
Abstract
Background
Diagnosis of West Nile virus (WNV) infections is often difficult due to the extensive antigenic cross-reactivity among flaviviruses, especially in geographic regions where two or more of these viruses are present causing sequential infections. The purpose of this study was to characterize a panel of monoclonal antibodies (MAbs) produced against WNV to verify their applicability in WNV diagnosis and in mapping epitope targets of neutralizing MAbs.
Methods
Six MAbs were produced and characterized by isotyping, virus-neutralization, western blotting and MAb-epitope competition. The MAb reactivity against various WNVs belonging to lineage 1 and 2 and other related flaviviruses was also evaluated. The molecular basis of epitopes recognized by neutralizing MAbs was defined through the selection and sequencing of MAb escape mutants. Competitive binding assays between MAbs and experimental equine and chicken sera were designed to identify specific MAb reaction to epitopes with high immunogenicity.
Results
All MAbs showed stronger reactivity with all WNVs tested and good competition for antigen binding in ELISA tests with WNV-positive equine and chicken sera. Four MAbs (3B2, 3D6, 4D3, 1C3) resulted specific for WNV, while two MAbs (2A8, 4G9) showed cross-reaction with Usutu virus. Three MAbs (3B2, 3D6, 4D3) showed neutralizing activity. Sequence analysis of 3B2 and 3D6 escape mutants showed an amino acid change at E307 (Lys → Glu) in the E protein gene, whereas 4D3 variants identified mutations encoding amino acid changed at E276 (Ser → Ile) or E278 (Thr → Ile). 3B2 and 3D6 mapped to a region on the lateral surface of domain III of E protein, which is known to be a specific and strong neutralizing epitope for WNV, while MAb 4D3 recognized a novel specific neutralizing epitope on domain II of E protein that has not previously been described with WNV MAbs.
Conclusions
MAbs generated in this study can be applied to various analytical methods for virological and serological WNV diagnosis. A novel WNV-specific and neutralizing MAb (4D3) directed against the unknown epitope on domain II of E protein can be useful to better understand the role of E protein epitopes involved in the mechanism of WNV neutralization.
Similar content being viewed by others


Background
West Nile virus is an arbovirus member of the Japanese Encephalitis virus (JEV) serocomplex of the genus Flavivirus of the Flaviviridae family. WNV infection is one of the most widespread arboviral infections and can cause encephalitis in humans. Its transmission cycle involves mosquito-vectors (mainly Culex spp.) and birds as amplifying reservoirs, but a wide variety of vertebrate species, including reptiles, amphibians and mammals, such as equines and humans, are also susceptible to infection [1].
The WNV genome is made up of a single stranded positive-sense RNA molecule that encodes three structural proteins (capsid (C); pre-membrane (prM); and envelope (E)) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) [2]. The envelope E protein is the major surface protein of flaviviruses and the primary immunogen that plays a central role in virus attachment and entry into a cell via membrane fusion [3]. Crystallographic analysis reveals that the E glycoprotein of flaviviruses folds into three distinct structural domains (I, II and III) [4–6]. In particular, domain III of WNV E protein (DIII) is the putative receptor-binding domain and is an important target for neutralizing antibodies and in vivo protection [7–11]. The recent outbreaks of West Nile Disease in humans and horses in Europe and the spread of the virus from North through South America during the last decade suggest that the epidemiology of this infection is evolving. In the Mediterranean basin, outbreaks of WNV infection in recent years have been reported in France (2004 and 2006), Italy (2008, 2009) Morocco (2010), Spain (2010) and Greece [12]. WNV was previously considered an exotic agent, while it is now regarded as an emerging problem for both human and veterinary public health. These outbreaks have stimulated research into virus detection and characterization, underlining the need for rapid assays. Although many methods have been developed for WNV diagnosis, it is commonly difficult due to the extensive antigenic cross-reactivity among flaviviruses, especially in geographic areas where two or more of these viruses are present causing sequential infections [13]. It has recently been shown that WNV and Usutu virus (USUV) have similar transmission cycles, with overlapping geographic distributions [14, 15]. In this context, MAbs having strong and specific reactivity to WNV antigens are the most suitable choice for the development of standardized diagnostic tools.
The purpose of this study was to characterize a panel of monoclonal antibodies produced against WNV to verify their applicability in WNV diagnosis and in mapping epitope targets of neutralizing MAbs.
