
Abstract
We report the first synthesis of a cobalt Cp diene complex wherein the diene is derived by microbial dearomatising dihydroxylation of an aromatic ring. The complex has been characterised crystallographically and its structure is compared to that of an uncomplexed diene precursor.

Similar content being viewed by others

Background
The dearomatising dihydroxylation of an aromatic substrate by a microorganism was first reported by Gibson in 1968 [1]. The production and utilisation of these arene-derived diols in synthesis has since become established methodology, not least because in most instances the oxidation of substituted arenes gives enantiopure diols. The field has been the subject of several extensive reviews [2–8]. To date, over 400 arene cis-diols have been reported. The great majority of these are produced by organisms expressing toluene dioxygenase (TDO), naphthalene dioxygenase (NDO) and biphenyl dioxygenase (BPDO) enzymes. These oxidise substituted arenes in a regio- and stereoselective fashion. A robust predictive model has been developed by Boyd et al. for these transformations [9] and the sense of enantioinduction is conserved across organisms and substrates (Scheme 1(a), ortho-meta oxygenation). However, organisms that express benzoate dioxygenase (BZDO) enzymes oxidise benzoic acids in a process that exhibits not only different regioselectivity, but also the opposite absolute sense of enantioinduction. For example, R. eutrophus B9 [10], P. putida U103 [11] and P. putida KTSY01 (pSYM01) [12] oxidise benzoic acid to benzoate 1,2-cis dihydrodiol 4 (Scheme 1(b), ipso-ortho oxygenation). Substituted benzoic acids can also be oxidised by BZDO [13–17].

Regio- and stereoselectivity of dioxygenases.
Diol acid 4 and its derivatives have been employed in several synthetic contexts. Widdowson was the first to establish the absolute configuration of 4 and employ it in [4+2] cycloadditions [18] and Mihovilovic subsequently reported tethered cycloadditions [19, 20]. Applications of 4 in carbohydrate chemistry have been reported by Parker [21] and by us [22]. Myers has reported the synthesis of a useful library of chirons from 4 [23] and their application in the total synthesis of tetracyclines [24, 25]. The Hudlický route to idesolide [26] and our route to grandifloracin [27] also employ 4 as a chiral pool starting material for total synthesis.
Apart from isolated uses of organotin derivatives in cross-coupling [28–30] and formation of a zinc metal-organic framework [31], the organometallic chemistry of microbial arene dihydrodiols has been dominated by iron complexes. The more common ortho-meta diols of type 2 have been employed by Stephenson [32–39], Pearson [40], Suemune [41] and Mombrú [42] in the formation of tricarbonyliron(0) diene complexes. These complexes have proven to be useful in synthesis [43, 44]. As shown in Scheme 2, the endo isomer (that in which the metal is complexed to the ligand face bearing the hydroxyl groups) is the only diastereoisomer obtained in each instance. This may be rationalised due to the Lewis basic nature of the hydroxyl groups, which serve to coordinate the incoming metal fragment, thus directing it to the same face of the diene.

Tricarbonyliron(0)diene complexes of ortho-meta dihydrodiols.
We have recently explored iron diene complexation in the context of ipso-ortho arene dihydrodiol 4 and its derivatives and found that complexation of a tricarbonyliron fragment to methyl ester 7 affords solely the isomer in which the hydroxyls are endo and the ester is exo (8, Scheme 3) [45]. This result is noteworthy, since in contrast to 5, ligand 7 presents Lewis basic functionality on both sides of the ring. Accordingly, we have termed 7 a "competition ligand" and concluded from the exclusive formation of 8 that a diol is a much more effective directing group than a methyl ester. Literature precedent for cyclohexadiene ligands able to act as competition ligands in tricarbonyliron complexation was limited to a single report from Berchtold [46]. It was reported that complexation of 10 gave 11 as the sole product, implying that the acetoxy functionality is a markedly better site of coordination for the incoming iron fragment than the methyl ether.

