
Abstract
Background
This study aims to determine the relationship between the antioxidant and anti-inflammatory activities of the thirteen herbs and two fungi extracts, and their total phenolic and flavonoid contents.
Methods
Antioxidant activities were evaluated by four assays: an antioxidant activity assay using Saccharomyces cerevisiae, a DPPH ((2, 2-diphenyl-1-picrylhydrazyl) assay to assess free radical scavenging, an assay assessing ferrous ions or iron (II) chelating ability, and a ferric reducing antioxidant power (FRAP) assay. Total phenolic and flavonoid contents were determined using the Folin-Ciocalteu and aluminium chloride methods, respectively. Anti-inflammatory activities were determined by measuring the inhibition of nitric oxide and TNF-α production in lipopolysaccharide- and interferon-γ-activated J774A.1 macrophages. Their cytotoxicities against macrophages were determined by MTT assay.
Results
A positive linear correlation between antioxidant activities and the total phenolic and flavonoid content of the plant extracts was found. The plant extracts with high phenolic and flavonoid content also exhibited significant anti-inflammatory activity with good cell viability.
Conclusion
The selected herbs could be a rich source of antioxidants and free radical scavenging compounds. The levels of phenolic and flavonoid compounds were correlated with the antioxidant and anti-inflammatory activities of the extracts from the herbs.
Similar content being viewed by others

Background
Reactive oxygen species (ROS) include free radicals, e.g., superoxide (O2●-) and the hydroxyl (OH●), hydroperoxyl (OOH●), peroxy (ROO●) and alkoxy (RO●) radicals, and non-free radicals, e.g., hydrogen peroxide (H2O2) and hypochlorous acid (HOCl), which are constantly produced in the human body during cell metabolism [1]. Others are reactive nitrogen species (RNS) consisting of nitric oxide (NO●), peroxynitrite (ONOO●) and nitrogen dioxide (NO2). Free radicals are important in the regulation of signal transduction, gene expression and activation of receptors [2]. However, an excess of free radicals is toxic to almost every biological molecule in living cells [3], and can cause oxidative damage to functional macromolecules such as DNA, proteins, and lipids if not eliminated quickly [4]. Excess generation of free radicals can lead to many diseases such as age-related disorders, cancer, atherosclerosis, neurodegenerative diseases and inflammation [5, 6]. Antioxidant compounds from plants can minimize the generation of free radicals [6, 7] and alleviate diseases caused by oxidative stress [8, 9]. The phenolics and flavonoids of medicinal herbs contribute to the antioxidant activities of plants [4, 6, 10], and act as anti-inflammatory agents [11]. Production of proinflammatory molecules such as TNF-α and nitric oxide (NO) can modulate inflammation. These inflammatory molecules react with free radicals; for example, NO reacts with O2●- to produce ONOO●, which can cause irreversible damage to cell membranes, leading to cell death and tissue damage [12].
Thirteen medicinal plants and two fungi (Table 1) were chosen in this study for the measurement of antioxidant and anti-inflammatory activities. These plants and fungi are currently used by Chinese medical practitioners, in clinical studies, or both (Table 1). The antioxidant and anti-inflammatory activities of selected thirteen TCM plants and two fungi were measured systematically with a series of assays, as well as their relationship with the total phenolic and flavonoid contents.
Methods
Plant materials
The dried plant materials were obtained from the Beijing Tong Ren Tang Chinese Herbal Medicine shop (Sydney, Australia). A voucher specimen of each sample has been deposited in the Herbal Analysis Laboratory, University of Western Sydney. The scientific names and family names of the herbs are shown in Table 1. The samples were ground to a fine powder in a grinder before extraction.
Chemicals and reagents
Gallic acid, quercetin, 2, 2-diphenyl-1-picrylhydrazyl (DPPH), dimethyl sulfoxide (DMSO), sodium carbonate, aluminium chloride, sodium nitrate, sodium hydroxide, H2O2, Folin-Ciocalteu (F-C) reagent, ascorbic acid, 95% ethanol, bovine serum albumin (BSA), lipopolysaccharide (LPS: E. coli serotype 0127:B8), EDTA, N-(1-1-napthyl) ethylenediamine dihydrochloride, penicillin G sodium benzyl, resazurin sodium 10%, streptomycin, sulfanilamide, tetramethyl benzidine (TMB) and trypan blue were purchased from Sigma (Australia) and Lomb Scientific Pty Ltd. (Australia). Antibiotics, Dulbecco’s modified Eagle’s medium (DMEM), foetal bovine serum (FBS) and glutamine were purchased from GIBCO. Interferon-γ (murine) and tumour necrosis factor-α (TNF-α) enzyme-linked immunosorbent assay (ELISA) kits were purchased from BD Bioscience (San Jose, CA, USA).
