
Abstract
Schnitzler’s syndrome is an autoinflammatory disorder characterized by the association of a monoclonal IgM (or IgG) gammopathy, a chronic urticarial rash, and signs and symptoms of systemic inflammation, including fever, arthralgias and bone pain. It was first described in 1972. This review summarizes the clinical features, efficacy of therapies, and follow-up data of the 281 cases that have been reported to date. Also, the results of skin histology, bone imaging, laboratory investigations, and studies of the pathogenesis will be discussed, including the pivotal role of interleukin-1 beta in this disorder.
Similar content being viewed by others

Introduction
Schnitzler’s syndrome (SchS) is an autoinflammatory disease characterized by the association of a monoclonal immunoglobulin M (IgM, or sometimes IgG) gammopathy (M-protein), a chronic urticarial rash, and signs and symptoms of systemic inflammation [1]. The first case was described by the French dermatologist Professor Dr. Liliane Schnitzler in 1972 [2]. As the phenotype is rather unspecific and many physicians are not familiar with this syndrome, SchS is highly under-diagnosed. This was underlined by the recent retrospective study at the Mayo Clinic, in which 46 undiagnosed cases were identified by cross-referencing cases from their dysproteinemia database with medical records from all patients with chronic urticaria at that institution [3].
This review summarizes the clinical features, efficacy of therapies, and follow-up data regarding the 281 cases that have been reported to date. Also, the results of skin histology, bone imaging, laboratory investigations, and studies on the pathophysiology will be discussed, including the pivotal role of interleukin-1 beta (IL-1β) in this disorder.
Review
Methods and diagnostic criteria
In August 2014, a PubMed search using the key words ‘Schnitzler syndrome’ and ‘Schnitzler’s syndrome’ was performed to retrieve all cases. Relevant articles in any language were analyzed, as well as additional cases that are included in the Schnitzler’s syndrome database, which we aim to convert to a registry. In an attempt to prevent double counting of the same patient, the case reports were carefully considered. Cases from the Mayo Clinic, for example, were described in three papers with different focuses. As it could be retrieved from the methods that they mostly overlapped, the cases with IgM monoclonal gammopathy were conservatively regarded as entirely overlapping, and the 4 IgG cases from the Sokumbi paper were added to the 62 IgM patients from the Jain paper [3–5]. Only patients that fulfilled the Strasbourg diagnostic criteria for SchS were included (Table 1). Hence, patients that lacked the paraprotein were excluded, even though they might have developed a monoclonal gammopathy after publication of the case reports.
Epidemiology
To date, 281 cases have been reported, with a male–female ratio of 1.5 [2–146]. (Kabashima, Schiff, Frerichs, Van Hoof, Relas, Björk, Fox, Croot: personal communications (p.c.)) As indicated above, SchS is likely to be under-diagnosed, as was illustrated by the study of Jain et al. [3] On the other hand, since awareness of this diagnosis is increasing, many cases are not reported unless there are novel findings. Further, as the diagnosis is one of exclusion, one has to carefully consider the differential diagnoses. The median age of onset is 51 years, based on available data on 174 patients. Initially, most cases were reported in French medical journals, but nowadays, patients have been reported in 25 countries including Japan, Australia and Brazil (Figure 1). Most are of Caucasian descent.

Number of reported cases per country.
Clinical features
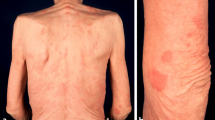
As a major diagnostic criterion, a chronic urticarial rash is present in all patients. In the majority of cases, this is the presenting symptom, and it often precedes the other symptoms several years. Urticaria were explicitly reported as the presenting symptom in 174 cases with a median age of onset of 51 years. The age of onset of the other symptoms was often not mentioned. The age of onset of fever was given in 66 cases, with a median of 52 years old. In 16 of these cases, urticaria preceded the fever (median 3 years; range 1–14 years); in one, fever preceded the urticaria one year; and in the rest, they occurred concurrently. The frequency of urticaria ranges from daily to a few times a year, and the extent of skin involvement differs greatly. Patients describe red skin lesions covering the trunk and extremities, sparing head, neck, palms and soles. Individual lesions persist less than 24 to 48 h. In only 21% of cases, the lesions become pruritic over time, and often the skin lesions are associated with a burning rather than pruritic sensation. Angioedema is rare (Table 2).
Additional symptoms frequently develop months to years after the disease onset, and often, but not exclusively, occur concurrently with the urticarial rash. The second most common symptom is intermittent fever in 72% of patients, without a periodic pattern. The frequency of fever attacks ranges from daily to a few times a year. Weight loss is reported in 16%. Two-thirds of patients complain of joint pain, mostly in the knees, hips and back, but sometimes also in the hands and feet. Bone pain affects over half of the patients. It typically affects the shins, but also other long bones, the hips and back. Neuropathy was reported in 7% (Table 2). It usually concerns a symmetrical sensory polyneuropathy, and in two patients, anti-myelin-associated-glycoprotein antibodies were found [81, 114]. Single case reports mentioned other symptoms, such as intercostal neuralgia and headache in one patient, and pancreatitis in another, which all ceased during IL-1 blocking treatment [76, 83]. It is unclear if these symptoms belong to the disease spectrum of SchS. In the latter case, this seems unlikely, as the pancreatitis preceded the onset of urticaria of SchS 15 years and the mother and son also suffered from pancreatitis but not from SchS [76]. The response to the IL-1 receptor antagonist (IL-1Ra) anakinra in both cases, suggests involvement of IL-1 in neuropathic pain and pancreatitis, respectively. Other incidentally reported symptoms include pseudoxanthoma elasticum [60, 90, 144], headache, vertigo and depression [60], and severe thrombophilia with antiphospholipid syndrome and hyperhomocysteinemia [51]. The family history is typically negative for the symptoms of SchS.
Physical examination shows a few to numerous nummular erythematous macules or slightly elevated plaques that may become confluent and that cover the trunk and extremities, mostly sparing head and neck, and never involving the palmoplantar skin. These urticarial lesions differ from the wheals of chronic spontaneous and inducible urticarias (CSU and CindU, respectively) in that they are rather symmetrically distributed, persist longer (<24-48 hours per individual lesion), are less edematous, and less itchy [147]. Fever may or may not be present at the time of assessment, and may measure up to 40°C. The affected joints may be painful during examination, but overt arthritis is rarely seen. In only a small number of patients, lymphadenopathy or hepatosplenomegaly are found (Table 2).
Laboratory investigations
Serum markers of systemic inflammation (erythrocyte sedimentation rate and C-reactive protein) are almost invariably elevated. Leukocytosis, usually neutrophilia, is found in three-quarters of the patients, and anemia in two-thirds (Table 2).
The second hallmark of SchS is a paraprotein, or monoclonal gammopathy, which is of the IgM kappa (IgMκ) subtype in 85% of the patients. Variant cases of the IgG subtype constitute 7% of the reported cases. The actual percentage of IgG cases may be higher, though, as it was initially not included in the definition, and exclusively IgM cases were included in the largest case series [3]. Interestingly, a biclonal gammopathy was present in 7 cases. In 2 of these cases, an IgA monoclonal gammopathy was found in addition to a IgMκ gammopathy. In 1 patient, 3 different IgM M-proteins were found. In 89% of cases, kappa was the light chain subtype. Bence Jones proteins were detected in 23% of the cases in whom they were assessed (Table 2).
As the differential diagnosis of chronic urticaria, a paraprotein, and systemic inflammation is extensive, several hematological, infectious and autoimmune diseases need to be excluded (Table 3). Therefore, laboratory investigations of a patient with these symptoms should include a complete blood count, leukocyte differential, blood cultures, serology for hepatitis C, streptococcal antibodies, rheumatoid factor, antinuclear antibodies, cold agglutinins, cryoglobulins, ferritin, complement factors and serum electrophoresis.
Histology of skin biopsies
SchS lesional skin is among the few diagnoses that usually classify as neutrophilic urticarial dermatosis (NUD) [67]. NUD is characterized by a perivascular and interstitial neutrophilic infiltrate with intense leukocytoclasia but without vasculitis and dermal edema. Indeed, in half of the cases, a perivascular and interstitial infiltrate of neutrophils in the absence of vasculitis is reported. Presumably, this is an underestimation, as in 19%, the infiltrate was not specified. Also, sampling errors, treatment effects, or different disease stages could account for the lack of neutrophils in over 10% of cases. Vasculitis was reported in 20% of cases, but this may be an overestimation, as re-evaluation of several of them revealed the absence of fibrinoid necrosis of the vessel walls and thus the lack of vasculitis [148]. Immune depositions consisting of IgM, C3 and sometimes IgG were found in 30% of cases (Table 2). However, the location of the depositions varied greatly, from an interstitial granular pattern to presence along the blood vessel walls (see also “Pathogenesis”).
Skeletal examinations
Usually, conventional radiographs are taken for the detection of bone abnormalities in SchS. These are normal in over half of the cases, and show osteosclerosis in almost all other cases. Nuclear scintigraphy shows increased uptake in bone in 85% of cases. In most cases, the radiographic and scintigraphic abnormalities are found concomitantly. In others, abnormalities on bone scintigraphy were not found by conventional radiographs, indicating that the former method is more sensitive for detection of the early disease stages [42]. A thorough study in 22 patients with SchS compared the accuracy of several imaging techniques for the detection of bone lesions. Only 38% of osseous abnormalities were identified by plain radiographs, and the remaining ones required the use of other modalities (nuclear scintigraphy, positron emission scanning, gallium scan, magnetic resonance imaging or computed tomography). In view of its high sensitivity, nuclear bone scintigraphy was proposed as the initial screening modality [5]. The most commonly involved sites are the proximal tibia, distal femur, and the innominate bone, but vertebrae, ribs and other bones could also be affected. The focal uptake in the proximal tibiae and distal femora was referred to as the ‘hot knees sign’, which is regarded as typical for both SchS and Erdheim-Chester disease [5]. Bone abnormalities are often found at the site of bone pain. Interestingly, radiographic abnormalities have been reported preceding development of typical clinical symptoms [15]. The few bone biopsies taken were in most cases normal or showed signs of sclerosis (Table 2).