Cyclohexadiene "competition ligands" in iron tricarbonyl complex formation.
Encouraged by the uses of arene dihydrodiols in iron complexation described above, we sought to explore the chemistry of group 9 complexes of these ligands. Specifically, a cobalt cyclopentadienyl complex of an arene dihydrodiol would be isolobal with the corresponding tricarbonyliron complex [47], yet display distinct reactivities in certain cases. Such group 9 complexes of arene dihydrodiols have not been reported previously. Vollhardt has reported cobalt complex 13 [49] and Maitlis has reported iridium complex 14 [49] (Figure 1), yet these were not accessed by direct complexation of a cyclohexadiene diol; rather, they derive from double nucleophilic additions of methoxide anion to the corresponding dicationic η6 complexes of benzene.

Co and Ir diene complexes analogous to those reported in this work.
Results
Four compounds were considered as potential ligands for cobalt (Scheme 4). Acetonide methyl ester 16 may be synthesised by two reported routes which differ in the order of events [15, 18, 23]. Formation of the methyl ester first gives 7, the diol of which may then be protected to give 16. Alternatively, formation of the acetonide gives 15, which may then be readily esterified to 16 (Scheme 4). Either route affords 16 in near-quantitative yield. We were able to crystallise 15 as its monohydrate and determine its molecular structure by single crystal x-ray diffraction (Figure 2, Table 1 Additional file 1).

Two routes to 16.

Solid state structure of 15. Ellipsoids are represented at 50% probability. H atoms are shown as spheres of arbitrary radius.
Dienes 4, 7, 15 and 16 are all viable ligands for CpCo complex formation. Our primary focus was acetonide ester 16, for two reasons. Firstly, we had previously observed free acid 4 to form an intractable mixture when we attempted the synthesis of its iron tricarbonyl complex, with methyl esters proving more amenable to complexation. Secondly, we wished to ascertain whether the presence of the acetonide in 16 would favour complexation on the upper ligand face, in contrast to 8 [51]. Furthermore, it should be noted that diene rearrangements upon complexation have been described for analogous cobalt complexes; Vollhardt has proposed the intermediacy of η5 cyclohexadienyl cobalt complexes to account for the rearrangement of tethered amines [52] and enol ethers [53], further developing this chemistry to effect a concise total synthesis of strychnine [54, 55].
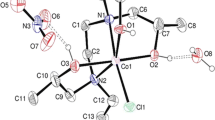
Our initial attempts at forming diene complexes involved CpCo(CO)2 as a precursor, which was exposed to UV light in the presence of diene ligand, in order to effect photodissociation of the carbonyl ligands. This approach did not furnish any products, however, and an approach employing ClCo(PPh3)3 and NaCp as precursors was similarly unsuccessful. We then turned to use of Jonas' reagent, CpCo(C2H4)2 (17), which is able to undergo ligand exchange with dienes and loss of two molar equivalents of ethylene [56]. Reaction of 16 and 17 in toluene at room temperature afforded novel cobalt complex 18, in which the metal was indeed complexed to the upper ligand face, but no alkene isomerisation had occurred (Scheme 5). Crystallisation of 18 from hexane afforded crystals suitable for X-ray structure determination (Figure 3, Table 1).

Solid state structure of 18. Ellipsoids are represented at 50% probability. H atoms are shown as spheres of arbitrary radius.

Use of Jonas' reagent 17 to access diene complex 18.
Discussion
With structures in hand for both uncomplexed and complexed dienes 15 and 18 we are able to compare the two. To our knowledge, this is the first time a direct comparison of a CpCo cyclohexadiene complex and an uncomplexed diene precursor has been made, since for other reported CpCo cyclohexadiene crystal structures, no structures of their precursors are available.