Preparation of water extracts
Sample powder (3 g) was autoclaved with 30 mL deionised water at 121°C for 1 h. The extracted samples were centrifuged at 10,447 × g (Sorvall Ultra pro 80, Kendro Instruments Australia Pty, Ltd) for 20 min and the supernatants were transferred to a 50-mL volumetric flask. The residues were rinsed two more times, and the pooled extract was adjusted to 50 mL with deionised water. The samples were stored at −20°C until analysis of phenolic and flavonoid contents.
Preparation of ethanol extracts
Sample powder (3 g) was extracted with 30 mL of 95% ethanol in a water bath at 70°C for 6 h. The extracted samples were centrifuged and the supernatants were transferred to a 50-mL volumetric flask. The residues were rinsed two more times and the pooled extracts were adjusted to 50 mL with 95% ethanol. The samples were stored at −4°C until analysis of phenolic and flavonoid content. All water and ethanol extracts were filtered through a 0.45-μm nylon filter before analysis.
Determination of total phenolic and flavonoid content
The total phenolic content was determined according to the Folin-Ciocalteu (F-C) colorimetric method [13]. Briefly, 50 μL of sample and 50 μL of F-C reagent were pipetted into an Eppendorf tube. The contents were vortexed for 10 s and then left to stand at room temperature for 2 min before the reaction was stopped by adding 500 μL of 5% (w/v) sodium carbonate solution and 400 μL of distilled water, and the volume was adjusted to 1 mL. The mixture was then vortexed and incubated at 45°C for 30 min before cooling rapidly with ice. The absorbance of the solution was measured at 760 nm. Gallic acid concentrations ranging from 25 to 300 μg/mL were prepared and a calibration curve was obtained using a linear fit [14]. The samples were analysed in duplicate.
The total flavonoid content was determined according to the aluminium chloride method [15]. Briefly, 0.5 mL of sample and 300 μL of NaNO2 (1:20 w/v) were pipetted into a test tube and the contents were vortexed for 10 s and left to stand at room temperature for 5 min. After standing, 300 μL of AlCl3 (1:10 w/v), 2 mL of NaOH (1 M) and 1.9 mL of distilled water were added to the reaction mixture, which was then vortexed for 10 s, and the absorbance was measured at 510 nm. Quercetin concentrations ranging from 0 to 1200 μg/mL were prepared and a standard calibration curve was obtained using a linear fit. The samples were analysed in duplicate.
Free radical DPPH scavenging assay
The DPPH assay was carried out according to procedures of Brand-William et al.[16], with minor modifications. Different volumes (10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 μL) of ethanol and water extracts were mixed with DPPH radical in methanol (2.2 mg/L, 200 μL) in a 96-well microplate. The final volume of each well was made up to 300 μL by adding the appropriate amount of methanol. The mixture was shaken gently on a microplate reader (Bio-Rad, Hercules, CA ,USA) and the absorbance at 515 nm was measured every 2 min for 30 min or until the absorbance reached its maximum value. The DPPH concentration in the reaction medium was calculated from a calibration curve derived from serial dilution of the DPPH standard. The control (containing all reagents except the test compound) and standards were subject to the same procedure. The free radical scavenging activity was expressed as the percentage inhibition of free radical generation by the sample, and calculated using the following formula:
where Acontrol is the absorbance of the control, and Asample is the absorbance of the sample at 515 nm. The samples were analysed in triplicate.
Assay for scavenging activity using Saccharomyces cerevisiae
The antioxidant activities of the ethanol and water extracts were also measured according to a S. cerevisiae-based high throughput assay [17]. S. cerevisiae BY4743 was cultured overnight in a 50-mL volume by inoculation of a single colony. The culture was then diluted to an optical density at 600 nm (OD600) of 0.2 in media, and 180 μL of the culture broth was added to each well in a 96-well microtitre plate to which 10 μL per well of each herbal extract had been added in duplicate. H2O2 (10 μL, 32% v/v) was added to give a final concentration of 4 mM. The initial OD600 reading was taken using a microplate reader (Multiskan EX, Thermo Electron, USA), and the plates were then incubated in an incubator at 30°C with shaking at 750 rpm. Yeast growth was monitored by reading the OD600 until 20 h. Ascorbate was used as the positive control. The samples were analysed in duplicate.