A recent study in serum of 13 SchS patients showed increased bone formation (high bALP, osteocalcin and osteoprotegerin) which was not balanced by an increase in bone resorption (normal CTX and sRANKL). Also, the mean vascular endothelial growth factor (VEGF) levels were almost 3.5-fold higher in SchS compared to controls. Successful treatment led to a significant reduction in VEGF [131].
Bone marrow examination
Bone marrow aspirates or histology were normal in the majority of cases; plasmacytosis was found in 9%, and a malignancy was found in 21% of the samples. The latter is an overestimation of the actual percentage of malignancies, as in multiple reports, the analyses of bone marrow aspirates were not mentioned. The hematological malignancies are specified in Table 4.
Differential diagnosis
As SchS is a disorder per exclusionem, several autoimmune, hematological, infectious, and other autoinflammatory diseases need to be excluded by means of an extensive workup (Table 3). Depending on the dominant clinical feature, the differential diagnosis can be tailored at either chronic urticaria, recurrent fever, arthralgia/arthritis and/or bone pain, a paraprotein, or a combination. The major considerations are mentioned below.
The most prevalent type of chronic urticaria is CSU, followed by CIU, e.g. cold-induced urticaria. As indicated above, the urticarial lesions in SchS differ from the wheals of these urticarias in that they are rather symmetrically distributed, persist longer (<24-48 hours per individual lesion), are less edematous, less itchy, and rarely associated with angioedema [147]. In contrast to CSU and most CIU, delayed-pressure urticaria can be accompanied by systemic symptoms including fever and arthralgias, but serum markers of inflammation are usually normal. Urticarial vasculitis should be excluded; even though vasculitis was reported in 20% of SchS cases, true fibrinoid necrosis of the vessel walls was absent in 5 re-examined cases, as indicated before. Histologically, NUD is typical for SchS, whereas in CSU and CIU, little leukocytoclasia and much more edema are seen in the dermis [4, 67]. The differential diagnosis of NUD includes other systemic inflammatory diseases, mainly adult-onset Still’s disease (AOSD), lupus erythematosus (LE), and hereditary autoinflammatory syndromes. AOSD is a chronic systemic inflammatory disease characterized by high spiking fever, evanescent rash, pharyngitis, polyarthralgia, lymphadenopathy, hepatosplenomegaly, serositis, and leukocytosis, as well as elevated liver enzymes, erythrocyte sedimentation rate, and serum ferritin associated with low glycosylated fraction of ferritin (<20%) [149]. In AOSD, a pink maculopapular rash occurs rather than the flat urticaria seen in SchS. Bone pain with hyperostosis and a paraprotein are typical for SchS, whereas very high serum ferritin concentrations are rather specific for AOSD. Finally, AOSD primarily affects young adults, in contrast to SchS, in which the median age of onset is 51 years. LE can be accompanied by urticaria, but in systemic LE, the main skin feature is a butterfly-shaped malar erythema, and polycyclic or erythematosquamous plaques in sun-exposed areas in subacute cutaneous LE. Skin biopsies show a lichenoid infiltrate in the majority of cases, but NUD can occur. Arthralgia and fever can occur in LE, but bone pain and a paraprotein are lacking. SchS shares many features with the hereditary autoinflammatory syndrome cryopyrin-associated periodic syndrome (CAPS), which is caused by NLRP3 mutations that lead to enhanced IL-1β production that induces chronic systemic inflammation, including fever and joint pain. CAPS constitutes a spectrum from cold-induced urticaria to severe neonatal-onset disease with meningitis and debilitating arthritis. In SchS, cold-sensitivity was only mentioned in 3 cases in the literature, but one expert reported this in one-third of their cases. The presence of NLRP3 variants is not a strong distinguishing factor, as part of the CAPS patients are mutation-negative, and certain NLRP3 variants were identified in a handful of SchS patients (see “Pathophysiology”). Still, germline NLRP3 mutations are commonly found in CAPS and have not been reported in SchS to date. Depending on the severity, disease onset of CAPS is generally during childhood, and often relatives are affected. Bone pain is usually lacking, and paraproteins have not been reported in CAPS.
The differential diagnosis of the osteosclerotic lesions seen in SchS on conventional radiographs includes Erdheim-Chester disease, metastases, and polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS) syndrome [5]. The so-called ‘hot knees’ sign on bone scintigraphs indicates focal uptake within the distal femora and proximal tibiae, and is very specific for Erdheim-Chester disease, but is often seen in SchS too. The former is a non-Langerhans cell histiocytosis in which several organ systems are affected, leading to neurological signs, bone pain, exophthalmos, and xanthelasmata, among others [150]. POEMS syndrome is characterized by polyradiculoneuropathy, a clonal plasma cell disorder, sclerotic bone lesions, elevated VEGF, the presence of Castleman’s disease, and a few minor criteria. Neuropathy is the dominant feature in POEMS, whereas this was only found in 7% of SchS patients. The paraprotein in POEMS is of the lambda subtype in 91%; in SchS, this is 11% [151]. This suggests notable skewing towards kappa light chain restriction in SchS.
Monoclonal gammopathy of unknown significance (MGUS) is a common disorder and its prevalence increases with age. As chronic urticaria is prevalent as well, one would expect concomitant MGUS in many cases. Kappa and lambda light chains are quite evenly distributed in MGUS, in contrast to the dominance of the kappa light chain in SchS. Jain et al. calculated a high odds ratio for a correlation between an IgM monoclonal protein and chronic urticaria (odds ratio 9801; P = 0.0001), and stated that chances are high that a patient who has both chronic urticaria and an IgMκ monoclonal gammopathy has SchS [3].
Naturally, the presence of hematopoietic malignancies needs to be excluded by bone marrow examination. Waldenström’s macroglobulinemia (WM) is an important differential diagnosis, but it is also a long-term complication of SchS (see ‘Prognosis’). Thus, even after the diagnosis of SchS is established, one has to remain alert to the development of a lymphoproliferative disorder. The concentration of VEGF cannot help distinguish between SchS, WM, POEMS and IgM MGUS, as it is elevated in all of these disorders [152].
Treatment
The effects of 35 different treatment modalities have been reported in SchS. Table 5 summarizes the efficacy of all of these, and indicates the number of patients in which it was tried. IL-1 blocking therapies are the most effective ones. Both the IL-Ra anakinra [3, 11, 12, 14, 16, 18, 27, 29, 33, 35, 39–41, 45, 47, 50, 55, 58, 59, 63, 68–71, 76, 77, 83, 92–94, 104, 106, 113, 115, 119–121, 127, 129, 131–133, 135, 136, 140, 153] (Van Hoof, Relas, Björk, Fox, Croot: p.c., and personal observations), which blocks IL-1α and IL-1β, and the anti-IL-1β antibody canakinumab [12, 41, 136, 154] (Fox: p.c.), which only inhibits IL-1β, induce a complete remission in more than 90% of cases. Rilonacept, a fusion protein including the IL-1R, is as effective in 50% of cases [71]. Interestingly, in the 3 cases in which anakinra was not effective, the IL-6 antibody tocilizumab proved highly effective [70]. The anti-CD-20 antibody rituximab, interferon-α, corticosteroids and thalidomide are very effective in about 20% of cases, followed by colchicine and pefloxacine. The latter is partially effective in two-thirds of patients [10, 32, 66, 104, 131]. Corticosteroids were only effective in high doses, the use of which is restricted by the side effects. Thalidomide had to be discontinued in a few cases due to the development of polyneuropathy. The majority of therapies tried were ineffective (Table 5). Antihistamines are invariably ineffective or hardly effective, whereas these drugs are beneficial in histamine-dependent urticarias.
The treatment of choice in SchS is anakinra, which is highly effective in 81 of 86 cases. It only suppresses inflammation, and does not cure the disease. Invariably, symptoms recur within 1 to (rarely) 6 days upon withdrawal of anakinra. The major side effect is an erythematous injection site reaction, and upper respiratory tract infections occur more often. As neutropenia may occur during IL-1 inhibition, quarterly leukocyte differential counts are recommended – and shorter intervals during the first weeks after the initiation of treatment. Interestingly, in two SchS cases, IL-1Ra was successfully reintroduced after early occurring treatment-induced neutropenias [104, 136]. As anakinra requires painful daily injections, longer-acting agents are needed. Canakinumab proved to be an effective and safe alternative, but was not registered for SchS [41]. Thus, we need other safe, long-acting IL-1 blocking treatment modalities.
Follow-up
The major complication of SchS is the development of a hematological malignancy. This was reported in 35 SchS patients at a median follow-up of 8 years after the onset of disease [3, 4, 8, 19, 21, 26, 34, 36, 40, 53, 61, 63, 64, 72, 91, 97, 99, 108, 112, 131, 133, 138, 141]. Two-thirds of these malignancies concerned WM, a lymphoplasmacytic lymphoma (Table 4). Most other cases also developed a lymphoma. Intriguingly, one patient developed acute myeloid leukemia [3]. As exact follow-up data on 74 patients are lacking, the risk of developing a hematological malignancy cannot reliably be indicated. One can only deduce that overall 35 out of 281 reported cases (12%) developed a hematological malignancy. To date, only 1 patient developed WM during IL-1 inhibition: after 2 years of anakinra in combination with methotrexate [63].
Accurate survival data are also lacking, as the duration from disease onset until death is only known for 8 out of the 25 deceased cases, with a median of 6 years. Eighteen patients survived over 20 years after the disease onset. Median follow-up was 7 years. From 58 cases, follow-up data of less than 5 years are available, on top of the 74 cases in which it is unknown. In our previous analysis of the first 94 reported cases, we found no reduction in survival [40]. Jain et al. reported that hemoglobin below 12.2 g/dL was the only adverse prognostic factor in their series of 62 patients [3].