It is immediately evident that complexation to Co effects a major change in the conformation of the cyclohexadiene ring. Whereas the uncomplexed ligand 15 possesses a near-planar cyclohexadiene ring, in 18 this ring adopts a boat conformation. Specifically, the C2-C3-C4-C5 diene motif remains near-coplanar (dihedral angle of 2.7(3)° in 18 and -6.5(2)° in 15), but the two sp3 hybridised atoms (C1 and C6) are significantly out of plane in 18 (dihedral angles of 41.8(2)° for C1-C2-C3-C4 and 42.9(3)° for C3-C4-C5-C6 in 18, compared with 1.7(2)° and 4.0(2)° in 15). A consequence of this is that in complex 18 the lower face of the cyclohexadiene ring is subject to additional steric hindrance due to the acetonide endo methyl group. This is evidenced by the shorter distance between this carbon and C3 or C4 (18 has C3-C11 4.572(3) Å and C4-C11 4.709(3) Å, whereas in 15 the corresponding distances are 5.160(2) Å and 5.296(2) Å). The effect on Co complexation on the C-C bond order in the cyclohexadiene ring is also clearly visible. In uncomplexed 15, C2-C3 and C4-C5 are both 1.327(2) Å, whereas the formally single C3-C4 bond is appreciably longer at 1.463(2) Å. In contrast, in complex 18, bond lengths are 1.424(3) Å for C2-C3 and C3-C4 and 1.418(3) for C4-C5, i.e. equivalent within 3σ, as is precedented [47]. It should also be noted that in complex 18, the η5-cyclopentadienyl and η4-diene ligands are not coplanar. Rather, the angle between the C2-C5 and C111-C115 planes is 8.40°.
Conclusions
We have prepared the first organocobalt complex derived from a product of microbial arene dihydroxylation and characterised it by crystallographic and other means. We are currently investigating synthetic applications of this complex and our results will be reported in due course.
Experimental
Synthesis of (+)-η5-Cyclopentadienyl(η4-(3aS,4S,7aS)-methyl 2,2-dimethyl-3a,7a-dihydrobenzo[d][1, 3]dioxole-3a-carboxylate)cobalt(I) 18
Diene 16 (301 mg, 1.43 mmol, 1 equiv) was dissolved in dry, degassed toluene (10 mL) in a side-armed Schlenk that had been purged and refilled with argon three times. The resulting solution was added via cannula to η5-cyclopentadienylbis(ethylene)cobalt 17 (258 mg, 1.43 mmol, 1 equiv) and the mixture was stirred for 30 min at room temperature until the evolution of ethylene had ceased. The solvent was removed in vacuo and the solid residue was redissolved in a minimal amount of hexane and left to crystallise at -28°C for 48 h. Complex 18 was isolated as red-orange crystals (124 mg, 26%); m. pt. 118-120°C; [α]D +42 (c = 1, CH2Cl2); 1H-NMR (300 MHz, C6D6, Additional file 2) δ 5.44 (1H, d, J = 5.0 Hz, O-CH-), 5.09-5.04 (2H, m, -CH=CH-CH=CH-), 4.41 (5H, s, Cp-H), 3.53 (3H, s, O-CH3), 3.14 (1H, dd, J = 5.5, 1.0 Hz,=CH-C-COOCH3), 2.82 (1H, td, J = 5.0, 2.0 Hz, -O-CH-CH=), 1.42 (3H, s, C-CH3), 1.28 (3H, s, C-CH3) ppm; 13C-NMR (75 MHz, CDCl3, Additional file 2) δ 174.9, 113.9, 82.1, 80.8, 79.8, 74.6, 51.6, 48.9, 48.6, 27.1, 25.6 ppm; νmax (film) 2986, 2937, 1732, 1436, 1370, 1307, 1259, 1229, 1206, 1166, 1109, 1064, 1009, 888, 821, 762 cm-1; HRMS (+ve ESI-TOF) m/z calcd for (C15H20CoO4+H)+, 335.0688, found 335.0694. Found: C, 57.58; H, 5.76. C15H20CoO4 requires C, 57.49; H, 5.73%).