Ferrous ion-chelating effect
The ferrous ion-chelating effects of the extracts were estimated according to the method of Chua et al.[18]. Briefly, 740 μL of methanol and 200 μL of sample were incubated with 20 μL of FeCl2 solution (2 mM). The reaction was initiated by adding 40 μL of ferrozine (5 mM) into the mixture, which was then allowed to stand at ambient temperature for 10 min. The absorbance of the reaction mixture was measured at 562 nm. Distilled water instead of ferrozine solution was used as a blank, to correct for any unequal colour of the sample solution. EDTA-Na2 was used as a reference standard. The ferrous ion-chelating ability was calculated as follows:
where Acontrol is the absorbance of the control, Asample is the absorbance of the sample or standard, and Ablank is the absorbance of the blank.
Ferric-reducing antioxidant power assay
The FRAP of the extracts was tested according to Oyaizu [19]. Different concentrations of the samples (1 mL) and the reference chlorogenic acid were added to 2.5 mL of phosphate buffer (0.1 M, pH 6.6) and 2.5 mL of potassium ferricyanide (1.0%, w/v). Each mixture was incubated at 50°C for 20 min and 2.5 mL of trichloroacetic acid (10%) was added. The mixture was shaken vigorously, and the solution (2.5 mL) was mixed with 2.5 mL of distilled water and 0.5 mL of FeCl3 (0.1%, w/v). After a 30 min incubation, absorbances were read at 700 nm. Each analysis was carried out in duplicate. Increased absorbance of the reaction mixture indicates stronger reducing power.
Maintenance, preparation and activation of J774A.1 macrophages
J774A.1 macrophages were incubated in 50-mL flasks in DMEM containing 10% FBS supplemented with antibiotics (1%) and glutamine (1%). The cells were maintained in 5% CO2 at 37°C, with the media being replaced every 3 days. Once the cells had grown to confluence in the culture flask, they were removed using a rubber policeman. The cell suspension was concentrated by centrifugation for 5 min at 180 × g, the supernatant was removed and the pellet was resuspended in a small volume of fresh DMEM (with 1% antibiotics and 10% FBS). The cell density was estimated using a Neubauer counting chamber (Neubauer Precicdor HBG, Germany). Cell concentration was adjusted using DMEM (with 1% antibiotics and 10% FBS) to obtain 75,000 cells/well, after which 180 μL of the cell suspension was dispensed into the 60 inner wells of a 96-well plate. Sterile distilled water was added to the outer row of wells. For assays of the extracts, 20 μL of the extract solution was added with or without activator (1 μg/mL LPS and 10 U/mL IFN-γ). In most cases, the maximum dose of extract used was 1 mg/mL with a minimum of five doses made by serial dilution. The cells were incubated for 24 h at 37°C and 5% CO2. Cells with media alone were used as negative controls and activated cells were used as positive controls.
Griess assay
NO level was determined with the Griess reagent [20], which quantifies nitrite, one of the stable reaction products. Briefly, the supernatant was mixed with an equal volume of Griess reagent (0.1% w/v naphthyl ethylenediamine and 1% w/v sulfanilamide in 5% v/v phosphoric acid) in the wells of a 96-well flat-bottomed microtitre plate. After standing for 5 min at room temperature, the absorbance was measured at 550 nm using a Bio-Rad microplate reader. The remaining supernatant was removed from each well for the TNF-α assay by ELISA.
MTT assay
Cell viability was assessed using a modified MTT assay [21]. Briefly, cells (7×105 cells/well) were seeded in a 96-well plate and treated with 20 μl of sample extract. After incubation for 24 h, the supernatant was replaced with 200 μL of media with MTT solution (500 μg/mL). After further 4 h incubation at 37°C, 200 μL of DMSO was added to each well to solubilise the deposited formazan. The optical density of each well was measured at 490 nm by a microplate reader.