The results suggest the applicability of these MAbs to various analytical methods for WNV diagnosis allowing the characterization of a novel WNV-specific and neutralizing epitope located on DII of E protein that has not previously been described with WNV MAbs.
Results
Selection and characterization of monoclonal antibodies
During the screening phase of hybridomas, several MAbs that were reactive to the WNV antigen were obtained. Six hybridomas (3B2, 3D6, 1C3, 4D3, 2A8, 4G9) showing a strong signal with indirect ELISA and immunofluorescence (IF) against homologous WNV were selected and cloned by limiting dilution to ensure monoclonality and stability. Positive clones that secreted a high titer of selected antibodies were further identified and MAbs were efficiently purified by protein-A chromatography and conjugated with horseradish peroxidase (HRP). The general characteristics of selected anti-WNV MAbs are summarized in Table 1.
All six MAbs showed good reactivity with Immunoperoxidase Monolayer Assays (IPMA) staining on all the eight WNVs tested. Four MAbs 3B2, 3D6, 4D3 and 1C3 resulted WNV-specific without any cross-reaction to USUVs and Tick-borne encephalitis virus (TBEV), and three of these (3B2, 3D6, 4D3) showed neutralizing activity at the same titers against both the homologous WNV Eg101 and the prototype lineage 2 WNV B956. MAbs 2A8 and 4G9 showed cross-reaction with the two USUVs tested, but not with TBEV. No neutralizing activity was observed. The heavy chain subclasses were determined as IgG2a for all MAbs with the exception of 4D3, which was determined as IgG2b. The light chains of all of these were kappa isotype.
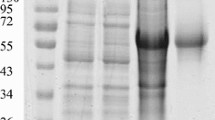
Two MAbs (3B2, 3D6) resulted positive at western blotting (WB), revealing a band with a molecular weight of 52Kd corresponding to E protein. The remaining MAbs did not recognize denatured WNV antigen at WB, suggesting that they are all directed to conformational-dependent epitopes (Figure 1A). Figure 1B shows IPMA assays performed with WNV, USUV and MAbs 3B2 and 2A8.

(A) Western blot analysis. Reactivity of six anti-WNV MAbs with denatured antigen. A MAb against influenza virus was used as negative control. M: protein molecular weight markers. Lane 1: sera from WNV infected horse. Lane 2: sera from WNV vaccinated horse. Lane 3: sera from WNV negative horse. (B) Detection of WNV and USUV-infected cells using IPMA (10X). WNV-infected Vero cells stained with MAb 3B2 (1) and 2A8 (2). USUV-infected VERO cells stained with MAb 3B2 (3) and 2A8 (4).
None of the MAbs showed any cross-reaction against Dengue virus serotype 1, 2, 3 and 4 (DENV 1, 2, 3, 4), Yellow fever virus (YFV), TBEV and Japanese Encephalitis virus (JEV) by indirect ELISA.
Competition binding ELISAs for MAb epitope studies
To determine if the selected MAbs would bind to overlapping epitopes, we tested MAbs using competitive binding ELISAs. For these studies, the saturating concentrations of unlabeled MAbs were incubated with partially purified WNV antigen bound to the ELISA plates, after which an HRP-conjugated "competitor" MAb was added. If preincubation of the antigen with an unlabeled MAb blocked subsequent binding of a conjugated MAb, we concluded that MAbs bind to similar or at least overlapping epitopes. The optimal dilution of each HRP-conjugated MAb was determined by testing serial two-fold dilution in WNV-coated ELISA plates and selecting this as the working dilution to generate an optical density of around 1.5. The selected working dilution was 1/8000 for MAb 3B2, 1/16000 for MAbs 3D6, 2A8, 4G9 and 1/4000 for 4D3, 1 C3. The results of competitive ELISAs are reported in Table 2.
HRP-conjugated 3B2 inhibited the binding of 3D6 to the antigen and vice versa, showing that 3B2 and 3D6 recognized overlapping epitopes. 4D3 and 1 C3 did not significantly compete with any of the other MAbs tested in this assay, which meant that 4D3 and 1 C3 recognized two distinct epitopes different from 3B2, 3D6, 2A8 and 4 G9. HRP-conjugated 2A8 inhibited the binding of 4G9 to the antigen and vice versa, indicating that 2A8 and 4G9 recognized overlapping epitopes.