Methods
Reactions were carried out under an atmosphere of argon. Solvents were dried and degassed by passing through anhydrous alumina columns using an Innovative Technology Inc. PS-400-7 solvent purification system. All reagents were purchased from the Sigma-Aldrich Chemical Co. or Fisher Scientific Ltd. and were used without further purification. IR spectra were recorded on a Perkin-Elmer Spectrum 100 FT IR spectrometer with universal ATR sampling accessory, with absorbances quoted as ν in cm-1. NMR spectra were run on a Brüker Avance 300 instrument at 298 K. Mass spectra were recorded with a micrOTOF electrospray time-of-flight (ESI-TOF) mass spectrometer (Brüker Daltonik). Specific rotations were recorded on an Optical Activity AA-10 Automatic polarimeter with a path length of 1 dm. Concentrations (c) are quoted in g/100 mL.
References
Gibson DT, Koch JR, Schuld CL, Kallio RE: Oxidative degradation of aromatic hydrocarbons by microorganisms. II. Metabolism of halogenated aromatic hydrocarbons. Biochem. 1968, 7: 3795-3802. 10.1021/bi00851a003.
Hudlický T: Recent chemoenzymatic total syntheses of natural and unnatural products: codeine, balanol, pancratistatin, and oseltamivir. Pure Appl Chem. 2010, 82: 1785-1796. 10.1351/PAC-CON-09-10-07.
Hudlický T, Reed JW: Celebrating 20 years of Synlett - Special account on the merits of biocatalysis and the impact of arene cis-dihydrodiols on enantioselective synthesis. Synlett. 2009, 685-703.
Austin KAB, Matveenko M, Reekie TA, Banwell MG: Chemoenzymatic methods for the enantioselective assembly of bioactive natural products. Chem Aust. 2008, 75: 3-7.
Boyd DR, Bugg TDH: Arene cis-dihydrodiol formation: from biology to application. Org Biomol Chem. 2006, 4: 181-192. 10.1039/b513226f.
Johnson RA: Microbial arene oxidations. Org React. 2004, 63: 117-264.
Hudlický T, Gonzales D, Gibson DT: Enzymatic dihydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichimica Acta. 1999, 32: 35-62.
Widdowson DA, Ribbons DW, Thomas SD: The use of substituted cyclohexadienediols as versatile chiral synthons. Janssen Chimica Acta. 1990, 8: 3-9.
Boyd DR, Sharma ND, Hand MV, Groocock MR, Kerley NA, Dalton H, Chima J, Sheldrake GN: Stereodirecting substituent effects during enzyme-catalyzed synthesis of cis-dihydrodiol metabolites of 1,4-disubstituted benzene substrates. J Chem Soc Chem Commun. 1993, 974-976.
Reiner AM, Hegeman GD: Metabolism of benzoic acid by bacteria. Accumulation of (-)-3,5-cyclohexadiene-1,2-diol-1-carboxylic acid by a mutant strain of Alcaligenes eutrophus. Biochem. 1971, 10: 2530-2536. 10.1021/bi00789a017.
Cass AEG, Ribbons DW, Rossiter JT, Williams SR: Carbon-13 nuclear magnetic resonance studies in vivo on the metabolism of [1-13C]benzoate by mutants of Pseudomonas putida. Biochem Soc Trans. 1986, 14: 1268-1269.
Sun SY, Zhang X, Zhou Q, Chen JC, Chen GQ: Microbial production of cis-1,2-dihydroxy-cyclohexa-3,5-diene-1-carboxylate by genetically modified Pseudomonas putida. Appl Microbiol Biotechnol. 2008, 80: 977-984. 10.1007/s00253-008-1603-2.
Griffen JA, le Coz AM, Kociok-Köhn G, Ali Khan M, Stewart AJW, Lewis SE: Expanding the chiral pool: oxidation of meta-bromobenzoic acid by R. eutrophus B9 allows access to new reaction manifolds. Org Biomol Chem. 2011, 9: 3920-3928. 10.1039/c1ob05131h.
