
Abstract
Background
Among the 20 natural amino acids histidine is the most active and versatile member that plays the multiple roles in protein interactions, often the key residue in enzyme catalytic reactions. A theoretical and comprehensive study on the structural features and interaction properties of histidine is certainly helpful.
Results
Four interaction types of histidine are quantitatively calculated, including: (1) Cation-π interactions, in which the histidine acts as the aromatic π-motif in neutral form (His), or plays the cation role in protonated form (His+); (2) π-π stacking interactions between histidine and other aromatic amino acids; (3) Hydrogen-π interactions between histidine and other aromatic amino acids; (4) Coordinate interactions between histidine and metallic cations. The energies of π-π stacking interactions and hydrogen-π interactions are calculated using CCSD/6-31+G(d,p). The energies of cation-π interactions and coordinate interactions are calculated using B3LYP/6-31+G(d,p) method and adjusted by empirical method for dispersion energy.
Conclusions
The coordinate interactions between histidine and metallic cations are the strongest one acting in broad range, followed by the cation-π, hydrogen-π, and π-π stacking interactions. When the histidine is in neutral form, the cation-π interactions are attractive; when it is protonated (His+), the interactions turn to repulsive. The two protonation forms (and pKa values) of histidine are reversibly switched by the attractive and repulsive cation-π interactions. In proteins the π-π stacking interaction between neutral histidine and aromatic amino acids (Phe, Tyr, Trp) are in the range from -3.0 to -4.0 kcal/mol, significantly larger than the van der Waals energies.

Similar content being viewed by others

Introduction
The 20 natural amino acids are the building blocks of three dimensional protein structures. Each of them has its unique structural characters and physicochemical properties, and plays irreplaceable role in biochemistry and biological functions of proteins. Among the 20 natural amino acids histidine (His, H) may be the most versatile actor in the protein architectures and bioactivities [1–4]. The versatility of histidine in molecular interactions arises from its unique molecular structure [5]. The side chain imidazole of histidine is an aromatic motif; an ionizable group with the acidic ionization constant around pKa=6.5; a coordinating ligand of metallic cations (for example, Ca2+ and Zn2+); and a hydrogen bond donor and acceptor. The unique structure of histidine makes it plays multiple roles in the molecular interactions. The roles of histidine in molecular interactions are even complicated by pH condition and its two protonation forms, the neutral form and the protonated form [6]. Figure 1 shows the optimized structures of histidine in the neutral form (A) and in the protonated form (B). The functional groups of histidine and their interaction functions are illustrated in Figure 1.

The optimized geometries of amino acids and the structures of four interaction types. (A) The optimized geometry and the interaction contributors of histidine (His). (B) The optimized geometry of protonated histidine (His+). The protonated imidazole is an organic cation in the cation-π interactions with other aromatic amino acids. (C) The protonated Lys is simplified as CH3NH3+. (D) The protonized Arg+ is simplified as CHNH2NH2+. (E) The protonated His+ is simplified as the protonated imidazole C3N2H5+. (F) The interaction structure of cation-π interaction. The cation could be at the upside or downside of the aromatic plane. (G) The interaction structure of π-π stacking interaction between Phe and His (simplified as the imidazole). In the π-π stacking interaction the two aromatic planes are stacking in parallel. (H) The hydrogen-π interaction structure between His (imidazole) and aromatic motif. The polar hydrogen atom of His perpendicularly points to the π-plane. (I) The coordinate bonding interaction structure between His and metallic cation.
The molecular interactions of histidine with other amino acids and metallic cations in proteins can be classified into the following five types. (1) Cation-π interaction[7–9]. The side chain imidazole of His is an aromatic ring. Histidine can take part in the cation-π interactions as the aromatic motif with metallic cations or organic cations (protonated amino acids, Lys+ and Arg+) [7, 9–11]. On the other hand, the protonated His+ is an organic cation, which can join the cation-π interactions as an organic cation with other aromatic amino acids (Phe, Tyr, and Trp) [12–16]. (2) π-π stacking interaction[17–20]. The imidazole structure of histidine side chain is a conjugative π-plane, which can make π-π stacking interactions with the aromatic side chains of other amino acids (Phe, Tyr, and Trp) [20, 21]. (3) hydrogen-π interaction[22, 23]. The polar hydrogen atom of histidine can form hydrogen-π bond with other aromatic amino acids in ‘T’ orientation. (4) Coordinate bond interaction[3, 24, 25]. The basic nitrogen atom in the imidazole of histidine has a lone electron pair that make it a coordinate ligand of metallic cations, such as Zn2+ and Ca2+[26, 27]. (5) Hydrogen bond interaction[28–31]. The polar hydrogen atom of the imidazole is a hydrogen-bond donor, and the basic nitrogen atom is a hydrogen-bond acceptor.