TNF-α determination by ELISA
A sandwich ELISA method was used as described previously [22] to determine the TNF-α concentration. The capture antibody was used with 0.1 M sodium carbonate buffer (pH 9.5). Serial dilutions of TNF-α standard from 0 to 1000 pg/mL in diluent (10% BSA in PBS) were used as the internal standard. TNF-α was detected using a biotinylated secondary antibody and an avidin peroxidase conjugate with TMB as the detection reagent. After 30 min, the reaction was stopped by 2 M sulfuric acid and the absorbance was measured at 450 nm.
Data presentation and analysis
The results were expressed as mean ± standard deviation (SD), calculated from duplicate determinations and the linear relationship was visually determined. The significance of differences among groups of data were determined with one-way analysis of variance (ANOVA) by using statistical software SPSS 18.0 for Windows (IBM, USA) and Duncan’s multiple range test [23]. The threshold for statistical significance was P < 0.05.
Results and discussion
Total phenolic and flavonoid contents
The yields of the hot water and ethanol extracts varied widely (Table 2). The yield of the water extracts (13.9–328.9 mg/g) was higher than that of the ethanol extracts (3.3–22.3 mg/g). The total phenolic and flavonoid contents of the extracts was measured using the F-C reagent and aluminium chloride methods, respectively. The results obtained with the water and ethanol extracts are presented in Tables 3 and 4, respectively.
Water extracts
The highest phenolic content was found in C. bonariensis (H15: 200.0 μg/mg), followed by A. argyi (H7: 137.3 μg/mg), E. brevicornum (H14: 101.9 μg/mg) and P. vulgaris (H9: 100.0 μg/mg), while the lowest content was found in P. grandiflora (H13: 5.21 μg/mg). In the other herbs the total phenolic content ranged 12.7 to 93.5 μg/mg. The flavonoid content varied greatly from 9.92 to 532.48 μg/mg (Table 3). The highest flavonoid content was found in A. argyi (H7: 532.48 μg/mg) followed by C. bonariensis (H15: 276.48 μg/mg), E. brevicornum (H14: 265.28 μg/mg), B. genistelloides (H1: 200.08 μg/mg) and M. alba (H8: 180.88 μg/mg). The water extracts contained a higher proportion (≥ 50%) of flavonoids than phenolics, except S. baicalensis, which contained 75% phenolic compounds (Figure 1A).

Proportional relation (%) of flavonoid content to phenolic content in the herbal extracts. A: Hot water extracts. B: Ethanol extracts.
Ethanol extracts
The flavonoid content of the herbs was in the range 17.6–449.6 μg/mg, with large variations between the plants (Table 4). The highest flavonoid content was found in L. gracile (H10: 449.6 μg/mg extract) followed by P. vulgaris (H9: 194.02 μg/mg extract), P. alkekengi (H2: 120.02 μg/mg) and S. baicalensis (H12: 117.82 μg/mg); while the lowest was in C. militaris (H11: 17.62 μg/mg). Phenolics and flavonoids neutralize free radicals by donating a hydrogen atom or an electron, and chelate metal ions [24]. The ethanol extracts contained a higher proportion of flavonoids than phenolics (Figure 1B).
Antioxidant activities
The antioxidant activities of the water and ethanol extracts in this study were evaluated using an antioxidant assay for scavenging activity with S. cerevisiae, a DPPH free radical scavenging assay, an assay assessing the iron (II) chelating ability, and a FRAP assay. As presented in Table 3, thirteen samples showed antioxidant activity in the yeast-based assay that utilises the oxidative stress-mediated cell cycle arrest to reveal the oxidant scavenging and intracellular antioxidant activity. Most of the water and ethanol extracts (fourteen samples out of fifteen) displayed significant antioxidant activity as measured by the DPPH method. The results from the antioxidant assay suggested that the water extracts showed higher activity than the ethanol extracts in general. The percentage inhibitions of the free radical DPPH were in the range 7.08–69.91% for the water extracts and 2.65–238.94% for the ethanol extracts. Significant free radical scavenging activities (≥ 50%) were observed in all water extracts, except for those of B. chinensis (H5), T. capilipes (H6) and P. grandiflora (H13), in which the inhibition of DPPH was less than 10%. The highest inhibition of DPPH activity was found in the ethanol extract of L. gracile (H10: 238.94%). The free radical scavenging activities were very low in the ethanol extracts of C. militaris (H11: 4.4%) and P. grandiflora (H13: 2.6%). The herbs displayed antioxidant activities owing to a combination of their total phenolic and flavonoid content as reported in other studies [6, 10].
