol–gel synthesis of lithium silicate nanoparticles under ambient conditions: scope for CO 2 sequestration. Chem. Eng. Sci. 2006, 61: 3415. 10.1016/j.ces.2005.11.065
Simoes AZ, Moura F, Onofre TB, Ramirez MA, Varela JA, Longo E: Microwave-hydrothermal synthesis of barium strontium titanate nanoparticles. J. Alloy Comp. 2010, 508: 620. 10.1016/j.jallcom.2010.08.143
Alemi A, Khademinia S, Dolatyari M, Bakhtiari A: Hydrothermal synthesis, characterization, and investigation of optical properties of Sb3+-doped lithium silicates nanostructures. Int. Nano Lett. 2012, 2: 20. 10.1186/2228-5326-2-20
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MJ, Refson K, Payne MC: Materials Studio CASTEP, version 5.0. San Diego: Accelrys; 2009.
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MJ, Refson K, Payne MC: First principles methods using CASTEP. J. Kristallography 2005, 220: 567. 10.1524/zkri.220.5.567.65075
Hamann DR, Schluter M, Chiang C: Norm-conserving pseudo-potentials. Phys. Rev. Lett. 1979, 43: 1494. 10.1103/PhysRevLett.43.1494
Monkhorst HJ, Furthmuller J: Special points for Brillouin-zone integrations. Phys. Rev. B. 1976, 13: 5188. 10.1103/PhysRevB.13.5188
Kalinkin AM, Kalinkina EV, Zalkind OA, Makarova TI: Mechanochemical interaction of alkali metal metasilicates with carbon dioxide: 2. The influence of thermal treatment on the properties of activated samples. J. Colloid 2008, 70: 42. 10.1134/S1061933X08010079
Beneke K, Thiesen P, Lagaly G: Synthesis and properties of the sodium lithium silicate silinaite. Inorg. Chem. 1995, 34: 900. 10.1021/ic00108a022
Gutierrez GM, Cruz D, Pfeiffer H, Bulbulian S: Low temperature synthesis of Li2SiO3: effect on its morphological and textural properties. Res. Lett. Mat. Sci. 2008, 2: 1.
Zhang B, Allan TW, Easteal J: Effect of HNO3 on crystalline phase evolution in lithium silicate powders prepared by sol–gel processes. J. Mat. Sci. 2008, 43: 5139. 10.1007/s10853-008-2736-5
Fuss T, Mogus A, Milankovic B, Ray CS, Lesher CE, Youngman R, Day DE: Ex situ XRD, TEM, IR, Raman and NMR spectroscopy of crystallization of lithium disilicate glass at high pressure. J. Non-Cryst. Solids 2006, 352: 4101. 10.1016/j.jnoncrysol.2006.06.038
Soares PC, Zanotto ED, Fokin VM, Jain H: TEM and XRD study of early crystallization of lithium disilicate glasses. J. Non-Cryst. Solids 2003, 331: 217. 10.1016/j.jnoncrysol.2003.08.075
Zheng X, Wen G, Song L, Huang XX: Effects of P 2 O 5 and heat treatment on crystallization and microstructure in lithium disilicate glass ceramics. Acta Mat. 2008, 56: 549. 10.1016/j.actamat.2007.10.024
Smith RI, Howie RA, West AR, Pina AA, Villafuerte-Castrejón ME: Hydrogen bonding in barium hydroxide trihydrate by neutron diffraction. Acta Crystal. Sect. C. 1990, 46: 363. 10.1107/S010827018900750X
Smith RI, West AR, Abrahams I, Bruce PG: Rietveld structure refinement of metastable lithium disilicate using synchrotron X-ray powder diffraction data from the Daresbury SRS 8.3 diffractometer. Powder Diffraction. 1990, 5: 137. 10.1017/S0885715600015566
Dejong BHWS, Supér HTJ, Spek AL, Veldman N, Nachtegaal G, Fischer JC: Mixed alkali systems: structure and 29Si MASNMR of Li 2 Si 2 O 5 and K 2 Si 2 O 5 . Acta Crystal. Sect. B. 1998, 54: 568. 10.1107/S0108768198001062
Zanotto ED: Metastable phases in lithium disilicate glasses. J. Non-Cryst. Solids 1997, 219: 42.
Liebau F: Untersuchungen an Schichtsilikaten des Formeltyps A m (Si 2 O 5 ) n . I. Die Kristallstruktur der Zimmertemperaturform des Li 2 Si 2 O 5 . Acta Crystal. 1961, 14: 389. 10.1107/S0365110X61001303
Rindome GE: Color-centers formed in glass by solar radiation. J. Am. Ceram. Soc. 1962, 45: 7. 10.1111/j.1151-2916.1962.tb11020.x
Engel K, Frischat G, Textures H: Texture formation in a glass ceramic of Li 2 O 2SiO 2 composition. Microstructures. 1995, 24: 155.
Deubener J: Spectroscopy and thermal properties of Ga 2 S 3 based glasses. J. Non-Cryst. Solids 2000, 274: 195. 10.1016/S0022-3093(00)00188-5
Paszkowicz W, Wolska A, Klepka MT, All SAE, Eldin FME: Combined X-ray diffraction and absorption study οf crystalline vanadium-doped lithium disilicate. Acta Phy. Pol.: A 2010, 117: 315.
Hesse KF: Refinement of the crystal structure of lithium polysilicate. Acta Crystal. Sect. B. 1977, 33: 901. 10.1107/S0567740877004932
Vollath D: Nanomaterials: an Introduction to Synthesis. Wiley, Weinheim: Properties and Applications; 2008.
Schaefer HE: Nanoscience: the Science of the Small in Physics. Chemistry, Biology and Medicine, Springer, Berlin: Engineering; 2010.
Ching WY, Li YP, Veal BW, Lam D: Electronic structures of lithium metasilicate and lithium disilicate. J. Phy. Rev. B 1985, 32: 1203. 10.1103/PhysRevB.32.1203
Tang T, Luo DL: Density functional theory study of electronic structures in lithium silicates: Li2SiO3 and Li4SiO4. J. At. Mol. Sci. 2010, 1: 185.
Fang RC: Solid State Spectroscopy. Hefei: Chinese Science Technology University Press; 2003.
Zhang Y, Shen WM: Basic of Solid Electronics. Hangzhou: Zhe Jiang University Press; 2005.
de Almeida JS, Ahuja R: Tuning the structural, electronic, and optical properties of Be x Zn 1-x Te alloys. Appl. Phys. Lett. 2006, 89: 061913. 10.1063/1.2219341
Bouhemadou A: Khenata, Comput, R: Ab initio study of the structural, elastic, electronic and optical properties of the antiperovskite SbNMg 3. Mat. Sci. 2007, 39: 803.
Saniz R, Ye LH, Shishidou T, Freeman A: Structural, electronic, and optical properties of NiAl 3 : first-principles calculations. J. Phy. Rev. B. 2006, 74: 014209.
Acknowledgment
The authors express their sincere thanks to the authorities of Tabriz University for financing the project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
All authors, AA, SK, SWJ, MD, and AB, participated in the experiments and read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Alemi, A., Khademinia, S., Joo, S.W. et al. Lithium metasilicate and lithium disilicate nanomaterials: optical properties and density functional theory calculations. Int Nano Lett 3, 14 (2013). https://doi.org/10.1186/2228-5326-3-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2228-5326-3-14