In protein interactions the roles of histidine are complicated by the five interaction types and two protonation forms. The unique behaviors of histidine have been discussed in literatures from different aspects [7, 32]. However, the quantitative interaction energies of five interaction types and the factors affecting the interaction energies still need more investigations. The influences of five interaction types to the protonation form (and pKa value) of histidine are still unclear. In this study the multiple roles of histidine in molecular interactions are quantitatively studied using quantum chemical calculations, and the factors, which influence the interaction energies and pKa value of histidine in proteins, are analyzed in detail.
Methods and materials
Quantum chemistry is a powerful tool in chemical and biochemical studies [33, 34]. The cation-π and π-π stacking interactions have been studied by many authors using several QM methods [35, 36]. The DFT method B3LYP is a widely used method in organic chemistry and biochemistry because of its higher accuracy and less computational workload [37, 38]. However, in past twenty years the DFT methods were found to fail in describing the molecular dispersion interactions [39–42]. On the other hand, the more advanced coupled-cluster with single and double CCSD and triple excitations CCSD(T) methods [43–46] are able to evaluate the dispersion interaction well. However, such sophisticated methods take much more computer cpu time than that of the DFT methods. A computational comparison of the three methods (B3LYP, CCSD, and CCSD(T)) to the five interaction types (mentioned in the introduction section) is performed. The results of the comparison calculations are listed in Table 1.
From the data in Table 1 we find that the DFT method B3LYP cannot yield attractive interaction energy in C6H6-C6H6 π-π stacking interaction, completely failing in describing the π-π stacking interactions, which are dispersion dominated phenomenon. On the other hand the higher level method CCSD calculation produces attractive C6H6-C6H6 π-π stacking energy −1.883 kcal/mol. In this study the energy differences between B3LYP and CCSD are used as the dispersion contribution in the molecular interaction energies. In the hydrogen-π interaction more than 50% interaction energy is from the dispersion contribution. The interaction energies of other three interaction types (cation-π interactions, common hydrogen bond interactions, and metal cation-His coordinate interactions), obtained by using B3LYP and CCSD methods, have no remarkable difference. In above three interaction types the electrostatic (charge) interactions and orbital coordinate interactions make the main contributions, and the contribution of dispersion interactions are less than 10% [8, 41, 42]. In the C6H6CH3-H3O+ cation-π calculations the CPU time of three methods (B3LYP, CCSD, and CCSD(T)) are 1.08 hours, 50 days, and 86 days, respectively. However, the energy difference of cation-π interaction between B3LYP and CCSD(T) is only 1.08 kcal/mol, less than 6%.
In this study the π-π stacking interactions and the hydrogen-π interactions are calculated using CCSD/6-31+G(d,p) method, and the B3LYP/6-31+G(d,p) is used in the calculations of cation-π interactions and ligand-cation coordinate interactions. In recent years great efforts are made to make up the shortcoming of DFT in dispersion interactions, including design of new functional [47], or empirical correction terms [41, 42, 48–50]. , In this study the missing dispersion energies in DFT calculations are corrected by an empirical method suggested by Du et al [42]. The interaction energies in solutions are calculated by using the polarizable continuum model (PCM) [50–53].
In this study most molecule monomers are optimized by using CCSD/6-31+G(d,p) methods. Some large amino acids, such as Tyr and Trp, first are optimized at B3LYP/6-31+G(d,p) level, then the side chains are optimized at CCSD/6-31+G(d,p) level. The geometry parameters of side-chain, obtained from CCSD calculations, are combined with the parameters of DFT optimizations. In this study the protonated His+ is simplified as the protonated imidazole (C3N2H4+), protonated Arg+ is simplified as CHNH2NH2+, and the protonated amino acid Lys+ is simplified as CH3NH3+, respectively, as shown in Figure 1C, D, and E. The structures of four interaction types (cation-π interaction, π-π stacking interaction, hydrogen-π interaction, and coordinate bond interaction) of His are shown in Figure 1F, G, H, and I, respectively. Usually amino acids have several stable structural conformations with different energies. In proteins the orientations of residue side chains and the structural conformations of peptide backbone are innumerous. The optimized structures of amino acids, shown in Figure 1, are only one of the possible conformations. In Figure 1 F the metallic cation can be put at the upside or at the downside of the aromatic planes. In the ‘Upside’ structure the cation-π interaction may be complicated by the interaction elements in peptide backbone. On the other hand, the ‘Downside’ structure is less affected by other interaction elements. In this study we focus on the ‘pure’ cation-π interactions, the ‘Downside’ structures. All calculations are performed on Sugon-5000A computer using Gaussian 09 software package [54]. The detailed geometrical parameters of optimized molecular structures are stored in supporting material (Optimized-Mol.zip).
Results
In this section all calculation results are reported and summarized using tables and figures. Brief comparisons and illustrations are provided following the calculation results. Four interaction types (cation-π, π-π stacking, hydrogen-π, and coordinate bond interaction) of histidine with other amino acids and metallic cations are calculated in gas phase and in solutions (water, acetonitrile, and cyclohexane). The hydrogen bonding interaction of histidine is not included in this study, because it is a familiar and well studied interaction type.