The ferrous ion/iron (II) chelating abilities of the samples are summarised in Tables 3 and 4. Iron is the chief peroxidant and is able to generate lipid peroxidation through the Fenton reaction or by accelerating the dissociation of lipid hydroperoxides to their respective peroxy and alkoxy radicals [25]. The ability of the extracts to bind Fe2+ in the presence of ferrozine was compared with that of EDTA [26]. The water extracts had a higher Fe2+ chelating effect than the ethanol extracts. The water extracts of A. argyi (H7), P. vulgaris (H9), L. gracile (H10) and E. brevicornum (H14) exhibited 143.5, 106.2, 138.0 and 241.1% chelation of Fe2+ equivalent to 83.2, 65.9, 84.0 and 144.0 μg/mL EDTA, respectively. The highest percentage chelating capacity of the ethanol extracts was found in L. Seu calvatia (H4: 99.9%), followed by T. capilipes (H6: 73.2%); while other ethanol extracts showed a low percentage chelating capacity in the range 0–42.9% compared with water extracts. Flavonoid and phenolic compounds are known to act as antioxidants, radical scavengers and metal chelators [27]. These extracts have appreciable amounts of flavonoid and phenolic compounds that may contribute to their chelating ability.
The antioxidant activities of the water and ethanol extracts measured by the FRAP assay are shown in Tables 3 and 4. A wide variation was observed. The water extract of P. vulgaris (H9) showed the highest FRAP (CPE 103.9 mg/g) while P. grandiflora (H13) showed the lowest FRAP (CPE 2.9 mg/g). The FRAP of the ethanol extracts was in the range 3.56–87.5 CPE mg/g (Table 4). FRAP was significant in some of the water and ethanol extracts owing to the presence of phenolic and flavonoid compounds and a similar trend is observed for many other plant extracts that have been studied [28]. The reducing properties are generally associated with compounds that can donate hydrogen atoms to free radicals and convert them into stable non-reactive molecules [29].
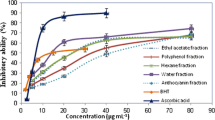
The anti-inflammatory properties of the water extracts were evaluated on the basis of their inhibition of NO and TNF-α production in LPS- and IFN-γ activated macrophages (Figure 2). NO is an important intracellular and intercellular regulator of multiple biological functions, including macrophage-mediated cytotoxicity, neurotransmission and smooth muscle relaxation [30, 31]. Overexpression of NO has been associated with oxidative stress [32, 33] and with the pathophysiology of various diseases such as arthritis, diabetes, autoimmune disease and chronic inflammation [34, 35]. It has also been shown that the production of TNF-α is crucial for the synergistic induction of NO synthesis in IFN-γ- and LPS-stimulated macrophages [36, 37]. The cell toxicity of the plant extracts was determined by MTT assay. The results indicated that most of the extracts showed significant cell viability at a concentration of 100 μg/mL (85% or higher), except for P. vulgaris (H9) and E. brevicornum (H14). Therefore the water extracts of these plants exhibited low toxicity. Most of the extracts (except for H7, H13, H14 and H15) also inhibited macrophage NO production (Figure 2B). H7, H13, H14 and H15 extracts activated macrophage NO production as well as TNF-α production (Figure 2C). The role of TNF-α production was suggested for the antitumor activity of phenols and flavonoids [38], but the underlying mechanisms by which different flavonoid and phenolic compounds affect TNF-α and NO production are to be investigated.

Effect of water extracts of herbs on LPS-stimulated macrophage production of NO and TNF-α, and cell viability. A: Cell viability. B: NO production. C: TNF-α production.
Conclusion
The selected herbs could be a rich source of antioxidants and free radical scavenging compounds. The levels of phenolic and flavonoid compounds were correlated with the antioxidant and anti-inflammatory activities of the herb extracts.
Abbreviations
- NO:
-
Nitric Oxide
- TNF-α:
-
Tumour Necrosis Factor-alpha
- DPPH:
-
2,2-diphenyl-1-picrylhydrazyl
- FRAP:
-
Ferric reducing antioxidant power
- LPS:
-
Lipopolysaccharide
- IFN-γ:
-
Interferon gamma
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FBS:
-
Foetal bovine serum.
References
Cheeseman KH, Slater TF: An introduction to free radical biochemistry. Br Med Bull. 1993, 49: 481-493.