One patient developed membranous nephropathy [65]. The development of AA amyloidosis was reported in 6 cases (2%): in one case 5 years after disease onset; in another 10 years, and after an unknown duration in the others [31, 33, 82, 89, 95, 101, 144]. Interestingly, although many SchS patients have been followed up for more than two decades, no cases of AL amyloidosis have been reported.
Pathogenesis
Theories concerning the pathophysiology of SchS include autoimmunity, a hematological origin, and more recently, autoinflammation. Currently, the latter is the dominant hypothesis.
Initially, most of the cases were reported in dermatological journals, since chronic urticaria is often the presenting symptom of SchS, and professor of Dermatology Liliane Schnitzler was the first to recognize and report the particular combination of chronic urticaria and a monoclonal gammopathy [2]. Consequently, initial investigations focused on the skin by means of histological and immunohistochemical studies. Histopathological examination shows a neutrophilic infiltrate in most cases (Table 1). Hence, a neutrophilic infiltrate on skin biopsy was added as a minor criterium for the diagnosis of SchS [1].
With a monoclonal gammopathy as the second disease hallmark, the presence of immune depositions was assessed in many skin biopsies. It was absent in 70% of the cases, but IgM, C3 and sometimes IgG skin depositions were found in others (Table 1). Still, the location of the depositions varied greatly. No clinical or histological changes were observed upon the injection of purified human IgMκ into rabbit skin [32]. In 2 out of 3 patients, Western blotting of epidermal extracts showed IgMκ anti-skin antibodies which recognized 2 different unknown antigens. This was not seen in dermal abstracts, whereas the immune infiltrate is mostly limited to the dermis [155]. Altogether, the heterogeneous findings render a major causal role for the M-protein in initiating the skin lesions unlikely.
The urticaria are histamine-independent, as antihistamines are invariably ineffective [1, 40], and wheals did not develop upon injection of serum into skin [134]. In one patient, IgG3 antibodies directed against endothelial cells and mast cells, and IgG2 antibodies specific for the alpha-chain of the IgE receptor were detectable, but these antibodies did not mediate histamine release in mast cells or basophils [126].
After the recognition of SchS as a potential autoinflammatory disorder, partly in view of its phenotypical similarities to CAPS, SchS research focused on the role of proinflammatory cytokines, IL-1β in particular [39, 40, 93, 113]. The effect of the IL-1Ra anakinra provided the first evidence for the crucial role IL-1 plays in the pathophysiology of SchS [39, 93]. Further, hypersecretion of IL-1β, IL-6 and TNFα by peripheral blood mononuclear cells (PBMCs) of a patient was found. This could be blocked by the in-vitro addition of the caspase-1 inhibitor YVAD, proving involvement of the inflammasome [113]. Others reported a case in which PBMCs sampled during active disease displayed an aberrant spontaneous release of IL-1β that was increased by 2-hour stimulation with LPS. Interestingly, subsequent stimulation of the patient’s cells with the P2X7R agonist BzATP failed to trigger the massive IL-1β release seen in PBMCs from healthy controls. Prednisolone treatment of the patient resulted in diminution of symptoms and normalized the cytokine responses [106]. Similar results were found in the CAPS in which systemic inflammation is caused by activating NLRP3 mutations. CAPS patient PBMCs constitutively produce IL-1β, and treatment with IL-1Ra results in both a dramatic clinical improvement and substantive down-regulation of LPS-induced IL-1β secretion by the patients’ cells in vitro[156]. Such a constitutive IL-1β production was not found by Launay et al., who however did report enhanced LPS-induced IL-1β and IL-6 production by PBMCs of two symptomatic patients, which was reduced by both in-vitro and in-vivo treatment with anakinra [77]. Migliorini et al. found that treatment with IL-1Ra significantly inhibited IL1B gene expression in monocytes [94], which indicates the presence of an auto-amplifying loop of IL-1β. Altogether, these data suggest that the inflammasome is primed in monocytes of SchS patients, requiring one rather than the usual two triggers for the activation of IL-1β. Normally, the maturation of IL-1β is tightly controlled, as it is a highly potent proinflammatory cytokine. It induces neutrophilia, anorexia, anemia, fever, chills, skin rash, elevated serum markers of systemic inflammation, joint destruction, and tissue remodeling. IL-1 also induces IL-6 production by e.g. endothelial cells, and IL-6 causes a rise in acute phase reactants, and platelet release from the bone marrow [157]. In 10 SchS cases, elevated serum concentrations of IL-6 were reported [9, 38, 85, 86, 96, 120, 127, 146].
The skin lesions in SchS may well be induced by local IL-1β production in the skin, as Volz et al. found IL-1β-expressing dermal cells in one patient [139].
It is not entirely clear how IL-1β overactivation leads to the characteristic bone abnormalities. Both IL-1 and IL-6 stimulate osteoclast function. Possibly, the osteoclerosis seen in SchS results from the elevated VEGF levels, as angiogenesis enhances osteogenesis [131].
The major question regarding the pathophysiology of SchS is if and how the monoclonal gammopathy correlates with the systemic inflammation. Initially, the M-protein was thought to cause the skin symptoms, but as indicated above, this is unlikely. Also, the lack of efficacy of rituximab in most cases shows that lowering the M-protein concentration rarely attenuates the systemic inflammation (Table 5). Further, the calculations of Jain et al. show that accidental concomitant occurrence of IgM monoclonal gammopathy and chronic urticaria is not probable [3]. The third option is that the systemic inflammation instigates the formation of a plasma cell clone. This hypothesis is supported by different lines of evidence. First, several cases have been reported in which a M-protein became detectable several years after the onset of symptoms. In a Norwegian patient, for example, this was 13 years [62]. Indeed, a few cases have been reported that lacked the M-protein [137, 158–160]. These were not included in this paper, as only those patients who fulfilled the Strasbourg diagnostic criteria were included, but these may well develop a monoclonal gammopathy over time. Further, in 9 cases, more than one M-protein was present (Table 2), which suggests a common factor capable of inducing plasma cell clones. IL-6 and IL-1β have been implicated in the development of hematological malignancies. IL-6 is a growth factor for B-lymphocytes, and crucial to the growth, proliferation and survival of myeloma cells. Through its stimulation of osteoclast function it also influences the tumor microenvironment in the bone marrow of patients with myeloma [161]. In myeloma, IL-1 was found to stimulate IL-6 release from marrow stromal cells, which stimulates the survival and proliferation of plasma cells. Anakinra decreased IL-6 levels but left numbers of myeloma cells unaffected. However, combination therapy of anakinra with dexamethasone (which induces apoptosis) induced myeloma cell death. Moreover, in some patients with smoldering or indolent multiple myeloma who were at risk of progression to active myeloma, concomitant treatment with IL-1Ra and dexamethasone decreased the myeloma proliferative rate [162]. Thus, speculatively, combination therapy of anakinra with dexamethasone in SchS might clear the malignant clone and M-protein. To date, only one case of reduction of the M-protein concentration during anakinra treatment has been reported in SchS [69]. Neither several years of monotherapy with IL-1Ra, nor several months of treatment with IL-1β antibodies or an IL-1R fusion protein led to a decrease in the monoclonal gammopathy in all other patients [41, 71]. As in general, M-protein levels remain rather stable during anti-IL-1 treatment, one could speculate that IL-1 inhibition is capable of blocking further growth of the plasma cell clone, but cannot induce its demise. The short follow-up during anti-IL-6 treatment did not show a reduction in M-protein concentrations [70]. Long-term follow-up in more patients is needed to determine if IL-6 inhibition can affect the plasma cell clones in SchS. Indeed, anti–IL-6 treatment has been successfully used in Castleman’s disease, a rare lymphoproliferative disorder [163]. Theoretically, chronic infections, such as Epstein Barr virus or cytomegalovirus infections, might play a role in the development of the monoclonal gammopathy, as described in multiple myeloma and WD, but the other symptoms and signs of SchS could not all be accounted for by such an infection. Still, it cannot be excluded as it has not been studied.
In view of the phenotypical similarities to CAPS, and the pivotal role of IL-1β overproduction in both CAPS and SchS, the involvement of the inflammasome in the latter is likely. Analysis of NLRP3 mutations was negative in 16 of 21 cases [45, 93, 106, 112, 120, 164] (Fox, Kabashima, Relas: p.c.) In two cases, a V198M variant was detected, but both had unaffected family members carrying this variant [88, 112]. It was also reported in families with classical CAPS phenotypes [112], and in patients with autoinflammatory phenotypes who concurrently had mutations in the Mediterranean fever gene [165] or a low-penetrance mutation in the TNFRSF1A gene [166]. The population allele frequency of this variant is about 0.5%, and as it shows variable expressivity and reduced penetrance, the pathophysiological significance of this variant remains to be determined [112]. In another patient, the Q703K polymorphism was found [127], which is the most common NLRP3 polymorphism with an allele frequency of 5% in healthy Caucasians [167]. This polymorphism is thus not suspected to play a major inflammation-initiating role, but it could modify inflammation under certain circumstances, as it was reported to lead to gain-of-function alterations [168]. Finally, a clear pathophysiological role of NLRP3 has just been described in two patients with IgG variant SchS [164]. Our group found in one patient an F523L mutation that was reported to cause a severe neonatal-onset phenotype in two patients with CAPS [169]. The late onset and milder phenotype in the SchS patient is probably due to the restricted occurrence of the NLRP3 mutation in 10% of the myeloid cells. In our most severely affected SchS patient, we identified a K435E variant in exon 3 of NLRP3 in about 30% of the myeloid cells. This variant is predicted to be pathogenic, and PBMCs of this patient spontaneously produced high amounts of IL-1β, as did the PBMCs of the patient with the known mutation. Interestingly, the variants were not present in T- or B-lymphocytes, keratinocytes, or fibroblasts of these patients, and presented as myeloid-lineage-restricted mosaicism [164]. Previously, somatic mosaicism of NLRP3 was reported in neonatal onset CAPS patients [170–173], but there was no significant difference in mutation frequency between several leukocyte subsets and buccal mucosa, which clearly differentiates these patients from the two SchS cases [170]. In a few SchS patients, no mutations were found in the NLRP1, NOD2, and TNFRSF1A genes [45, 93, 98] (Kabashima, p.c.).