Cation-π interactions of Histidine
The cation-π interaction energies of His are summarized in Table 2. In the up part of Table 2 the His is the aromatic motif in neutral form. The cation-π interaction energies are different when the cations are at the downside and upside of the aromatic π-plane, because the interaction environments are different at the two sides (see Figure 1F). The cation-π energy (−147.4 kcal/mol) of Zn2+ is much larger than other cations, because the 3d valence orbitals of Zn2+ can make stronger bonding MO with the imidazole π-MO of histidine. In gas phase the cation-π interaction energies of His with organic cations (protonated amino acids Lys+ and Arg+) are in the range −8 to −9 kcal/mol, stronger than the common hydrogen bonds of water (−5 to −6 kcal/mol) [55, 56]. However, the cation-π interaction energies of His are smaller than that of other three aromatic amino acids (Phe, Tyr, and Trp) because of the smaller π-system size [57, 58]. In the lower part of Table 2 the protonated histidine (His+) is the cation in the cation-π interactions. The cation-π interaction energy of His+-Trp is −13.6 kcal/mol, larger than other two interaction pairs (His+-Tyr and His+-Phe), because of the larger aromatic size of Trp.
The cation-π interaction energies in three solvents (water, acetonitrile, and cyclohexane) are listed in Table 3. The cation-π interaction energies decrease sharply with the increase of solvent dielectric constants ε. Generally speaking, in gas phase cation-π interaction energies of metallic cations are larger than that of organic cations (protonated amino acids), however, in the aqueous solution, the cation-π interaction energies of organic cations are larger than that of metallic cations.
The cation-π interaction energies are distance and orientation dependent. Figure 2 shows the cation-π energy curves of several cation-π interaction pairs as the functions of distance (R) and orientation angle (θ). Figure 2A and B are the interaction energies of His-Na+ and His-K+ as the functions of distance (from the cation to the center of imidazole ring) and the orientation angle (between the interaction direction and the perpendicular direction). The strongest interaction is at the perpendicular direction (θ=0°) to the π-plane. Figure 2C and D show the interaction energy curves of His-Ca2+, His-Zn2+, His-Lys+ and His-Arg+ at the perpendicular direction. The curve shapes of cation-π interaction energies are similar to the common van der Waals interactions, however, the attractive interaction regions are broader, and at the short distance the repulsive interactions are softer than the common van der Waals interactions.

The cation-π interaction energies of histidine (His) with metallic cations and organic cations. (A) The cation-π interaction energies of His–Na+ as the function of distance R and orientation angle θ. (B) The cation-π interaction energies of His–K+ as the function of distance R and orientation angle θ. The cation-π interactions are distance and orientation dependent. The most favorable direction is perpendicular to the center of π-plane. (C) The cation-π interaction energies of His–Ca2+ and His–Zn2+ as the function of distance between cation and the aromatic center of His. (D) The cation-π interaction energies of His–Lys2+ and His–Arg+ as the function of distance between cation and the aromatic center of His. All calculations are performed by using B3LYP/6-31+G(d,p) method.
After the His is protonated (His+) there are no attractive cation-π interactions between His+ and the metallic cations (e.g., Na+, K+, and Ca2+) and the organic cations (Arg+ and Lys+), instead of repulsive interactions. Figure 3 shows the repulsive cation-π interactions between the protonated histidine (His+) and cations as the function of distance R. In protein structures the histidine frequently and reversibly transforms from neutral form to protonated form [6, 59, 60]. Based on our calculations the proton transformation in His may switch the cation-π interactions from attractive to repulsive. Actually the His+–cation interactions are the combination of repulsive electrostatic interaction between two cations and the attractive cation-π interaction between cation and aromatic motif. In Figure 3A in short distance the repulsive interactions of His+ with Na+ and K+ are steep, because the strong repulsive electrostatic forces exceed the attractive cation-π forces. However, the repulsive interactions of His+ with Ca2+ and Zn2+ are softer at the short distance, because the attractive cation-π forces of Ca2+ and Zn2+ are larger than that of Na+ and K+. Particularly at longer distance the interaction of His+–Zn2+ turns to attractive, which may arise from the long interaction range of 3d valence orbitals of Zn2+ in cation-π interactions.

The repulsive cation-π interactions between protonated histidine (His+) and cations. (A) The repulsive cation-π interactions between protonated histidine (His+) and cation Na+ and K+. At short distance the repulsive interaction energies are very strong, then the energies decrease with the distance R. (B) The repulsive cation-π interactions between protonated histidine (His+) and cation Ca2+ and Zn2+. At short distance the curves are softer than that of Na+ and K+. At long distance (>5Å) the interaction of His+–Zn2+ turns to attractive, which may arise from the long interaction range of 3d valence orbitals of Zn2+. (C) The repulsive cation-π interactions between protonated histidine (His+) and organic cations Lys+ and Arg+. All calculations are performed by using B3LYP/6-31+G(d,p) method.