Ajith TA, Janardhanan KK: Indian medicinl mushrooms as a source of antioxidant and antitumor agents. J Clin Biochem Nutr. 2007, 40: 157-162. 10.3164/jcbn.40.157.
Liu H, Visner GA: Oxidants and antioxidants. Molecular Pathology of Lung Diseases. Edited by: Zander DS, Popper HH, Jagirdar J, Haque AK, Cagle PT, Barrios R. 2008, Springer, 470-475.
Zhang L, Ravipati AS, Koyyalamudi SR, Jeong SC, Reddy N, Smith PT, Bartlett J, Shanmugam K, Münch DG, Wu MJ: Antioxidant and anti-inflammatory activities of selected medicinal plants containing phenolic and flavonoid compounds. J Agr Food Chem. 2011, 59: 12361-12367. 10.1021/jf203146e.
Tang SY, Whiteman M, Peng ZF, Jenner A, Yong EL, Halliwell B: Characterization of antioxidant and antiglycation properties and isolation of active ingredients from traditional Chinese medicines. Free Radic Biol Med. 2004, 36: 1575-1587. 10.1016/j.freeradbiomed.2004.03.017.
Cai Y, Luo Q, Sun M, Corke H: Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sci. 2004, 74: 2157-2184. 10.1016/j.lfs.2003.09.047.
Dragland S, Senoo H, Wake K, Holte K, Blomhoff R: Several culinary and medicinal herbs are important sources of dietary antioxidants. J Nutr. 2003, 133: 1286-1290.
Akinmoladun AC, Obuotor EM, Farombi EO: Evaluation of antioxidant and free radical scavenging capacities of some Nigerian indigenous medicinal plants. J Med Food. 2010, 13: 444-451. 10.1089/jmf.2008.0292.
Özen T, Çöllü Z, Korkmaz H: Antioxidant properties of Urtica pilulifera root, seed, flower, and leaf extract. J Med Food. 2010, 13: 1224-1231. 10.1089/jmf.2009.1303.
Hendra R, Ahmad S, Oskoueian E, Sukari A, Shukor MY: Antioxidant, Anti-inflammatory and Cytotoxicity of Phaleria macrocarpa (Boerl.) Scheff Fruit. BMC Complem Altern M. 2011, 11: 110-10.1186/1472-6882-11-110.
Talhouk R, Karam C, Fostok S, El-Jouni W, Barbour E: Anti-inflammatory bioactivities in plant extracts. J Med Food. 2007, 10: 1-10. 10.1089/jmf.2005.055.
Wang C, Schuller Levis GB, Lee EB, Levis WR, Lee DW, Kim BS, Park SY, Park E: Platycodin D and D3 isolated from the root of Platycodon grandiflorum modulate the production of nitric oxide and secretion of TNF-[alpha] in activated RAW 264.7 cells. Int J Immunopharmacol. 2004, 4: 1039-1049. 10.1016/j.intimp.2004.04.005.
Cicco N, Lanorte MT, Paraggio M, Viggiano M, Lattanzio VA: Reproducible, rapid and inexpensive Folin-Ciocalteu micro-method in determining phenolics of plant methanol extracts. Microchem J. 2009, 91: 107-110. 10.1016/j.microc.2008.08.011.
Cai Y, Sun M, Xing J, Corke H: Antioxidant phenolic constituents in roots of Rheum officinale and Rubia cordifolia: structure-radical scavenging activity relationships. J Agr Food Chem. 2004, 52: 7884-7890. 10.1021/jf0489116.
Zhishen J, Mengcheng T, Jianming W: The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem. 1999, 64: 555-559. 10.1016/S0308-8146(98)00102-2.
Brand-Williams W, Cuveleir ME, Berset C: Use of a free radical method to evaluate antioxidant activity. Lebensm Wiss Technol. 1995, 28: 25-30. 10.1016/S0023-6438(95)80008-5.
Wu MJ, O’Doherty PJ, Fernandez HR, Lyons V, Rogers PJ, Dawes IW, Higgins VJ: An antioxidant screening assay based on oxidant induced growth arrest in Saccharomyces cerevisiae. FEMS Yeast Res. 2011, 11: 379-387. 10.1111/j.1567-1364.2011.00726.x.
Chua MT, Tung YT, Chang ST: Antioxidant activities of ethanolic extracts from twigs of Cinnamomum osmophleum. Bioresource Technol. 2008, 99: 1918-1925. 10.1016/j.biortech.2007.03.020.