Discussion
Since our previous review in 2007, the number of reported SchS cases has tripled due to increasing awareness of the disease and the remarkable efficacy of IL-1 inhibition. The focus of research on the pathophysiology of SchS has shifted from hypothetical autoimmune properties of the paraprotein to the autoinflammatory nature of excessive IL-1β production. Further, bone abnormalities have been studied in more detail [5], and an international consensus paper specified the diagnostic criteria, recommended therapies and follow-up [1]. Novel techniques such as NGS provide the opportunity for scrutinizing genetic susceptibility.
We still do not know what triggers the chronic systemic inflammation, but the presence of mosaicism of NLRP3 mutations in myeloid cells of two variant SchS patients suggests that (mosaicism of) mutations of genes in the IL-1 pathway may be responsible for disease in other cases [164]. NGS will facilitate the detection of even low percentages of mutant cells. The efficacy of anti-IL-6 treatment in 3 patients who were unresponsive to IL-1 inhibition suggests that in some cases, the defect is downstream of IL-1 [70]. This could include aberrant IL-1R signaling or overproduction of IL-6.
The presence of the monoclonal gammopathy is the most puzzling aspect of SchS. Accumulating data suggest that the monoclonal gammopathy is caused by the systemic inflammation rather than vice versa (see ‘Pathophysiology’). In a landmark paper in 2012, Treon et al. described a L265P mutation in the MyD88 gene in bone marrow samples from 49 out of 54 patients with WM. MyD88 is a crucial adaptor protein for the function of many toll-like receptors and the IL-1R, and the L265P mutation triggered IRAK-mediated NF-κB signaling. In addition, it was associated with a more severe phenotype [174, 175]. As 12% of SchS cases develop WM, the MyD88 L265P mutation might be present in a subset of SchS patients as well.
Intriguingly, in CAPS, long-term excessive IL-1β signaling does not result in a monoclonal gammopathy. I can only speculate that the high age of onset makes the SchS bone marrow cells more vulnerable for malignant conversion. Hypothetically, the presence of mutant (myeloid) cells in the bone marrow of SchS patients produces high local concentrations of IL-1β and IL-6, facilitating the development of a lymphoproliferative disorder.
During the past 42 years, SchS has evolved from an elusive little-known disorder to an autoinflammatory disorder that is recognized by increasing numbers of dermatologists, rheumatologists, allergologists, hematologists and other specialists. Diagnostic criteria have been revisited, effective treatments have been identified (IL-1 (and IL-6) inhibition), as well as the risk of development of lymphoproliferative disorders, and novel genetic techniques have partially shed light on the pathophysiology of SchS. Presumably, during the next decade, (mosaicism of) mutations of genes in the IL-1 pathway in several other cases of SchS will be uncovered. Finally, long-term follow-up will teach us if IL-1 inhibition is capable of preventing the development of lymphoproliferative disorders.
Abbreviations
- Ig:
-
Immunoglobulin
- IL-1β:
-
Interleukin-1 beta
- IL-1Ra:
-
Interleukin-1 receptor antagonist
- MGUS:
-
Monoclonal gammopathy of unknown significance
- NGS:
-
Next-generation sequencing
- NLRP3:
-
Nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3
- POEMS:
-
Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes
- SchS:
-
Schnitzler’s syndrome
- TNFα:
-
Tumor necrosis factor alpha
- VEGF:
-
Vascular endothelial growth factor
- WM:
-
Waldenström’s macroglobulinemia.
References
Simon A, Asli B, Braun-Falco M, De Koning H, Fermand JP, Grattan C, Krause K, Lachmann H, Lenormand C, Martinez-Taboada V, Maurer M, Peters M, Rizzi R, Rongioletti F, Ruzicka T, Schnitzler L, Schubert B, Sibilia J, Lipsker D: Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013, 68 (5): 562-568. 10.1111/all.12129.
Schnitzler L: Lésions urticariennes chroniques permanentes (érythème pétaloïde?). J Dermatol Angers. 1972
Jain T, Offord CP, Kyle RA, Dingli D: Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. 2013, 98: 1581-1585. 10.3324/haematol.2013.084830.
Sokumbi O, Drage LA, Peters MS: Clinical and histopathologic review of Schnitzler syndrome: the mayo clinic experience (1972–2011). J Am Acad Dermatol. 2012, 67: 1289-1295. 10.1016/j.jaad.2012.04.027.
Niederhauser BD, Dingli D, Kyle RA, Ringler MD: Imaging findings in 22 cases of Schnitzler syndrome: characteristic para-articular osteosclerosis, and the “hot knees” sign differential diagnosis. Skeletal Radiol. 2014, 43: 905-915. 10.1007/s00256-014-1857-y.
Adam Z, Krejci M, Pour L, Neubauer J, Prasek J, Hajek R: Schnitzler syndrome–report on a fourteen-year course of the disease and an overview of information on the disease. Vnitr Lek. 2008, 54: 1140-1153.
Akimoto R, Yoshida M, Matsuda R, Miyasaka K, Itoh M: Schnitzler’s syndrome with igg kappa gammopathy. J Dermatol. 2002, 29: 735-738. 10.1111/j.1346-8138.2002.tb00212.x.
Altmeyer PWS: Makroglobulinaemie waldenstrom assoziiert mit einem chronisch rezidivierenden urtikariellen exanthem. Akt Dermatol. 1977, 3: 71-76.
Asahina A, Sakurai N, Suzuki Y, Narushima K: Schnitzler’s syndrome with prominent neutrophil infiltration misdiagnosed as sweet’s syndrome: a typical example of urticarial neutrophilic dermatosis. Clin Exp Dermatol. 2010, 35: e123-e126. 10.1111/j.1365-2230.2009.03746.x.
Asli B, Bienvenu B, Cordoliani F, Brouet JC, Uzunhan Y, Arnulf B, Malphettes M, Rybojad M, Fermand JP: Chronic urticaria and monoclonal igm gammopathy (Schnitzler syndrome): report of 11 cases treated with pefloxacin. Arch Dermatol. 2007, 143: 1046-1050.
Asli B, Brouet JC, Fermand JP: Spontaneous remission of schnitzler syndrome. Ann Allergy Asthma Immunol. 2011, 107: 87-88. 10.1016/j.anai.2011.04.006.
Badawi AH, Gierer S, Fraga GR: Schnitzler syndrome. Allergy Asthma Proc. 2014, 35: 75-77. 10.2500/aap.2014.35.3703.
Barriere H, Schnitzler L, Moulin G, Grolleau Y: chronic urticarial lesions and macroglobulinemia. Apropos of 5 cases. Sem Hop. 1976, 52: 221-227.
Benitez Gutierrez LMP S, Tutor de Ureta P, Yebra Bango M: schnitzler’s syndrome: a case report and literature review of the response to treatment with anakinra. Med Clin (Barc ). 2013, 140: 427-428. 10.1016/j.medcli.2012.09.003.
Bertrand A, Feydy A, Belmatoug N, Fantin B: Schnitzler’s syndrome: 3-year radiological follow-up. Skeletal Radiol. 2007, 36: 153-156.
Besada E, Nossent H: Dramatic response to il1-ra treatment in longstanding multidrug resistant schnitzler’s syndrome: a case report and literature review. Clin Rheumatol. 2010, 29: 567-571. 10.1007/s10067-010-1375-9.
Bhattacharyya J, Mihara K, Morimoto K, Takihara Y, Hide M: Elevated interleukin-18 secretion from monoclonal igm + b cells in a patient with schnitzler syndrome. J Am Acad Dermatol. 2012, 67: e118-e120. 10.1016/j.jaad.2011.10.002.
Billey T, Beldjerd M, Popa L, Lassoued S: Schnitzler syndrome: a dramatic improvement with anakinra. Presse Med. 2010, 39: 1338-1339. 10.1016/j.lpm.2010.07.009.
Blaise S, Vallat JM, Tabaraud F, Bonnetblanc JM: Sensitive chronic inflammatory demyelinating polyradiculoneuropathy in schnitzler’s syndrome. Ann Dermatol Venereol. 2003, 130: 348-351.
Bonnetblanc JM, Drouet M, Laplaud P, Bedane C, Bernard P: Urticaria with macroglobulinaemia. Disease activity associated alterations in immunoglobulins profile and bone marrow hypodiploidy. Dermatologica. 1990, 181: 41-43. 10.1159/000247858.
Borradori L, Rybojad M, Puissant A, Dallot A, Verola O, Morel P: Urticarial vasculitis associated with a monoclonal igm gammopathy: Schnitzler’s syndrome. Br J Dermatol. 1990, 123: 113-118. 10.1111/j.1365-2133.1990.tb01831.x.
Bodar EJ, Simon A, van der Meer JW: Effects of the histone deacetylase inhibitor itf2357 in autoinflammatory syndromes. Mol Med. 2011, 17: 363-368.
Bozeman S, Lewis S: Schnitzler syndrome successfully treated with methotrexate. Ann Allergy Asthma Immunol. 2008, 101: 219-10.1016/S1081-1206(10)60213-X.
Carbone J, Paravisini A, Sarmiento E, Rodriguez-Molina J, Fernandez-Cruz E: Partial response to cyclosporine in a patient with schnitzler’s syndrome. Allergol Immunopathol (Madr). 2007, 35: 71-73. 10.1157/13101341.