Engesser KH, Schmidt E, Knackmuss HJ: Adaptation of Alcaligenes eutrophus B9 and Pseudomonas sp. B13 to 2-fluorobenzoate as growth substrate. Appl Environ Microbiol. 1980, 39: 68-73.
Reineke W, Otting W, Knackmuss HJ: Cis-dihydrodiols microbially produced from halo- and methylbenzoic acids. Tetrahedron. 1978, 34: 1707-1714. 10.1016/0040-4020(78)80206-3.
Reineke W, Knackmuss HJ: Chemical structure and biodegradability of halogenated aromatic compounds. Substituent effects on 1,2-dioxygenation of benzoic acid. Biochim Biophys Acta. 1978, 542: 412-423. 10.1016/0304-4165(78)90372-0.
Knackmuss HJ, Reineke W: Effect of chloro substituents on the oxygenation of benzoate by Alcaligenes eutrophus B9. Chemosphere. 1973, 2: 225-230. 10.1016/0045-6535(73)90045-3.
Jenkins GN, Ribbons DW, Widdowson DA, Slawin AMZ, Williams DJ: Synthetic application of biotransformations: absolute stereochemistry and Diels-Alder reactions of the (1S,2R)-1,2-dihydroxycyclohexa-3,5-diene-1-carboxylic acid from Pseudomonas putida. J Chem Soc Perkin Trans 1. 1995, 2647-2655.
Fischer TCM, Leisch HG, Mihovilovic MD: Intramolecular Diels-Alder cyclization of biodihydroxylated benzoic acid derivatives towards novel heterocyclic scaffolds. Monatsh Chem. 2010, 141: 699-707. 10.1007/s00706-010-0291-7.
Mihovilovic MD, Leisch HG, Mereiter K: Microwave-mediated intramolecular Diels-Alder cyclization of biodihydroxylated benzoic acid derivatives. Tetrahedron Lett. 2004, 45: 7087-7090. 10.1016/j.tetlet.2004.07.109.
Parker MH, Maryanoff BE, Reitz AB: Synthesis of carba-β-L-fructopyranose and carbacyclic analogs of topiramate, an anticonvulsant agent. Synlett. 2004, 2095-2098.
Pilgrim S, Kociok-Köhn G, Lloyd MD, Lewis SE: "Inosaminoacids": novel inositol-amino acid hybrid structures accessed by microbial arene oxidation. Chem Commun. 2011, 47: 4799-4801. 10.1039/c1cc10643k.
Myers AG, Siegel DR, Buzard DJ, Charest MG: Synthesis of a broad array of highly functionalized, enantiomerically pure cyclohexanecarboxylic acid derivatives by microbial dihydroxylation of benzoic acid and subsequent oxidative and rearrangement reactions. Org Lett. 2001, 3: 2923-2926. 10.1021/ol010151m.
Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG: A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science. 2005, 308: 395-398. 10.1126/science.1109755.
Charest MG, Siegel DR, Myers AG: Synthesis of (-)-tetracycline. J Am Chem Soc. 2005, 127: 8292-8293. 10.1021/ja052151d.
Adams DR, Aichinger C, Collins J, Rinner U, Hudlický T: Chemoenzymatic synthesis of idesolide from benzoic acid. Synlett. 2011, 5: 725-729.
Palframan MJ, Kociok-Köhn G, Lewis SE: Total synthesis of (+)-grandifloracin by iron complexation of a microbial arene oxidation product. Org Lett. 2011, 13: 3150-3153. 10.1021/ol201057r.
Ley SV, Redgrave AJ, Taylor SC, Ahmed S, Ribbons DW: Microbial oxidation in synthesis: preparation of novel 3-substituted cis-cyclohexa-3,5-diene-1,2-diol derivatives from (1S,2S)-3-bromocyclohexa-3,5-diene-1,2-diol. Synlett. 1991, 741-742.
Gonzalez D, Schapiro V, Seoane G, Hudlický T: New metabolites from toluene dioxygenase dihydroxylation of oxygenated biphenyls. Tetrahedron: Asymmetry. 1997, 8: 975-977. 10.1016/S0957-4166(97)00059-1.