One-way partial competition observed for some MAbs, as for 4D3 HRP, could be due to antibody-induced conformational changes in E protein upon antibody binding or steric hindrance. MAbs were thus categorized into four groups. MAbs binding to the same epitopes or overlapping epitopes were put into the same group, while non-competing MAbs were grouped individually (Table 2). Group one consisted of MAbs 3B2 and 3D6, group two included MAb 1 C3, group three included MAb 4D3 and group four consisted of MAbs 2A8 and 4G9.
Antigen capture ELISA (AC-ELISA)
All possible combinations of trapping and conjugated MAbs were evaluated to establish a sensitive AC-ELISA for WNV detection. Each pair of MAbs was able to detect all WNVs when used in sandwich ELISA without significant differences in sensitivity. No reactivity to TBEV was observed while pairs of MAbs formed by 2A8 and 4 G9 allowed the detection of USUVs (data not shown). Good WNV-specificity combined with the highest signal was obtained using 3B2 both as capture antibody and HRP-conjugated as detector antibody. To determine the detection limit of AC-ELISA, serial dilutions of WNV Eg101 culture supernatant (105.1 TCID50/100 μl) were used to construct the binding curve (Figure 2). An uninfected cell culture supernatant was used as negative control. According to the cut-off threshold, calculated as the mean of optical density (OD) of negative control plus two standard deviations, the detection limit for the WNV AC-ELISAs was deduced to be 10 3.8 TCID50/100 μl.

Sensitivity of AC-ELISA using MAb 3B2 and WNV cell culture supernatant. Detection limit from 200- to 300-fold dilution of cell culture supernatant which was 103,8TCID50/0,1 ml. The broken line indicates the absorbance ratio cut-off value.
Competitive binding assays with experimental sera
This experiment was designed to evaluate the capability of known experimental sera to inhibit the binding of anti-WNV MAbs to the antigen and then to verify the possibility of using them in competitive ELISA tests for serological diagnosis in various animal species.
Sera from SPF chickens experimentally infected with WNV and equine sera immunized using inactivated vaccine were tested in competitive ELISAs with each HRP-conjugate MAb at a predetermined dilution. The results, expressed as percent inhibition of MAb binding by competition with serum antibodies, are shown in Table 3. Good competition for antigen binding between all MAbs and sample sera collected from vaccinated horse, as well as infected chicken, was observed.
Escape mutant analysis for MAb epitope mapping
To identify residues that might define epitopes recognized by the neutralizing monoclonal antibodies, potential neutralization-resistant escape mutants (EM) were selected by growing WNV Eg101 in Vero cells in the presence of MAbs 3B2, 3D6 and 4D3.
Four or five neutralization-resistant escape mutants were identified for each MAb. Virus neutralization assays showed that 3B2 and 3D6 were unable to neutralize both EM3B2 and EM3D6, but retained strong neutralizing activity against EM4D3. On the contrary, MAb 4D3 was unable to neutralize EM4D3, but retained strong neutralizing activity against both EM3B2 and EM3D6. Comparative sequence analysis of viral genome corresponding to E genes of escape mutants and wild-type WNV Eg101 was performed. A similar single-nucleotide mutation was identified on the E-protein genes of both EM3B2 and EM3D6 at residue 919 (AAG → GAG) encoding an amino acid change at E307 (Lys → Glu). Various nucleotide mutations were identified on the E-protein gene of EM4D3 at residue 827 (AGC → ATC) encoding an amino acid change at E276 (Ser → Ile) or at residue 833 (ACT → ATT) encoding an amino acid change at E278 (Thr → Ile). Tridimensional macromolecular structure of E protein showing these amino acid residues are reported in Figure 3. None of these mutations was associated with measurable differences for growth in cell cultures.

3D macromolecular structure of WNV E protein based on the crystal structure available in Molecular modelling database (PDB: 2I69-A) visualized by Cn3D version 4.3. Mutated amino acid residues important for MAbs binding 3B2, 3D6 (E307) and 4D3 (E276, E278) are evidenced in yellow.
Based on the sequence of in vitro neutralization escape variants, MAb 3B2 and 3D6 map to a region on the lateral surface of DIII, which has been previously described as an important neutralizing epitope for WNV [7, 16], while MAb 4D3 appears to recognize a novel neutralizing epitope on DII that has not previously been described with WNV MAbs.