Carlesimo M, La Pietra M, Arcese A, Orsini D, Marmo G, Fidanza L, Mari E, Camplone G: Chronic urticaria and igg paraproteinaemia: unusual spectrum of Schnitzler syndrome. Clin Ter. 2011, 162: e85-e87.
Carlioz R, Haas C, Jaubert F, Allouche C, Lowenstein W, Le Jeunne C, Durand H: Waldenstrom’s disease with diffuse large immunoblastic cell lymphoma and chronic urticaria. Ann Med Interne. 1989, 140: 51-52.
Cascavilla N, Bisceglia M, D’Arena G: Successful treatment of schnitzler’s syndrome with anakinra after failure of rituximab trial. Int J Immunopathol Pharmacol. 2010, 23: 633-636.
Cascavilla N, D’Arena G, Dell’Olio M: Schnitzler syndrome. Br J Haematol. 2008, 143: 152-10.1111/j.1365-2141.2008.07287.x.
Chiu H-YT T-F: Long-term control of refractory Schnitzler syndrome with anakinra: a case report. Dermatol Sin. 2010, 28: 82-86. 10.1016/S1027-8117(10)60017-1.
Cianchini G, Colonna L, Bergamo F, Angelo C, Puddu P: Efficacy of psoralen-uv-a therapy in 3 cases of schnitzler syndrome. Arch Dermatol. 2001, 137: 1536-1537.
Claes K, Bammens B, Delforge M, Evenepoel P, Kuypers D, Vanrenterghem Y: Another devastating complication of the Schnitzler syndrome: Aa amyloidosis. Br J Dermatol. 2008, 158: 182-184.
Clauvel JP, Brouet JC, Danon F, Leibowitch M, Seligmann M: Chronic urticaria with monoclonal igm–a report of five cases. Clin Immunol Immunopathol. 1982, 25: 348-353. 10.1016/0090-1229(82)90199-4.
Conlon NP, Hayden P, Barnes L, Doran M, O’Shea F, Feighery C: Schnitzler’s syndrome; a case highlighting the complications of long-standing acquired autoinflammation. Eur J Dermatol. 2014, 24: 405-406.
Cream JJ, Porter D: Urticaria in waldenstrom’s macroglobulinaemia. J R Soc Med. 1979, 72: 858-859.
Crouch R, Akhras V, Sarkany R: Schnitzler’s syndrome: successful treatment with anakinra. Australas J Dermatol. 2007, 48: 178-181. 10.1111/j.1440-0960.2007.00375.x.
Dalle S, Balme B, Sebban C, Pariset C, Berger F, Thomas L: Schnitzler syndrome associated with systemic marginal zone b-cell lymphoma. Br J Dermatol. 2006, 155: 827-829. 10.1111/j.1365-2133.2006.07417.x.
de Castro FR, Masouye I, Winkelmann RK, Saurat JH: Urticarial pathology in schnitzler’s (hyper-igm) syndrome. Dermatology. 1996, 193: 94-99. 10.1159/000246220.
de Kleijn EM, Telgt D, Laan R: Schnitzler’s syndrome presenting as fever of unknown origin (fuo). The role of cytokines in its systemic features. Neth J Med. 1997, 51: 140-142. 10.1016/S0300-2977(97)00041-7.
de Koning HD, Bodar EJ, Simon A, van der Hilst JC, Netea MG, van der Meer JW: Beneficial response to anakinra and thalidomide in schnitzler’s syndrome. Ann Rheum Dis. 2006, 65: 542-544. 10.1136/ard.2005.045245.
de Koning HD, Bodar EJ, van der Meer JW, Simon A: Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007, 37: 137-148. 10.1016/j.semarthrit.2007.04.001.
de Koning HD, Schalkwijk J, van der Ven-Jongekrijg J, Stoffels M, van der Meer JW, Simon A: Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in schnitzler’s syndrome. Ann Rheum Dis. 2012
de Raedt NML: Schnitzler’s syndrome: an unusual case of bone pain with suggestive nuclear imaging features. Médecine Nucléaire. 2009, 33: 655-657.
De Saint-Pierre V, Ehrhart A, Baron D, Plantin P, Le GP: systemic urticaria, sclerosing osteopathy, monoclonal gammopathy (schnitzler syndrome). Apropos of a case. Rev Rhum Mal Osteoartic. 1992, 59: 288-292.
De WS, Lecouvet FE, Malghem J, Jamar F, Lambert M: Schnitzler’s syndrome: an unusual cause of bone pain with suggestive imaging features. AJR Am J Roentgenol. 2000, 175: 1325-1327. 10.2214/ajr.175.5.1751325.
Devlin LA, Wright G, Edgar JD: A rare cause of a common symptom, anakinra is effective in the urticaria of Schnitzler syndrome: a case report. Cases J. 2008, 1: 348-10.1186/1757-1626-1-348.
Dupuy O, Pinede L, Coppere B, Duhaut P, Pasquier J, Ninet J: Schnitzler syndrome with stable course over a 18-year period. Report of a case. Presse Med. 1995, 24: 1402-
Eiling E, Moller M, Kreiselmaier I, Brasch J, Schwarz T: Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol. 2007, 57: 361-364. 10.1016/j.jaad.2007.03.036.
Eiling E, Schroder JO, Gross WL, Kreiselmaier I, Mrowietz U, Schwarz T: The schnitzler syndrome: chronic urticaria and monoclonal gammopathy–an autoinflammatory syndrome?. J Dtsch Dermatol Ges. 2008, 6: 626-631. 10.1111/j.1610-0387.2008.06627.x.
Elsendoorn A, Voultoury J, Gaubert AS, Roblot P, Paccalin M: Chronic urticaria in elderly people: a case of Schnitzler syndrome. J Am Geriatr Soc. 2008, 56: 1152-1153. 10.1111/j.1532-5415.2008.01694.x.
Fairris C, Darvay A: Schnitzler’s syndrome: a diagnostic conundrum. BMJ Case Reports. 2014, 2014: 2014-
Famularo G, Barracchini A, Minisola G: Severe thrombophilia with antiphospholipid syndrome and hyperhomocysteinemia in a patient with schnitzler’s syndrome. Clin Exp Rheumatol. 2003, 21: 366-368.
Famularo G, Minisola G, De SC: Schnitzler’s syndrome: a true auto-inflammatory disorder?. Semin Arthritis Rheum. 2008, 38: 163-10.1016/j.semarthrit.2008.01.002.
Ferrando FJ, Pujol J, Hortells JL, Navarro M, Pinol J, Carapeto FJ: Schnitzler’s syndrome: report of a case with bone osteolysis. J Investig Allergol Clin Immunol. 1994, 4: 203-205.
Florez AF, Gallardo AE, Garcia-Barredo R, Landeras Alvaro RM, Gorrino AM, Martinez-Taboada VM, Fernandez Echevarria MA: Radiological aid to clinical diagnosis of schnitzler’s syndrome: multimodality imaging approach. Clin Rheumatol. 2008, 27: 107-110.
Frischmeyer-Guerrerio PA, Rachamalla R, Saini SS: Remission of schnitzler syndrome after treatment with anakinra. Ann Allergy Asthma Immunol. 2008, 100: 617-619. 10.1016/S1081-1206(10)60064-6.
Gallo R, Sabroe RA, Black AK, Greaves MW: Schnitzler’s syndrome: No evidence for autoimmune basis in two patients. Clin Exp Dermatol. 2000, 25: 281-284. 10.1046/j.1365-2230.2000.00642.x.
Germain P, Fach J, Bui N, Traissac T, Delbrel X, Le BM, Longy-Boursier M: Schnitzler syndrome: a rare cause of systemic urticaria. Rev Med Interne. 2000, 21: 285-289. 10.1016/S0248-8663(00)80049-1.
Giannobile JV: Schnitzler’s Syndrome: American College of Allergy. 2009, Miami, USA: Asthma & Immunology
Gilson M, Abad S, Larroche C, Dhote R: Treatment of schnitzler’s syndrome with anakinra. Clin Exp Rheumatol. 2007, 25: 931-
Gossrau G, Pfeiffer C, Meurer M, Reichmann H, Lampe JB: Schnitzler’s syndrome with neurological findings. J Neurol. 2003, 250: 1248-1250. 10.1007/s00415-003-0169-2.
Govindaraju S, Brochot P, Ringot AC, Paternotte L, Morrone A, Eschard JP, Etienne JC: Chronic urticaria-macroglobulinemia (Schnitzler syndrome): developing to igm myeloma. Apropos of a case. Rev Med Interne. 1993, 14: 780-783.
Gran JT, Midtvedt O, Haug S: A woman with recurrent urticaria, joint pain and fever. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. 2011, 131: 135-136.
Gran JT, Midtvedt O, Haug S, Aukrust P: Treatment of schnitzler’s syndrome with anakinra: report of three cases and review of the literature. Scand J Rheumatol. 2011, 40: 74-79. 10.3109/03009742.2010.493894.
Herraez Albendea MM, Lopez Rodriguez M, Lopez de la Guia A, Canales Albendea MA: Schnitzler syndrome. Reumatologia Clinica. 2013, 9: 384-10.1016/j.reuma.2013.02.005.
Iwafuchi Y, Morita T, Hata K, Nakamura A, Miyazaki S: Schnitzler syndrome complicated by membranous nephropathy. Clin Nephrol. 2012, 78: 497-500. 10.5414/CN107135.
Kastritis E, Katoulis A, Terpos E, Panayiotides I, Gavriatopoulopu M, Dimopopoulos MA: Schnitzler’s syndrome: increased levels of bone formation and angiogenesis factors are reduced after successful pefloxacin treatment. Clin Lymphoma Myeloma. 2008, 8: 359-362. 10.3816/CLM.2008.n.053.
Kieffer C, Cribier B, Lipsker D: Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore). 2009, 88: 23-31. 10.1097/MD.0b013e3181943f5e.