π-π stacking interactions of histidine
The π-π stack is referred to the face to face interactions between two or more aromatic molecules. In the 20 natural amino acids 4 of them contain aromatic side chains (Phe, Tyr, Trp, and His). The π-π stacking interactions in proteins are a controversial research topic [17–21]. Our and other authors’ comparison calculations revealed that π-π stacking interactions are dispersion-dominated phenomenon [43–46]. For the neutral (uncharged and non polar) π-π stacking interactions the B3LYP method cannot yields attractive energies, and DFT fails in describing dispersion energies. Our calculation results using CCSD/6-31+G(d,p) are summarized in Table 4. In proteins the strength of the π-π stacking interactions between neutral His and other aromatic amino acids (Phe, Tyr, and Trp) are in the range from −3.0 to −4.0 kcal/mol, higher than the C6H6-C6H6 π-π stacking energy (−1.88 kcal/mol) [57], because the π-π interaction energies between aromatic amino acids may contain the contributions of hydrogen-π interactions [61, 62], which will be discussed in next section. The π-π stacking interaction energies between the protonated histidine (His+) and other aromatic amino acids are in the range from −3.6 to −8.4 kcal/mol, remarkably larger than that of neutral His.
The π-π stacking energies increase with the size of π-system. In Table 4 the π-π stacking energy (−4.035 kcal/mol) of His-Trp is larger than that of His-Phe and His-Tyr because of the larger π-system of Trp. In DNA the π-π stacking interactions have larger contributions than in proteins [41, 47–49]. The protonated amino group (CHNH2NH2+) of Arg+ forms a π-plane, and the larger π-π stacking energy (−5.0432 kcal/mol) of His-Arg+ may partially from the cation-π interaction. The lower part of Table 4 lists the π-π stacking interaction energies between the protonated His+ and three aromatic amino acids (Phe, Tyr, and Trp), which are remarkably larger than that in the up part of Table 4.
Hydrogen-π interactions of histidine
The hydrogen-π interaction is the interaction between polar hydrogen atom and π-electron density of aromatic molecule [61, 62]. In contract to the hydrogen-π interaction, the common hydrogen bond is the interaction between polar hydrogen and electron density of electronegative elements (such as oxygen and nitrogen). Unlike π-π stacking interactions, in which the π-planes of two aromatic amino acids are in parallel orientation, the hydrogen-π interactions of histidine and other aromatic amino acids take ‘T’ orientation. In proteins when the polar hydrogen atom of histidine perpendicularly points to the aromatic ring of other amino acids, the hydrogen-π interaction happens. The hydrogen-π interaction energies of His with other aromatic amino acids (Phe, Tyr, and Trp) are listed in Table 5.
Comparing the data in Table 4 and Table 5, the energies of hydrogen-π interactions are larger than the corresponding energies of π-π stacking interactions. In proteins the energies of hydrogen-π interactions are in the range −5 to −8 kcal/mol, comparable to the common hydrogen bond interactions (−4 to −6 kcal/mol). Actually, the π-π stacking interaction energies of aromatic amino acids contain the contributions of hydrogen-π interactions from the polar hydrogen atoms in His and in Tyr.
Coordinate bonding interactions between His and cations
The basic nitrogen atom in imidazole ring of His bears a lone electron pair, which may form coordinate bond with metallic cations, such as Ca2+, Zn2+, and Cu2+. The coordinate interaction of His is a unique molecular interaction type in the 20 natural amino acids. The coordinate interaction energies of His with some metallic cations (Na+, K+, Ca2+, and Zn2+) are listed in Table 6. The energy curves of His-cation coordinate interactions as the function of distance are shown in Figure 4. Comparing the Table 6 with Table 2, 4 and 5, the His-cation coordinate interaction energies are much stronger than the cation-π interactions and the π-π stacking interactions. The interaction energy (−195.2164 kcal/mol) of His-Zn2+ is higher than all other interaction pairs, because the 3d valence orbitals of Zn2+ can make stronger bonding MO with the lone electron pair of nitrogen atom in imidazole. The metallic cations often play important role in the catalytic activity of enzymes in biology. Therefore the coordinate interactions of His with metallic cations are significantly important in protein science and biochemistry.

The coordinate interaction energies of His with metallic cations as the function of distance R. (A) The coordinate bonding interaction curves of His–Na+ and His–K+. (B) The coordinate bonding interaction curves of His–Ca2+ and His–Zn2+. The interaction energies of coordinate bonding interactions are larger than other three interaction types (cation-π interaction, hydrogen-π interaction, and π-π stacking interaction). The coordinate interaction of His–Zn2+ is a long range interaction, and the energy is as high as −195 kcal/mol. All results are calculated at B3LYP/6-31+G(d,p) level.