Discussion
West Nile virus is not only a human pathogen, but is also a major veterinary pathogen. One of the principal factors preventing the development of clinical disease due to WNV infection is the presence of protective neutralizing antibodies. However, despite much progress, many unanswered questions remain concerning WNV neutralization mechanisms.
The majority of potent murine neutralizing MAbs described to date against WNV bind to domain III, more specifically to an epitope on the lateral surface of DIII recognized by specific antibodies with the strongest neutralizing activity in vitro and in vivo [7, 9, 10, 16, 17]. In this study, six monoclonal antibodies produced against WNV lineage 1 were characterized and their applicability in laboratory diagnosis was assessed. Furthermore, through selection of neutralizing-MAb escape mutants, we defined the molecular basis of two epitopes recognized by our neutralizing MAbs.
We showed that all six MAbs (3B2, 3D6, 4D3, 1C3, 2A8, 4G9) had strong reactivity in IPMA for all the eight WNVs tested, including reference strains belonging to lineages 1 and 2 as well as field strains isolated during the Italian epidemics. This finding suggests that all MAbs recognized conserved regions among WNVs. We also showed the cross-reactivity of 2A8 and 4G9, which recognize the overlapping epitopes shared between WNV and USUV while no MAbs exhibited any cross-reactions with other related flaviviruses tested. Considering that WNV, USUV and TBEV are the flaviviruses currently circulating in Europe, a good knowledge of MAb reactivity against them is very important for their implementation as diagnostic tools.
MAbs 3B2, 3D6 and 4D3 neutralized lineage 1 and lineage 2 WNV. Moreover, they neither neutralized nor recognized USUV and TBEV. MAb-binding sites of neutralizing MAbs have been characterized by the selection and sequencing of MAb escape mutants. We showed that 3B2 and 3D6 recognized the same epitopes located on the distal lateral surface of DIII. Specifically, sequence analysis of 3B2 and 3D6 escape variants showed a single-nucleotide mutation in the E protein gene that encodes an amino acid change at E307 (Lys → Glu). This residue defines an epitope that has previously been described as an important epitope recognized by WNV-specific antibodies with the strongest neutralizing activity [7, 9, 10, 16, 17]. Recent studies report that recombinant DIII peptides containing virus-specific epitopes could be used for specific serological diagnosis of flaviviral infections [18–20]. Considering the extensive antigenic cross-reactivity among flaviviruses, we speculated that joining recombinant DIII peptides to anti-DIII MAbs such as 3B2 and 3D6 for the development of serological tests for WNV diagnosis could provide good results as regards specificity. Additionally, Oliphant et al. (2005) showed that MAbs against WNV-DIII have therapeutic potential against this virus, while DIII E peptides could be used as subunit flavivirus vaccines [18, 21–23].
Numerous studies on neutralizing monoclonal antibodies outside DIII have been reported [24–27]. We saw that MAb 4D3 recognized a neutralizing epitope on domain II that is a JEV complex epitope, already seen in other studies [28] but which has not been previously described in WNV. Nucleotide mutations were identified on the E gene of 4D3 variants encoding amino-acid changes at E276 (Ser → Ile) or E278 (Thr → Ile). The WNV inhibiting role of this specific neutralizing epitope located on DII needs further investigation to verify its role as a candidate for the development of diagnostic tools, as well as for use as an antiviral vaccine and for therapeutics. The mechanisms of neutralization and the role of the different epitopes of the WNV envelope protein have not been fully understood. According to Sanchez et al. [29], the neutralizing response to WNV generated in infected horses is both variable and polyclonal in nature, with epitopes both within and outside DIII playing important roles. The same study reports that in some horses that were naturally infected with WNV, antibodies directed against DIII epitopes were not consistently detected, and DII may be an important target for neutralizing antibodies in these animals. The anti-domain II MAb 4D3 could be used in competitive tests, in parallel to MAbs directed against DIII, to reveal WNV-neutralizing antibodies in animal species. This is supported by our results obtained with competitive ELISAs designed using experimental horse and chicken sera and HRP-conjugated MAbs. Good competition for antigen binding between MAb and sample sera from vaccinated horse, as well as infected chicken, was observed. Additionally, good competition was also observed in competitive ELISAs between 3B2, 3D6 and infected horse sera collected during the WNV Italian epidemics (data not shown). These results also indicate that all MAbs herein described react to epitopes with high immunogenicity and that they could be used for the development of competitive tests for the detection of different types of anti-WNV antibodies in animal species, e.g. WNV-specific and neutralizing antibodies directed to DIII (MAbs 3B2 and 3D6) or DII (MAb 4D3), WNV-specific non-neutralizing antibodies (Mab 1C3) or flavivirus cross-reactive non-neutralizing antibodies (MAbs 2A8 and 4 G9).