Klemmer N, Lenain P, Balguerie X, Le LX: Effectiveness of anti-il1 in schnitzler’s syndrome. Joint Bone Spine. 2007, 74: 509-510. 10.1016/j.jbspin.2006.11.018.
Kluger N, Riviere S, Guillot B, Bessis D: Efficacy of interleukin 1 receptor antagonist (anakinra) on a refractory case of schnitzler’s syndrome. Acta Derm Venereol. 2008, 88: 287-288.
Krause K, Feist E, Fiene M, Kallinich T, Maurer M: Complete remission in 3 of 3 anti-il-6-treated patients with schnitzler syndrome. J Allergy Clin Immunol. 2011
Krause K, Weller K, Stefaniak R, Wittkowski H, Altrichter S, Siebenhaar F, Zuberbier T, Maurer M: Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy. 2012, 67: 943-950. 10.1111/j.1398-9995.2012.02843.x.
Kropp J-DC BM: Urticaria-vasculitis und schnitzler-syndrom. Allergologie. 1994, 17: S17-S20.
Kuenzli S, Buchet S, Saurat JH: Successful treatment of schnitzler’s syndrome with interferon alfa-2b. Dermatology. 2002, 205: 74-10.1159/000063142.
Kurian A, Lee JK, Vadas P: Schnitzler syndrome with cold-induced urticaria. J Dermatological Case Reports. 2010, 4: 50-53.
Lagana B, Podesta E, Picchianti DA, Salerno G, Di RR, D’Amelio R: Schnitzler’s syndrome with biclonal gammopathy successfully treated with hydroxychloroquine and low dose steroids. Clin Exp Rheumatol. 2008, 26: 1161-
Larocca CA, McEvoy JW, Ellis CL, Junkins-Hopkins J, Kolb T, Baer AN, Garibaldi BT: Schnitzler’s syndrome associated with pancreatitis: a disease of il-1 dysregulation. Clin Rheumatol. 2012, 31: 169-174. 10.1007/s10067-011-1804-4.
Launay D, Dutoit-Lefevre V, Faure E, Robineau O, Hauspie C, Sobanski V, Hachulla E, Labalette M, Hatron PY, Dubucquoi S: Effect of in vitro and in vivo anakinra on cytokines production in schnitzler syndrome. PLoS One. 2013, 8: e59327-10.1371/journal.pone.0059327.
Lautenschlager S, Itin PH: Schnitzler syndrome. Hautarzt. 1993, 44: 781-784.
Lauwers A, Chouvy V, Mosnier JF, Misery L, Alexandre C: A case of schnitzler’s syndrome with nodular regenerative hyperplasia of the liver. Rev Rhum Engl Ed. 1999, 66: 281-283.
Lazarevic V, Markuszewska A, Stenberg B, Haggroth J: Schnitzler syndrome–unknown, rare but treatable. Lakartidningen. 2008, 105: 3348-3351.
Lebbe C, Rybojad M, Klein F, Oksenhendler E, Catala M, Danon F, Morel P: Schnitzler’s syndrome associated with sensorimotor neuropathy. J Am Acad Dermatol. 1994, 30: 316-318. 10.1016/S0190-9622(94)70031-1.
Lecompte M, Blais G, Bisson G, Maynard B: Schnitzler’s syndrome. Skeletal Radiol. 1998, 27: 294-296. 10.1007/s002560050385.
Lee KY, Grattan CE: Intracostal neuralgia as a previously undescribed symptom of schnitzler’s syndrome. Br J Dermatol. 2012, 167: 1392-1393. 10.1111/j.1365-2133.2012.11057.x.
Lim W, Shumak KH, Reis M, Perez-Ordonez B, Sauder D, Fam A, Imrie KR: Malignant evolution of schnitzler’s syndrome–chronic urticaria and igm monoclonal gammopathy: report of a new case and review of the literature. Leuk Lymphoma. 2002, 43: 181-186. 10.1080/10428190210181.
Lin RY, Jagannath S: Treatment of schnitzler’s syndrome with colchicine: a case report. Internet J Asthma Allergy Immunol. 2004, 4: 1-
Lipsker D, Veran Y, Grunenberger F, Cribier B, Heid E, Grosshans E: The schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore). 2001, 80: 37-44. 10.1097/00005792-200101000-00004.
Loock J: Schnitzler syndrome. Z Rheumatol. 2012, 71: 181-186. 10.1007/s00393-011-0857-8.
Loock J, Lamprecht P, Timmann C, Mrowietz U, Csernok E, Gross WL: Genetic predisposition (nlrp3 v198m mutation) for il-1-mediated inflammation in a patient with schnitzler syndrome. J Allergy Clin Immunol. 2010, 125: 500-502. 10.1016/j.jaci.2009.10.066.
Lozano GF, Aguirre PA, Rivera Civico JM, Nevado SM: The schnitzler syndrome. A case report. Med Clin (Barc). 1999, 112: 158-159.
Machet L, Vaillant L, Machet MC, Esteve E, De Muret A, Khallouf R, Arbeille B, Muller C, Lorette G: Schnitzler’s syndrome (urticaria and macroglobulinemia) associated with pseudoxanthoma elasticum. Acta Derm Venereol. 1992, 72: 22-24.
Machet L, Vaillant L, Machet MC, Reisenleiter M, Goupille P, Lorette G: Schnitzler’s syndrome (urticaria and macroglobulinemia): evolution to waldenstrom’s disease is not uncommon. Acta Derm Venereol. 1996, 76: 413-
Markotic A, Skerk V, Cvetko KL, Beus A: Is the clinical picture of schnitzler syndrome always schnitzler syndrome?. Clin Exp Rheumatol. 2009, 27: 507-509.
Martinez-Taboada VM, Fontalba A, Blanco R, Fernandez-Luna JL: Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum. 2005, 52: 2226-2227. 10.1002/art.21101.
Migliorini P, Corso ID, Tommasi C, Boraschi D: Free circulating interleukin-18 is increased in Schnitzler syndrome: a new autoinflammatory disease?. Eur Cytokine Netw. 2009, 20: 108-111.
Mittal N, Renaut P, Sharma R, Robbie M: Gastrointestinal amyloidosis associated with schnitzler’s syndrome. Pathology. 2013, 45: 424-426. 10.1097/PAT.0b013e328360e00c.
Morita A, Sakakibara S, Yokota M, Tsuji T: A case of urticarial vasculitis associated with macroglobulinemia (schnitzler’s syndrome). J Dermatol. 1995, 22: 32-35.
Murota H, Shoda Y, Ishibashi T, Sugahara H, Matsumura I, Katayama I: Improvement of recurrent urticaria in a patient with schnitzler syndrome associated with b-cell lymphoma with combination rituximab and radiotherapy. J Am Acad Dermatol. 2009, 61: 1070-1075. 10.1016/j.jaad.2008.12.040.
Nakajima K, Takahashi M, Yamamoto M, Takahashi A, Sano S, Kodama H, Arima K, Ida H: Successful treatment of schnitzler syndrome with cyclosporine. Int J Dermatol. 2014, 53: e361-e363. 10.1111/ijd.12300.
Nilsson G: mb schnitzler in sweden--yet another case. Lakartidningen. 2008, 105: 3672-
Oborilova A, Adam Z: Schnitzler’s syndrome. Vnitr Lek. 1998, 44: 423-427.
Pacchiano GDB A, Palladini G, Merlini G: "Schnitzler syndrome: Caso clinico raro": V Congresso interregionale Societa Italiana di Nefrologia. 2004
Pascual-Lopez M, Hernandez-Nunez A, Sanchez-Perez J, Fernandez-Herrera J, Garcia-Diez A: Schnitzler’s syndrome with monoclonal igg kappa gammopathy: good response to cyclosporin. J Eur Acad Dermatol Venereol. 2002, 16: 267-270. 10.1046/j.1468-3083.2002.00459.x.
Patel S, Sindher S, Jariwala S, Hudes G: Chronic urticaria with monoclonal igg gammopathy: a clinical variant of Schnitzler syndrome?. Ann Allergy Asthma Immunol. 2012, 109: 147-148. 10.1016/j.anai.2012.06.001.
Perrin F, Neel A, Graveleau J, Ruellan AL, Masseau A, Hamidou M: Two cases of anakinra-induced neutropenia during auto-inflammatory diseases: drug reintroduction can be successful. Presse Med. 2014, 43: 319-321. 10.1016/j.lpm.2013.06.028.
Peterlana D, Puccetti A, Tinazzi E, Simeoni S, Lunardi C: Schnitzler’s syndrome treated successfully with intravenous pulse cyclophosphamide. Scand J Rheumatol. 2005, 34: 328-330. 10.1080/03009740510017733.
Pizzirani C, Falzoni S, Govoni M, La CR, Donadei S, Di VF, Trotta F, Lo MA: Dysfunctional inflammasome in schnitzler’s syndrome. Rheumatology (Oxford). 2009, 48: 1304-1308. 10.1093/rheumatology/kep222.
Puddu P, Cianchini G, Girardelli CR, Colonna L, Gatti S, de Pita O: Schnitzler’s syndrome: report of a new case and a review of the literature. Clin Exp Rheumatol. 1997, 15: 91-95.
Pujol RM, Barnadas MA, Brunet S, de Moragas JM: Urticarial dermatosis associated with waldenstrom’s macroglobulinemia. J Am Acad Dermatol. 1989, 20: 855-857. 10.1016/S0190-9622(89)80127-6.
Ramadan KM, Eswedi HA, El-Agnaf MR: Schnitzler syndrome: a case report of successful treatment using the anti-cd20 monoclonal antibody rituximab. Br J Dermatol. 2007, 156: 1072-1074. 10.1111/j.1365-2133.2007.07799.x.