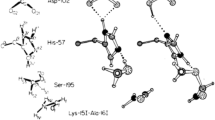
Although alkali metallic cations (Na+ and K+) are the most abundant elements in living systems, the transition metallic cations (e.g., Zn2+, Cu2+, and Fe2+) often play important role in the bioactivities of proteins and enzymes [3, 63]. In many cases the transition metallic cations bind at the host proteins through the coordinate bonds. For example, the extracellular lipase (T1 lipase) from Geobacillus zalihae strain T1 is a thermoalkalophilic enzyme [13, 64]. In the crystal structure of T1 lipase (PDB code 1JI3) a zinc cation Zn2+ binds with two histidines (His81 and His87) in the host pocket, as shown in Figure 5, which is one of the most conservative motif in the lipase family. The interaction structure between Zn2+ and two His is in the best orientation and distances for the coordinate interaction. The coordinate bond lengths of Zn2+ with His81 and His87 are 2.12 Å and 1.99 Å, respectively, very close to the optimized distance (1.9519 Å) in Table 6.

The coordinate bonding interaction between His and Zn2 +in T1 lipase (PDB code: 1JI3). (A) The location of His81, His87, and Zn2+ in the T1 lipase structure. (B) The coordinated bonds between His81 and Zn2+, and between His87 and Zn2+. The coordinate bond lengths of His81–Zn2+ and His87–Zn2+ are 2.12 Å and 1.99 Å, respectively, very close to the optimized distance (1.9519 Å).
Discussion
Histidine is an ionizable amino acid with the acidic ionization constant around pKa=6.5, very close to neutral. An interesting finding in this study is that the protonation of histidine has closely relationship with the interaction types. The cation-π interactions of neutral histidine (His) are attractive, and the cation-π interactions of protonated histidine (His+) are repulsive. A reasonable deduction is that pH condition can reversibly switch the cation-π interactions of histidine from attractive to repulsive. Vice versa, the cation-π interactions can affect the two protonation forms of histidine. In proteins the pKa value of His can change in a broad range due to the influence of interaction environment, and histidine can play the roles of both proton donor or acceptor [58, 65, 66]. The stronger attractive cation-π interaction can make the pKa value of His lower, and the lower pH condition may turn the cation-π interaction from attractive to repulsive. For the same reason, other interaction types (coordinate interaction, hydrogen-π interaction, hydrogen bond and the π-π stacking interaction) may also affect the pKa value of histidine to some degree [60].
In protein hydrolysis reactions the pKa value of His is a critically important property. In the catalytic triads of lipase, the basic nitrogen of histidine is used to abstract a proton from threonine, serine, or cysteine to activate it as a nucleophile. In carbonic anhydrases, a histidine proton shuttle is utilized to rapidly transport protons away from a zinc-bound water molecule to quickly regenerate the active form of the enzyme [67, 68]. In the histidine proton shuttle, histidine abstracts a proton with its basic nitrogen to make a positively-charged intermediate, and then use another molecule, a buffer, to extract the proton from its acidic nitrogen. Our study illustrates that in the proton shuttle procedure, the histidine is not working by itself alone, but with the collaboration of environmental residues through the multiple interactions that affect the pKa value of histidine.
Conclusion
Based on our calculation results the energy order of five interaction types (cation-π interaction, π-π stacking interaction, hydrogen-π interaction, hydrogen-bond interaction, and coordinate bond interaction) is as follows, Ecoor>Ecation-π>EH-π≈EH-b>Eπ-π. The coordinate interaction (Ecoor) of His with metallic cations is the strongest interaction with long interaction distance, followed by the cation-π interaction (Ecation-π). In the cation-π interactions, when His is in neutral form (unprotonated), interaction energy is attractive. However, when His is protonated, the interaction energy turns to repulsive. The π-π stacking interactions are the π-plane to π-plane interactions, with much more interaction conformations than other interaction types. In proteins the energies of π-π stacking interactions (Eπ-π) can change in a broad range, because of different interaction orientations. The π-π stacking interactions between neutral His and aromatic amino acids (Phe, Tyr, and Trp) are in the range −3.0 to −4.0 kcal/mol, significantly larger than the van der Waals interactions. However, the π-π stacking energies of protonated histidine (His+) are much larger than the energies of neutral His.
The interaction strength of cation-π interactions in solutions is a controversial research topic [17, 65, 69]. Based on our calculations by using PCM method, the energies of cation-π interactions decrease sharply with the increase of the dielectric constant ε of solvents. In gas phase the cation-π interaction energies of metallic cations are larger than that of organic cations (Lys+ and Arg+). However, in solutions of polar solvents (water and acetonitrile) the cation-π interaction energies of organic cations (protonated amino acids) are lager than that of metallic cations. The PCM is a continuum medium model [50–53]. The calculated values of PCM may be not very accurate, but the qualitative order is meaningful. In aqueous solution the cation-π interactions between protonated amino acids and aromatic amino acids may be more important than that of metallic cations [17, 69, 70]. However, this does not mean that the cation-π interactions of metallic cations are not important in solutions. In aqueous solution the hydrophilic residues are explored on the surface, and the hydrophobic residues are hidden in the core region of protein structures. In the hydrophobic pockets of proteins the dielectric constants are smaller than that in bulk solution. Therefore, the cation-π interactions are still working in the hydrophobic pockets and in core region of proteins.