The availability of MAbs with strong reactivity to WNV is also a crucial component for AC-ELISA development usable for large-scale screening in surveillance programs. Various MAb combinations were evaluated for WNV detection using AC-ELISA. It was found that a combination of 3B2 as catcher and tracer MAb gave the highest signal combined with good specificity. The detection limit of 103.8TCID50/100 μl for WNV-infected cell culture supernatant obtained in the present study is comparable with that found in previously published AC-ELISA [30]. Considering that AC-ELISAs performed with pairs of MAbs made up of 2A8 and 4G9 allowed the detection of USUVs, these tests could be useful for viral detection in pathological specimens or cell cultures in areas where WNV and USUV spread concomitantly.
Conclusions
Six MAbs against WNV were produced and characterized. The results suggest the applicability of these MAbs to various analytical methods for antigen or antibody detection in areas such as the Mediterranean basin, where there is contemporary circulation of WNV, USUV and TBEV. Additionally, in this study the novel WNV-specific neutralizing MAb 4D3 directed against the unknown epitope on domain II of the E protein of WNV was generated and characterized.
Materials and methods
Viruses
The virus used in this study as an immunogen for MAb production was the WNV reference strain Eg101.
Other WNVs used in this study included: B956 ATCC, NY99 ATCC, Central African Republic 1967, 203204/08 isolated from magpies (Pica pica) during the 2008 Italian epidemic and three other Italian field strains (225677/09, 208659/09, 204913/09) isolated in our lab in 2009 from a jay (Garrulus glandarius), a horse and mosquitoes (Culex pipiens) respectively. Other flaviviruses used included three strains of USUV, reference strain SAAR 1776, Vienna 2001-blackbird and 200092/2010 (Italian field strain isolated in our lab from a blackbird), and TBEV strain 103457/2009 (Western TBEV group) isolated in our lab from the brain of dog that had been in an endemic area (Hungary) for a short period in April 2009. Virus stocks were prepared in Vero cells that were cultured at 37°C with 5% CO2 in minimum essential medium (MEM) containing 10% of fetal serum bovine, 0.1 mM of nonessential amino acids, 0.15% sodium bicarbonate, 2 mM L-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml). Virus titers were determined by endpoint assays in Vero cells.
Antigen preparation
For its use as an immunogen for MAb production in Balb/c mice and for the preparation of ELISA coating antigen, WNV Eg101 was partially purified as follows. WNV was grown on a preformed monolayer of Vero cells and incubated at 37°C until the cytopathic effect (CPE) is complete. After a freeze-thaw step, virus inactivation was carried out by the addition of 0.05% (v/v) Betapropiolactone to the culture fluids, followed by incubation at 37°C for 2 h plus overnight incubation at 4°C. Virus inactivation in the culture fluid was verified following three blind passages in Vero cell monolayers without CPE development. Inactivated supernatant fluid was clarified by centrifugation at 4000 rpm for 30 min, added to 8% of PEG 6000 and NaCl 0.5 M then placed overnight at 4°C in agitation. The suspension was centrifuged at 5000 rpm for 30 min and the pellet was re-suspended in phosphate-buffer saline solution pH 7.4 (PBS) at 20X concentration compared to the initial volume. Following further clarification by centrifugation (5000 rpm for 30 min), the viral suspension was purified by ultracentrifugation at 35000 rpm for 2 h (rotor TST41 Kontron) through a 25% (w/w) sucrose cushion and the pellet was re-suspended in PBS. This concentrated antigen was kept at -70°C until use.
Experimental sera production
Immune chicken sera
Five SPF chickens were used. Three of these (positive control) were experimentally infected intramuscularly with 1 ml containing 106.5TCID50 of WNV Eg101 grown on BHK21, while the remaining two chickens (negative control) were injected intramuscularly with a cell suspension of BHK21. Blood samples were taken 7, 15, 24, 42 days post-infection.