Rizzi R, Curci P, Rinaldi E, Rinaldi F, Cimmino A, Ricco R, Specchia G, Liso V: Schnitzler’s syndrome: monoclonal gammopathy associated with chronic urticaria. Acta Haematol. 2008, 120: 1-4. 10.1159/000143499.
Romanini E, Magaletti M, D’Aviera L, Antonelli S: Severe heterotopic ossification after total hip arthroplasty in schnitzler syndrome. Hip Int. 2010, 20: 284-286.
Rowczenio DM, Trojer H, Russell T, Baginska A, Lane T, Stewart NM, Gillmore JD, Hawkins PN, Woo P, Mikoluc B, Lachmann HJ: Clinical characteristics in subjects with nlrp3 v198m diagnosed at a single uk center and a review of the literature. Arthritis Res Ther. 2013, 15: R30-10.1186/ar4171.
Ryan JG, de Koning HD, Beck LA, Booty MG, Kastner DL, Simon A: Il-1 blockade in Schnitzler syndrome: Ex vivo findings correlate with clinical remission. J Allergy Clin Immunol. 2008, 121: 260-262. 10.1016/j.jaci.2007.09.021.
Rybojad M, Moraillon I, Cordoliani F, Lebbe C, Baccard M, Flageul B, Weiss L, Morel P: schnitzler syndrome with genetic c4 deficiency. 2 cases. Ann Dermatol Venereol. 1993, 120: 783-785.
Saiz E, Galvez J, Mora A, Contreras L: Anakinra and schnitzler’s syndrome. Med Clin (Barc). 2008, 130: 358-359. 10.1157/13117359.
Sanchez G, Ano M, Garcia-Aviles C, Dieguez I: Schnitzler syndrome: a case study. J Investig Allergol Clin Immunol. 2000, 10: 41-43.
Saurat JH, Schifferli J, Steiger G, Dayer JM, Didierjean L: Anti-interleukin-1 alpha autoantibodies in humans: characterization, isotype distribution, and receptor-binding inhibition–higher frequency in schnitzler’s syndrome (urticaria and macroglobulinemia). J Allergy Clin Immunol. 1991, 88: 244-256. 10.1016/0091-6749(91)90335-L.
Schartz NE, Buder S, Sperl H, Audring H, Paus R, Tebbe B, Kruger K, Sterry W: Report of a case of schnitzler’s syndrome treated successfully with interferon alpha 2b. Dermatology. 2002, 205: 54-56. 10.1159/000063139.
Schneider SW, Gaubitz M, Luger TA, Bonsmann G: Prompt response of refractory schnitzler syndrome to treatment with anakinra. J Am Acad Dermatol. 2007, 56: S120-S122. 10.1016/j.jaad.2006.05.057.
Schrodl K, Nigg A, Treitl M, Flaig M, Jansson A, Schulze-Koops H, Reindl C: Schnitzler’s disease as an important differential diagnosis of chronic recurrent multifocal osteomyelitis: a case report. Case Reports in Rheumatology. 2012, 2012: 318791-
Schuster C, Kranke B, Aberer E, Arbab E, Sturm G, Aberer W: Schnitzler syndrome: response to anakinra in two cases and a review of the literature. Int J Dermatol. 2009, 48: 1190-1194. 10.1111/j.1365-4632.2009.04151.x.
Sediva A, Polouckova A, Podrazil M, Fronkova E, Kalina T: Characterization of the b-cell compartment in a patient with schnitzler syndrome. Scand J Rheumatol. 2011, 40: 158-160. 10.3109/03009742.2010.507219.
Shibolet O, Schatz O, Krieger M, Maly A, Caraco Y: Schnitzler syndrome: chronic urticaria, fever and immunoglobulin m monoclonal gammopathy. Isr Med Assoc J. 2002, 4: 466-467.
Singh B, Ezziddin S, Rabe E, Biersack HJ, Ahmadzadehfar H: Bone scan appearance supportive of schnitzler’s syndrome: report of two new cases. Clin Nucl Med. 2006, 31: 151-153. 10.1097/01.rlu.0000200783.42211.b7.
Soubrier M: Schnitzler syndrome. Joint Bone Spine. 2008, 75: 263-266. 10.1016/j.jbspin.2007.07.014.
Sperr WR, Natter S, Baghestanian M, Smolen J, Wolff K, Binder BR, Muller MR, Lechner K, Valenta R, Valent P: Autoantibody reactivity in a case of schnitzler’s syndrome: evidence for a th1-like response and detection of igg2 anti-fcepsilonrialpha antibodies. Int Arch Allergy Immunol. 2000, 122: 279-286. 10.1159/000024410.
Szturz P, Adam Z, Klabusay M, Fojtik Z, Kadanka Z, Stehlikova O, Chovancova J, Kalvodova L, Corbova D, Stary K, Neubauer J, Prasek J, Koukalova R, Rehak Z, Hajek R, Mayer J: Schnitzler syndrome: Case report, the experience with glucocorticoid and anakinra (kineret) therapies and monitoring of systemic cytokine response. Vnitr Lek. 2011, 57: 97-112.
Szturz P, Adam Z, Sediva A, Fojtik Z, Corbova D, Neubauer J, Prasek J, Hajek R, Mayer J: Schnitzler syndrome: Diagnostics and treatment. Klinicka onkologie: casopis Ceske a Slovenske onkologicke spolecnosti. 2011, 24: 271-277.
Szturz P, Sediva A, Zurek M, Adam Z, Stork J, Cermakova Z, Steyerova P, Vokacova A, Hrbek J, Sykora M, Spicka I, Mechl Z, Mayer J: Anakinra treatment in schnitzler syndrome - results of the first retrospective multicenter study in six patients from the czech republic. Klinicka onkologie: casopis Ceske a Slovenske onkologicke spolecnosti. 2014, 27: 111-126.
Tanneberger O, Buchner S, Zimmerli LU: Schnitzler’s syndrome with urticaria vasculitis. Internist (Berl). 2007, 48: 1432-1435. 10.1007/s00108-007-1956-0.
Terpos E, Asli B, Christoulas D, Brouet JC, Kastritis E, Rybojad M, Bengoufa D, Dimopoulos MA, Fermand JP: Increased angiogenesis and enhanced bone formation in patients with igm monoclonal gammopathy and urticarial skin rash: new insight into the biology of Schnitzler syndrome. Haematologica. 2012, 97: 1699-1703. 10.3324/haematol.2012.067306.
Thonhofer R, Uitz E, Graninger W: Schnitzler’s syndrome–exacerbation after anti-tnf treatment. Rheumatology (Oxford). 2007, 46: 1041-1042. 10.1093/rheumatology/kem048.
Tinazzi E, Puccetti A, Patuzzo G, Sorleto M, Barbieri A, Lunardi C: Schnitzler syndrome, an autoimmune-autoinflammatory syndrome: report of two new cases and review of the literature. Autoimmun Rev. 2011, 10: 404-409. 10.1016/j.autrev.2011.01.003.
Tomkova H, Shirafuji Y, Arata J: Schnitzler’s syndrome versus adult onset still’s disease. Eur J Dermatol. 1998, 8: 118-121.
Vandenhende MA, Bentaberry F, Morlat P, Bonnet F: Anakinra: an effective treatment in the Schnitzler syndrome. Joint Bone Spine. 2011, 78: 636-637. 10.1016/j.jbspin.2011.03.021.
Vanderschueren S, Knockaert D: Canakinumab in schnitzler syndrome. Semin Arthritis Rheum. 2013, 42 (4): 413-416. 10.1016/j.semarthrit.2012.06.003.
Varella TC, Nishimura MY, Machado MC, de Moraes-Vasconcelos D, Rivitti EA: Schnitzler’s syndrome without monoclonal gammopathy. Acta Derm Venereol. 2005, 85: 272-273.
Verret JL, Leclech C, Rousselet MC, Hurez D, Schnitzler L: schnitzler syndrome and waldenstrom disease. Fatal outcome of the original case. Ann Dermatol Venereol. 1993, 120: 459-460.
Volz T, Wolbing F, Fischer J, Braun M, Maggoschitz I, Schaller M, Berneburg M, Biedermann T: Dermal interleukin-1 expression and effective and long-lasting therapy with interleukin-1 receptor antagonist anakinra in schnitzler syndrome. Acta Derm Venereol. 2012, 92: 393-394. 10.2340/00015555-1307.
Wastiaux H, Barbarot S, Gagey-Caron V, Berthelot JM, Hamidou M, Stalder JF: Schnitzler syndrome: a dramatic improvement with anakinra. J Eur Acad Dermatol Venereol. 2009, 23: 85-87. 10.1111/j.1468-3083.2008.02708.x.
Welsh B, Tate B: Schnitzler’s syndrome: report of a case with progression to waldenstrom’s macroglobulinaemia. Australas J Dermatol. 1999, 40: 201-203. 10.1046/j.1440-0960.1999.00361.x.
Wendling D, Prati C, Hoen B, Toussirot E, Streit G, Dumoulin G: Symptomatic treatment of a case of schnitzler’s syndrome with pamidronate. Joint Bone Spine. 2008, 75: 507-508. 10.1016/j.jbspin.2007.12.005.
Westhoff TH, Zidek W, Uharek L, Steinhoff-Georgieva J, van der Giet M: Impairment of renal function in schnitzler’s syndrome. J Nephrol. 2006, 19: 660-663.
Wilhelmi ML Ocana Wilhelmi J, Ocana Sierra J: Schnitzler’s syndrome and pseudoxanthoma elasticum: 3rd Congress of EADV. Copenhagen. 1993, 20: 159-
Winckelmann G, Nagel HG, Maier R, Reuther G: The schnitzler syndrome as a cause of recurrent fever of unknown origin. Dtsch Med Wochenschr. 1996, 121: 860-864. 10.1055/s-2008-1043079.
Worm M, Kolde G: Schnitzler’s syndrome: successful treatment of two patients using thalidomide. Br J Dermatol. 2003, 148: 601-602. 10.1046/j.1365-2133.2003.05209_9.x.