Sünnemann HW, Banwell MG, de Meijere A: Diversity-oriented synthesis of enantiomerically pure steroidal tetracycles employing Stille/Diels-Alder reaction sequences. Chem Eur J. 2008, 14: 7236-7249. 10.1002/chem.200800601.
Sbircea L, Sharma ND, Clegg W, Harrington R, Horton PN, Hursthouse MB, Apperley DC, Boyd DR, James SL: Chemoenzymatic synthesis of chiral 4,4'-bipyridyls and their metal-organic frameworks. Chem Commun. 2008, 5538-5540.
Howard PW, Stephenson GR, Taylor SC: Transition metal mediated asymmetric synthesis. VII. 6-Methoxycyclohexadienyliron complexes: access to synthetic equivalents of cyclohexadiene dications. J Organomet Chem. 1988, 339: C5-C8. 10.1016/S0022-328X(00)99399-9.
Howard PW, Stephenson GR, Taylor SC: Convenient access to homochiral tricarbonyliron complexes. J Chem Soc Chem Commun. 1988, 1603-1604.
Howard PW, Stephenson GR, Taylor SC: Transition metal mediated asymmetric synthesis. X. Homochiral π-complexes with planar chirality: synthetic equivalents of chiral cyclohexadiene dications. J Organomet Chem. 1989, 370: 97-109. 10.1016/0022-328X(89)87278-X.
Howard PW, Stephenson GR, Taylor SC: Evidence for an anomalous microbial oxidation of acetophenone: new access to optically active tricarbonyliron complexes. J Chem Soc Chem Commun. 1990, 1182-1184.
Howard PW, Stephenson GR, Taylor SC: Regioselective access to tricarbonyliron complexes: controlled preparation and reactions of trifluoromethyl substituted complexes. J Organomet Chem. 1991, 419: C14-C17. 10.1016/0022-328X(91)86186-T.
Stephenson GR, Howard PW, Taylor SC: Assignment of absolute configurations from the circular dichroism spectra of cyclic η4-diene complexes of iron tricarbonyl. J Chem Soc Chem Commun. 1991, 127-129.
Stephenson GR, Howard PW: Circular dichroism spectra of tricarbonyliron π-complexes. J Chem Soc Perkin Trans 1. 1994, 2873-2880.
Stephenson GR, Anson CE, Swinson GJ: Biphenyl-cis-diol chemistry to access enantiopure aryl-substituted organoiron complexes. Tetrahedron Lett. 2011, 52: 3547-3550. 10.1016/j.tetlet.2011.03.088.
Pearson AJ, Gelormini AM, Pinkerton AA: Preparation of optically pure tricarbonylcyclohexadienyliron complexes: use of a trifluoromethyl group as a regiodirector during hydride abstraction. Organometallics. 1992, 11: 936-938. 10.1021/om00038a065.
Watanabe A, Kamahori T, Aso M, Suemune H: Asymmetric synthesis of C 2 -symmetric 5,6-bis(benzyloxy)cyclohexa-1,3-diene and a tricarbonyliron complex. J Chem Soc Perkin Trans 1. 2002, 2539-2543.
Russi S, Suescun L, Mombrú A, Pardo H, Mariezcurrena RA, Cavalli G, Seoane G: (S)-Tricarbonyl[(1,2,3,4-η)-(5R,6S)-1-chloro-5,6-dimethoxycyclohexa-1,3-diene]iron(0). Acta Cryst. 2000, C56: 820-821.
Astley ST, Meyer M, Stephenson GR: Hippeastrine synthesis: a combined bio-dioxygenation/organoiron chirality relay approach. Tetrahedron Lett. 1993, 34: 2035-2038. 10.1016/S0040-4039(00)60339-8.
Boyd DR, Sharma ND: Enzymatic and chemoenzymatic synthesis of arene trans-dihydrodiols. J Mol Catal B. 2002, 19-20: 31-42.