References
Martínez A: Evidence for a functionally important histidine residue in human tyrosine hydroxylase. Amino Acids. 1995, 9: 285-292. 10.1007/BF00805959.
Uchida K: Histidine and lysine as targets of oxidative modification. Amino Acids. 2003, 25: 249-257. 10.1007/s00726-003-0015-y.
Remko M, Fitz D, Rode BM: Effect of metal ions (Li+, Na+, K+, Mg2+, Ca2+, Ni2+, Cu2+ and Zn2+) and water coordination on the structure and properties of l-histidine and zwitterionic l-histidine. Amino Acids. 2010, 39: 1309-1319. 10.1007/s00726-010-0573-8.
Li F, Fitz D, Fraser DG, Rode BM: Catalytic effects of histidine enantiomers and glycine on the formation of dileucine and dimethionine in the salt-induced peptide formation reaction. Amino Acids. 2010, 38: 287-294. 10.1007/s00726-009-0249-4.
Agnieszka M, Janina KW, Katarzyna KK: Five-membered heterocycles. Part III. Aromaticity of 1,3-imidazole in 5+n hetero-bicyclic molecules. J Mol Struc. 2003, 655: 397-403. 10.1016/S0022-2860(03)00282-5.
Doğan A, Özel AD, Kılıç E: The protonation equilibria of selected glycine dipeptides in ethanol–water mixture: solvent composition effect. Amino Acids. 2009, 36: 373-379. 10.1007/s00726-008-0054-5.
Priyakumar UD, Punnagai M, Krishna Mohan GP, Sastry GN: A computational study of cation-π interactions in polycyclic systems: exploring the dependence on the curvature and electronic factors. Tetrahedron. 2004, 60: 3037-3043. 10.1016/j.tet.2004.01.086.
Reddy AS, Sastry GN: Cation [M = H+, Li+, Na+, K+, Ca2+, Mg2+, NH4+, and NMe4+] interactions with the aromatic motifs of naturally occurring amino acids: A theoretical study. J Phys Chem A. 2005, 109: 8893-8903. 10.1021/jp0525179.
Engerer LK, Hanusa TP: Geometric Effects in Olefinic Cation−π Interactions with Alkali Metals: A Computational Study. J Org Chem. 2011, 76: 42-49. 10.1021/jo101307z.
Hunter CA, Lawson KR, Perkins J, Urch CJ: Aromatic interactions. J Chem Soc Perkin Trans. 2001, 2: 651-669.
Crowley PB, Golovin A: Cation–π interactions in protein–protein interfaces. Proteins. 2005, 59: 231-239. 10.1002/prot.20417.
Vijay D, Sastry GN: Exploring the size dependence of cyclic and acyclic π-systems on cation-π binding. Phys Chem Chem Phys. 2008, 10: 582-590. 10.1039/b713703f.
Matsumura H, Yamamoto T, Leow TC, Mori T, Salleh AB, Basri M, Inoue T, Kai Y, Zaliha RN, Rahman RA: Novel cation-π interaction revealed by crystal structure of thermoalkalophilic lipase. Proteins. 2008, 70: 592-598.
Reddy AS, Zipse H, Sastry GN: Cation-π Interactions of Bare and Coordinatively Saturated Metal Ions: Contrasting Structural and Energetic Characteristics. J Phys Chem B. 2007, 111: 11546-11553. 10.1021/jp075768l.
Schottel BL, Chifotides HT, Dunbar KR: Anion-π interactions.Chem Soc Rev. 2008, 37: 68-83. 10.1039/b614208g.
Burley SK, Petsko GA: Amino-aromatic interactions in proteins. FEBS Lett. 1986, 203: 139-143. 10.1016/0014-5793(86)80730-X.
Stefan G: Do special noncovalent π-π stacking interactions really exist?. Angew Chem Int Ed. 2008, 47: 3430-3434. 10.1002/anie.200705157.
Mignon P, Loverix S, Steyaert J, Geerlings P: Influence of the π–π interaction on the hydrogen bonding capacity of stacked DNA/RNA bases. Nucl Acids Res. 2005, 33: 1779-1789. 10.1093/nar/gki317.
Petitjean A, Khoury RG, Kyritsakas N, Lehn JM: Dynamic devices, shape switching and substrate binding in ion-controlled nanomechanical molecular tweezers. J Am Chem Soc. 2004, 126: 6637-6647. 10.1021/ja031915r.
Sygula A, Fronczek FR, Sygula R, Rabideau PW, Olmstead MM: A Double Concave Hydrocarbon Buckycatcher. J Am Chem Soc. 2007, 129: 3842-3843. 10.1021/ja070616p.
Janiak C: A critical account on π-π stacking in metal complexes with aromatic nitrogen-containing ligands. J Chem Soc Dalton Trans. 2000, 3885-3896.
Meyer EA, Castellano RK, Diederich F: Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003, 42: 1210-1250. 10.1002/anie.200390319.