Krause K, Grattan CE, Bindslev-Jensen C, Gattorno M, Kallinich T, de Koning HD, Lachmann HJ, Lipsker D, Navarini AA, Simon A, Traidl-Hoffmann C, Maurer M: How not to miss autoinflammatory diseases masquerading as urticaria. Allergy. 2012, 67: 1465-1474.
Lipsker D: The schnitzler syndrome. Orphanet J Rare Dis. 2010, 5: 38-10.1186/1750-1172-5-38.
Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, Broussolle C, Seve P: Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014, 93: 91-99. 10.1097/MD.0000000000000021.
Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, Ferrarini M, Abdel-Wahab O, Heaney ML, Scheel PJ, Feeley NK, Ferrero E, McClain KL, Vaglio A, Colby T, Arnaud L, Haroche J: Consensus guidelines for the diagnosis and clinical management of erdheim-chester disease. Blood. 2014, 124: 483-492. 10.1182/blood-2014-03-561381.
Dispenzieri A: Poems syndrome: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014, 89: 214-223.
Terpos E, Anagnostopoulos A, Kastritis E, Bamias A, Tsionos K, Dimopoulos MA: Abnormal bone remodelling and increased levels of macrophage inflammatory protein-1 alpha (mip-1alpha) in waldenstrom macroglobulinaemia. Br J Haematol. 2006, 133: 301-304. 10.1111/j.1365-2141.2006.06017.x.
Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T, Gudjonsson JE, Kang HM, Allen MH, McManus R, Novelli G, Samuelsson L, Schalkwijk J, Stahle M, Burden AD, Smith CH, Cork MJ, Estivill X, Bowcock AM, Krueger GG, Weger W, Worthington J, Tazi-Ahnini R, Nestle FO, Hayday A, Hoffmann P, Winkelmann J: Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012, 44: 1341-1348. 10.1038/ng.2467.
de Koning HD, Schalkwijk J, van der Meer JW, Simon A: Successful canakinumab treatment identifies il-1beta as a pivotal mediator in schnitzler syndrome. J Allergy Clin Immunol. 2011, 128: 1352-1354. 10.1016/j.jaci.2011.05.023.
Lipsker D, Spehner D, Drillien R, Schmitt P, Cribier B, Heid E, Humbel RL, Grosshans E: Schnitzler syndrome: heterogeneous immunopathological findings involving igm-skin interactions. Br J Dermatol. 2000, 142: 954-959. 10.1046/j.1365-2133.2000.03477.x.
Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, D’Osualdo A, Buoncompagni A, Alpigiani MG, Alessio M, Martini A, Rubartelli A: Pattern of interleukin-1beta secretion in response to lipopolysaccharide and atp before and after interleukin-1 blockade in patients with cias1 mutations. Arthritis Rheum. 2007, 56: 3138-3148. 10.1002/art.22842.
Dinarello CA: Interleukin-1beta and the autoinflammatory diseases. N Engl J Med. 2009, 360: 2467-2470. 10.1056/NEJMe0811014.
Dybowski F, Sepp N, Bergerhausen HJ, Braun J: Successful use of anakinra to treat refractory schnitzler’s syndrome. Clin Exp Rheumatol. 2008, 26: 354-357.
Treudler R, Kauer F, Simon JC: Striking effect of the il-1 receptor antagonist anakinra in chronic urticarial rash with polyclonal increase in iga and igg. Acta Derm Venereol. 2007, 87: 280-281.
Husak R, Nestoris S, Goerdt S, Orfanos CE: Severe course of chronic urticaria, arthralgia, fever and elevation of erythrocyte sedimentation rate: Schnitzler’s syndrome without monoclonal gammopathy?. Br J Dermatol. 2000, 142: 581-582. 10.1046/j.1365-2133.2000.03394.x.
Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F, Janz S: Preclinical validation of interleukin 6 as a therapeutic target in multiple myeloma. Immunol Res. 2014, 59: 188-202. 10.1007/s12026-014-8528-x.
Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, Geyer SM, Campbell ME, Kyle RA, Rajkumar SV, Greipp PR, Kline MP, Xiong Y, Moon-Tasson LL, Donovan KA: Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009, 84: 114-122. 10.4065/84.2.114.
Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T: Humanized anti-interleukin-6 receptor antibody treatment of multicentric castleman disease. Blood. 2005, 106: 2627-2632. 10.1182/blood-2004-12-4602.
de Koning HD, van Gijn ME, Stoffels M, Jongekrijg J, Zeeuwen PL, Elferink MG, Nijman IJ, Jansen PA, Neveling K, van der Meer JW, Schalkwijk J, Simon A: Myeloid lineage-restricted somatic mosaicism of nlrp3 mutations in patients with variant schnitzler syndrome. J Allergy Clin Immunol. 2014, (e-pub ahead of print)
Singh-Grewal D, Chaitow J, Aksentijevich I, Christodoulou J: Coexistent mefv and cias1 mutations manifesting as familial mediterranean fever plus deafness. Ann Rheum Dis. 2007, 66: 1541-10.1136/ard.2007.075655.
Touitou I, Perez C, Dumont B, Federici L, Jorgensen C: Refractory auto-inflammatory syndrome associated with digenic transmission of low-penetrance tumour necrosis factor receptor-associated periodic syndrome and cryopyrin-associated periodic syndrome mutations. Ann Rheum Dis. 2006, 65: 1530-1531. 10.1136/ard.2006.054312.
Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD, Moak Z, Chuang M, Austin F, Goldbach-Mansky R, Hoffman HM, Kastner DL: The clinical continuum of cryopyrinopathies: novel cias1 mutations in north american patients and a new cryopyrin model. Arthritis Rheum. 2007, 56: 1273-1285. 10.1002/art.22491.
Verma D, Sarndahl E, Andersson H, Eriksson P, Fredrikson M, Jonsson JI, Lerm M, Soderkvist P: The q705k polymorphism in nlrp3 is a gain-of-function alteration leading to excessive interleukin-1beta and il-18 production. PLoS One. 2012, 7: e34977-10.1371/journal.pone.0034977.
Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, Rosenberg HF, Austin F, Remmers EF, Balow JE, Rosenzweig S, Komarow H, Shoham NG, Wood G, Jones J, Mangra N, Carrero H, Adams BS, Moore TL, Schikler K, Hoffman H, Lovell DJ, Lipnick R, Barron K, O’Shea JJ, Kastner DL: De novo cias1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (nomid): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46: 3340-3348. 10.1002/art.10688.
Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, Nishikomori R, Morimoto T, Kambe N, Goldbach-Mansky R, Aksentijevich I, de Saint BG, Neven B, van Gijn M, Frenkel J, Arostegui JI, Yague J, Merino R, Ibanez M, Pontillo A, Takada H, Imagawa T, Kawai T, Yasumi T, Nakahata T, Heike T: High incidence of nlrp3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an international multicenter collaborative study. Arthritis Rheum. 2011, 63: 3625-3632. 10.1002/art.30512.
Arostegui JI, Lopez Saldana MD, Pascal M, Clemente D, Aymerich M, Balaguer F, Goel A, Fournier del Castillo C, Rius J, Plaza S, Lopez Robledillo JC, Juan M, Ibanez M, Yague J: A somatic nlrp3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: Novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 2010, 62: 1158-1166. 10.1002/art.27342.
Saito M, Nishikomori R, Kambe N, Fujisawa A, Tanizaki H, Takeichi K, Imagawa T, Iehara T, Takada H, Matsubayashi T, Tanaka H, Kawashima H, Kawakami K, Kagami S, Okafuji I, Yoshioka T, Adachi S, Heike T, Miyachi Y, Nakahata T: Disease-associated cias1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008, 111: 2132-2141. 10.1182/blood-2007-06-094201.
Saito M, Fujisawa A, Nishikomori R, Kambe N, Nakata-Hizume M, Yoshimoto M, Ohmori K, Okafuji I, Yoshioka T, Kusunoki T, Miyachi Y, Heike T, Nakahata T: Somatic mosaicism of cias1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2005, 52: 3579-3585. 10.1002/art.21404.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, Arcaini L, Pinkus GS, Rodig SJ, Sohani AR, Harris NL, Laramie JM, Skifter DA, Lincoln SE, Hunter ZR: Myd88 l265p somatic mutation in waldenstrom’s macroglobulinemia. N Engl J Med. 2012, 367: 826-833. 10.1056/NEJMoa1200710.
Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR: Somatic mutations in myd88 and cxcr4 are determinants of clinical presentation and overall survival in waldenstrom macroglobulinemia. Blood. 2014, 123: 2791-2796. 10.1182/blood-2014-01-550905.
Acknowledgments
I am grateful for the contribution of additional cases by Dr. K. Kabashima, Department of Dermatology, Kyoto University Graduate School of Medicine, Kyoto, Japan; Dr. A. van Hoof, Department of Internal Medicine, General Hospital Sint-Jan, Brugge, Belgium; Dr. E. Schiff, Department of Internal Medicine, Bnai Zion Medical Center, Haifa, Israel; Dr. A-L Björk, Department of Internal Medicine, Näl, Trollhättan, Sweden; Dr. D.A. Fox, Department of Internal Medicine, University of Michigan, USA; Dr. L.M. Croot, Department of Rheumatology, Barnsley Hospital NHS Foundation Trust, Barnsley, United Kingdom; Dr. F. Frerichs, Department of Internal Medicine, Alkmaar Medical Center, Alkmaar, The Netherlands; and Dr. H. Relas, Department of Internal Medicine, University of Helsinki, Finland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author received research support from Novartis for an investigator-initiated trial in 2010 and took part in 2012 in an international workshop on Schnitzler’s syndrome that was sponsored by SOBI.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
de Koning, H.D. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy 4, 41 (2014). https://doi.org/10.1186/2045-7022-4-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-7022-4-41