Ali Khan M, Mahon MF, Stewart AJW, Lewis SE: Iron(0) tricarbonyl complexes of microbially derived cyclohexadiene ligands containing quaternary stereocentres. Organometallics. 2010, 29: 199-204. 10.1021/om9009069.
Ashworth RW, Berchtold GA: Preparation of the endo-6-acetoxy and endo-6-hydroxy derivatives of η5-(1,3-cyclohexadienyl)iron tricarbonyl fluoroborate. J Am Chem Soc. 1977, 99: 5200-5201. 10.1021/ja00457a061.
Chinn JW, Hall MB: Comparison of isolobal fragments: bonding of tricarbonyliron and cyclopentadienylcobalt to cyclobutadiene and cyclopentadienone. Organometallics. 1984, 3: 284-288. 10.1021/om00080a021.
Lai YH, Tam W, Vollhardt KPC: Transition metal activation of π-complexed benzene: double nucleophilic additions. J Organomet Chem. 1981, 216: 97-103. 10.1016/S0022-328X(00)84005-X.
Grundy SL, Smith AJ, Adams H, Maitlis PM: The selective reduction of benzene to cyclohexene mediated by platinum metal complexes: x-ray crystal structure of (2-6-η-1-nitromethylcyclohexadienyl)(1-5-η-pentamethylcyclopentadienyl)iridium(III) tetrafluoroborate. J Chem Soc Dalton Trans. 1984, 1747-1754.
Flack HD: On enantiomorph-polarity estimation. Acta Cryst. 1983, A39: 876-881.
Ali Khan M, Lowe JP, Johnson AL, Stewart AJW, Lewis SE: Accessing the antipodal series in microbial arene oxidation: a novel diene rearrangement induced by tricarbonyliron(0) complexation. Chem Commun. 2011, 47: 215-217. 10.1039/c0cc01169j.
Grotjahn DB, Vollhardt KPC: An unprecedented propellane-to-spirofused skeletal rearrangement upon oxidative demetalation of cyclopentadienyl cobalt-complexed polycyclic dienes: synthesis of a pentacyclic, potential precursor to strychnine. J Am Chem Soc. 1990, 112: 5653-5654. 10.1021/ja00170a047.
Boese R, Harvey DF, Malaska MJ, Vollhardt KPC: [2+2+2] Cycloadditions of alkynes to furans and thiophenes: A cobalt-mediated "enol ether walk". J Am Chem Soc. 1994, 116: 11153-11154. 10.1021/ja00103a039.
Eichberg MJ, Dorta RL, Lamottke K, Vollhardt KPC: The formal total synthesis of (±)-strychnine via a cobalt-mediated [2+2+2] cycloaddition. Org Lett. 2000, 2: 2479-2481. 10.1021/ol006131m.
Eichberg MJ, Dorta RL, Grotjahn DB, Lamottke K, Schmidt M, Vollhardt KPC: Approaches to the synthesis of (±)-strychnine via the cobalt-mediated [2+2+2] cycloaddition: rapid assembly of a classic framework. J Am Chem Soc. 2001, 123: 9324-9337. 10.1021/ja016333t.
Jonas K, Deffense E, Habermann D: Synthese und reaktionen von η5-cyclopentadienylbis(ethen)cobalt. Angew Chem. 1983, 95: 729-
Acknowledgements
We thank Prosidion Ltd, EPSRC, CIKTN and the University of Bath for funding. We also thank Prof. A. G. Myers (Harvard) for a gift of R. eutrophus B9 cells.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SEL, ALJ and AJWS conceived the experiments. DvdW, TP and MAK synthesised all reported compounds. ALJ performed crystallographic studies. SEL wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van der Waals, D., Pugh, T., Ali Khan, M. et al. A cobalt complex of a microbial arene oxidation product. Chemistry Central Journal 5, 80 (2011). https://doi.org/10.1186/1752-153X-5-80
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-5-80