Hughes RM, Waters ML: Effects of lysine acylation in a β-hairpin peptide: comparison of an amide-π and a cation-π interaction. J Am Chem Soc. 2006, 128: 13586-13591. 10.1021/ja0648460.
Kang SO, Hossain MA, Bowman-James K: Influence of dimensionality and charge on anion binding in amide-based macrocyclic receptors. Coord Chem Rev. 2000, 250: 3038-3052.
Miessler GL, Tarr DA: Inorganic Chemistry. 2003, Upper Saddle River, NJ: Pearson Prentice Hall, 3
Smith MB, March J: March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 2007, New York: Wiley-Interscience, 6
Jackson WG, Josephine AM, Silvia C: Alfred Werner's inorganic counterparts of racemic and mesomeric tartaric acid: A milestone revisited. Inorg Chem. 2004, 43: 6249-6254. 10.1021/ic040042e.
Sirois SW, Proynov EI, Truchon JF, Tsoukas CM, Salahub DR: A density functional study of the hydrogen-bond network within the HIV-1 protease catalytic site cleft. J Comput Chem. 2003, 24: 1110-1119. 10.1002/jcc.10176.
Du QS, Li DP, Liu PJ, Huang RB: Molecular potential energies in dodecahedron cell of methane hydrate and dispersion correction for DFT. J Mol Graph Model. 2008, 27: 140-146. 10.1016/j.jmgm.2008.03.008.
Henry M: Thermodynamics of hydrogen bond patterns in supramolecular assemblies of water molecules. Chem Phys Chem. 2002, 3: 607-616. 10.1002/1439-7641(20020715)3:7<607::AID-CPHC607>3.0.CO;2-A.
Henry M: Nonempirical quantification of molecular interactions in supramolecular assemblies. Chem Phys Chem. 2002, 3: 561-569. 10.1002/1439-7641(20020715)3:7<561::AID-CPHC561>3.0.CO;2-E.
Andrews LJ, Keefer RM: Molecular complexes in organic chemistry. 1964, San Francisco: Holden-Day
Mezey PG: Macromolecular density matrices and electron densities with adjustable nuclear geometries. J Math Chem. 1995, 18: 141-168. 10.1007/BF01164655.
Mezey PG: Quantum similarity measures and Löwdin's transform for approximate density matrices and macromolecular forces. Int J Quantum Chem. 1997, 63: 39-48. 10.1002/(SICI)1097-461X(1997)63:1<39::AID-QUA8>3.0.CO;2-3.
Sayyed FB, Suresh CH: Accurate prediction of cation−π interaction energy using substituent effects. J Phys Chem A. 2012, 116: 5723-5732. 10.1021/jp3034193.
Mohan N, Vijayalalakshmi KP, Koga N, Suresh CH: Comparison of aromatic NH…π, OH…π, and CH…π interactions of alanine using MP2, CCSD, and DFT methods. J Comput Chem. 2010, 31: 2874-2882.
Gresh N, Kafafi SA, Truchon JF, Salahub DR: Intramolecular interaction energies in model alanine and glycine tetrapeptides. Evaluation of anisotropy, polarization, and correlation effects. A parallel ab initio HF/MP2, DFT, and polarizable molecular mechanics study. J Compt Chem. 2004, 25: 823-834. 10.1002/jcc.20012.
Jurecka P, Cerný J, Hobza P, Salahub DR: Density functional theory augmented with an empirical dispersion term. Interaction energies and geometries of 80 noncovalent complexes compared with ab initio quantum mechanics calculations. J Comput Chem. 2007, 28: 555-569. 10.1002/jcc.20570.
Van Mourik T, Gdanitz RJ: A critical note on density functional theory studies on rare-gas dimers. J Chem Phys. 2002, 116: 9620-9623. 10.1063/1.1476010.
Morgado C, Vincent MA, Hillier IH, Shan X: Can the DFT-D method describe the full range of noncovalent interactions found in large biomolecules?. Phys Chem Chem Phys. 2007, 9: 448-451. 10.1039/b615263e.
Von Lilienfeld OA, Tavernelli I, Rothlisberger U, Sebastiani D: Optimization of effective atom centered potentials for London dispersion forces in density functional theory. Phys Rev Lett. 2004, 93: 153004-153007.
Du Q-S, Liu P-J, Deng J: Empirical correction to molecular interaction energies in density functional theory (DFT) for methane hydrate simulation. J Chem Theory Comput. 2007, 3: 1665-1672. 10.1021/ct700026d.
Purvis GD, Bartlett RJ: A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J Chem Phys. 1982, 76: 1910-1919. 10.1063/1.443164.
Lee TJ, Rice JE: An efficient closed-shell singles and doubles coupled-cluster method. Chem Phys Lett. 1988, 23: 406-415.
Scuseria GE, Schaefer HF: Is coupled cluster singles and doubles (CCSD) more computationally intensive than quadratic configuration interaction (QCISD)?. J Chem Phys. 1989, 90: 3700-3703. 10.1063/1.455827.