Oyaizu M: Studies on product of browning reaction prepared from glucosamine. Jpn J Nutr. 1986, 44: 307-315. 10.5264/eiyogakuzashi.44.307.
Cui S, Reichner JS, Mateo RB, Albina JE: Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or independent mechanisms. Cancer Res. 1994, 54: 2462-2467.
Behl C, Davis J, Cole GM, Schubert D: Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun. 1992, 186: 944-950. 10.1016/0006-291X(92)90837-B.
Zhang L, Koyyalamudi SR, Jeong SC, Reddy N, Smith PT, Ananthan R, Longvah T: Antioxidant and immunomodulatory activities of polysaccharides from the roots of Sanguisorba officinalis. Int J Biol Macromol. 2012, 51: 1057-1062. 10.1016/j.ijbiomac.2012.08.019.
Duncan DB: Multiple range tests for correlated and heteroscedastic means. Biometrics. 1957, 13: 164-176. 10.2307/2527799.
Petti S, Scully C: Polyphenols, oral health and disease: a review. J Dent. 2009, 37: 413-423. 10.1016/j.jdent.2009.02.003.
Gioti E, Fiamegos Y, Skalkos D, Stalikas C: Antioxidant activity and bioactive components of the aerial parts of Hypericum perforatum L. from Epirus, Greece. Food Chem. 2009, 117: 398-404. 10.1016/j.foodchem.2009.04.016.
Chang HF, Yang LL: Radical-scavenging and rat liver mitochondria lipid peroxidative inhibitory effects of natural flavonoids from traditional medicinal herbs. J Med Plants Res. 2012, 6: 997-1006.
Mira L, Fernandez MT, Santos M, Rocha R, Florencio MH, Jennings KR: Interactions of flavonoids with iron and copper ions: a mechanism for their antioxidant activity. Free Radic Res. 2002, 36: 1199-1208. 10.1080/1071576021000016463.
Katalinic V, Milos M, Kulisic T, Jukic M: Screening of 70 medicinal plant extracts for antioxidant capacity and total phenols. Food Chem. 2006, 94: 550-557. 10.1016/j.foodchem.2004.12.004.
Gordon MH: The mechanism of antioxidant action in vitro. Food antioxidants. Edited by: Hudson BJF. 1990, London: Elsevier Applied Science, 1-18.
Ignarro LJ: Regulation of cytosolic guanylyl cyclase by porphyrins and metalloporphyrins. Adv Pharmocol. 1994, 26: 35-65.
Beckman JS, Koppenol WH: Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996, 271: C1424-C1437.
Sies H, Mehlhorn R: Mutagenicity of nitroxide-free radicals. Arch Biochem Biophys. 1986, 251: 393-396. 10.1016/0003-9861(86)90087-1.
Ji Y, Akerboom TP, Sies H, Thomas JA: S-nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys. 1999, 362: 67-78. 10.1006/abbi.1998.1013.
Moncada S, Palmer RM, Higgs EA: Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991, 43: 109-142.
Arteel GE, Briviba K, Sies H: Protection against peroxynitrite. FEBS Lett. 1999, 445: 226-230. 10.1016/S0014-5793(99)00073-3.
Green SJ, Crawford RM, Hockmeyer JT, Meltzer MS, Nacy CA: Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J Immunol. 1990, 145: 4290-4297.
Jun CD, Choi BM, Kim HM, Chung HT: Involvement of protein kinase C during taxol-induced activation of murine peritoneal macrophages. J Immunol. 1995, 154: 6541-6547.
Peng B, Hu Q, Sun L, Chen Y, Liu X, Li J, Chang Q, Wang L, Tang J: Duchesnea phenolic fraction inhibits tumor growth through restoting the Th1/Th2 balance in U14 cervical cancer bearing mice. Chin Med. 2012, 3: 42-45. 10.4236/cm.2012.31007.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SRK, SCJ and CK designed the study and wrote the manuscript. PD, SCJ, SRK and SL performed the experiments. PD, SRK, SCJ, CK, and SL analysed the data. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Diaz, P., Jeong, S.C., Lee, S. et al. Antioxidant and anti-inflammatory activities of selected medicinal plants and fungi containing phenolic and flavonoid compounds. Chin Med 7, 26 (2012). https://doi.org/10.1186/1749-8546-7-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1749-8546-7-26