Scuseria GE, Janssen CL, Schaefer HF: An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J Chem Phys. 1988, 89: 7382-7388. 10.1063/1.455269.
Grimme S: Semiempirical hybrid density functional with perturbative second-order correlation. J Chem Phys. 2006, 124: 034108-10.1063/1.2148954.
Zimmerli U, Parrinello M, Koumoutsakos P: Dispersion corrections to density functionals for water aromatic interactions. J Chem Phys. 2004, 120: 2693-2699. 10.1063/1.1637034.
Grimme S: Accurate description of van der Waals complexes by density functional theory including empirical corrections. J Comput Chem. 2004, 25: 1463-1473. 10.1002/jcc.20078.
Miertus S, Scrocco E, Tomasi J: Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981, 55: 117-129. 10.1016/0301-0104(81)85090-2.
Amovilli C, Barone V, Cammi R, Cances E, Cossi M, Mennucci B, Pomelli CS, Tomasi J: Recent advances in the description of solvent effects with the polarizable continuum model. Adv Quant Chem. 1998, 32: 227-262.
Cossi M, Barone V: Analytical second derivatives of the free energy in solution by polarizable continuum models. J Chem Phys. 1998, 109: 6246-6254. 10.1063/1.477265.
Foresman JB, Keith TA, Wiberg KB, Snoonian J, Frisch MJ: Solvent effects. 5. influence of cavity shape, truncation of electrostatics, and electron correlation on ab initio reaction field calculations. J Phys Chem. 1996, 100: 16098-16104. 10.1021/jp960488j.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA: Gaussian 09, Revision B,01. 2010, Wallingford CT: Gaussian Inc
Zielkiewicz J: Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J Chem Phys. 2005, 123: 104501-10.1063/1.2018637.
Markovitch O, Agmon N: Structure and energetics of the hydronium hydration shells. J Phys Chem A. 2007, 111: 2253-2256. 10.1021/jp068960g.
Du QS, Long SY, Meng JZ, Huang RB: Empirical formulation and parameterization of cation-π interactions for protein modeling. J Compt Chem. 2012, 33: 153-162. 10.1002/jcc.21951.
Du QS, Liao SM, Meng JZ, Huang RB: Energies and Physicochemical Properties of Cation-π Interactions in Biology Structures. J Mol Graph Model. 2012, 34: 38-45.
Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH: PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011, 7: 525-537. 10.1021/ct100578z.
Huang RB, Du QS, Wang CH, Liao SM, Chou KC: A fast and accurate method for predicting pKa of residues in proteins. Protein Eeng Des Sel. 2010, 23: 35-42. 10.1093/protein/gzp067.
Ottiger P, Pfaffen C, Leist R, Leutwyler S, Bachorz RA, Klopper W: Strong N−H···π Hydrogen Bonding in Amide−Benzene Interactions. J Phys Chem B. 2009, 113: 2937-2943. 10.1021/jp8110474.
Steiner T, Koellner G: Hydrogen bonds with pi-acceptors in proteins: frequencies and role in stabilizing local 3D structures. J Mol Biol. 2001, 305: 535-557. 10.1006/jmbi.2000.4301.
Trakhanov S, Quiocho FA: Influence of divalent cations in protein crystallization. Protein Sci. 1995, 4: 1914-1919. 10.1002/pro.5560040925.
Fischer M, Pleiss J: The Lipase Engineering Database: a navigation and analysis tool for protein families. Nucleic Acids Res. 2003, 31: 319-321. 10.1093/nar/gkg015.
Bas DC, Rogers DM, Jensen JH: Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008, 73: 765-783. 10.1002/prot.22102.
Li H, Robertson AD, Jensen JH: Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005, 6: 704-721.
Badger MR, Price GD: The role of carbonic anhydrase in photosynthesis. Annu Rev Plant Physio Plant Mol Bio. 1994, 45: 369-392. 10.1146/annurev.pp.45.060194.002101.
Lindskog S: Structure and mechanism of carbonic anhydrase. Pharmacol Ther. 1997, 74: 1-20. 10.1016/S0163-7258(96)00198-2.
Biot C, Buisine E, Rooman M: Free-energy calculations of protein-ligand cation-π and amino-π interactions: From vacuum to protein-like environments. J Am Chem Soc. 2003, 125: 13988-13994. 10.1021/ja035223e.
Crowley PB, Golovin A: Cation-π interactions in protein–protein interfaces. Proteins. 2005, 59: 231-239. 10.1002/prot.20417.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
We declare that there are no any competing interests.
Authors’ contributions
All authors contributed equality for the development of the manuscript. RBH and QSD designed the research scheme and wrote the article. SML did most calculations, JZM and ZWP performed the data collection and analysis. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liao, SM., Du, QS., Meng, JZ. et al. The multiple roles of histidine in protein interactions. Chemistry Central Journal 7, 44 (2013). https://doi.org/10.1186/1752-153X-7-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-7-44