Immune horse sera
Two ponies tested seronegative for WNV using a virus neutralization test were vaccinated intramuscularly with 2 doses of commercial inactivated vaccine. Thirty-five days after the first vaccination, a second immunization with two additional doses was performed. Blood samples were taken 15, 21, 28 days after the first vaccination and 31 days after the second vaccination.
Animal care and all procedures were performed in accordance with guidelines and regulations of the Italian animal protection laws and under EC policy related issues (D.L. 116/1992 amending Dir. CEE n. 609/86).
Monoclonal antibody production
Two Balb/c mice were primed subcutaneously with 50 μg of Betapropiolactone-inactivated WNV with complete Freund's adjuvant and boosted intraperitoneally with the same antigen in PBS (phosphate-buffered saline pH 7.4) once or twice at one month intervals of one month. Three days after the last boost, the mice were sacrificed and hybridomas were generated following fusion of splenocytes with NS0 myeloma cells and selected cultures were grown following the standard procedure [31]. Hybridomas were screened for secretion of desired antibodies with indirect ELISA using the homologous WNV and IF with infected and non-infected Vero cells. Positive hybridoma cells were cloned using limiting dilution to obtain antibodies from a single cell. Hybridoma culture supernatants or ascitic fluids harvested after in vivo culture of hybridoma were used as a MAb source. MAbs were purified from ascites using affinity chromatography through protein A Sepharose (Pharmacia, Milan, Italy) in the presence of 3 M Nacl and 1.5 M glycine pH 8.9 [32], and conjugated with HRP using a modified form of the method described by Tjissen and Kurstak 1984 [33]. The immunoglobulin subclass was determined using a Mouse Immunoglobulin Isotyping ELISA kit (BD Pharmingen) following the manufacturer's instructions.
Immunofluorescence (IF)
Confluent monolayers of Vero cells grown in 96-well microplates were infected with 100TCID50 of WNV Eg101. After incubation for 48/72 hours at 37°C, or following the appearance of a mild cytopathic effect, infected cells were fixed in acetone for 1 h at -20°C, dried and incubated with hybridoma supernatants at 1:2 dilution in PBS for 1 h at 37°C. Cells were washed with PBS before a 30-min incubation with FITC-labeled goat anti-mouse immunoglobulin (produced in-house). The cells were washed again, stained with Evans Blue and observed using fluorescence microscopy. Cells showing a strong green fluorescent signal were recorded as positive. The uninfected cells were used as a negative control.
ELISAs
All ELISAs were performed in 96-well Nunc Maxisoarp ELISA plates.
Indirect ELISAs
For hybridoma screening, ELISA plates were coated with 50 μl per well of partially purified WNV antigen (produced as previously described) at saturating concentration by incubation overnight at 4°C in ELISA coating buffer (0.05 M carbonate/bicarbonate buffer pH 9.6). Plates were washed three times with 250 μl of wash buffer (PBS containing 0.05% Tween 20) with an automatic plate washer after which 50 μl of undiluted hybridoma culture supernatants were added to each well and incubated for 1 h at 37°C. After three washes, an HRP-conjugated goat anti-mouse immunoglobulin antibody (produced in-house) was added at dilution 1/500 in dilution buffer (PBS with 0.05% Tween 20 and 1% yeast extract) to each well and incubated again for 1 h at 37°C. After a final wash cycle, 50 μl of substrate solution (orthophenylenediamine 0.5 mg/ml and 0.02% H2O2 in 50 mM phosphate citrate buffer pH 5) was added. After 10 min, the colorimetric reaction was stopped by the addition of 2 N sulfuric acid; absorbance values were read at 492 nm using an ELISA reader.
To investigate the MAbs-specificity, the reactivity of all MAbs was evaluated against TBEV, DENV1, DENV 2, DENV 3, DENV 4, YFV, JEV by indirect ELISAs.
Competitive binding assays for MAb epitope studies
Competition binding ELISAs were performed in WNV-coated immunoplates. Fifty microliters of unlabeled hybridoma culture supernatants were added at saturating dilution. After incubation for 15 min at 37°C, 25 μl of HRP-conjugated "competitor" MAb was added at pre-determined optimal dilution (which had given an absorbance value of 1-1.5 in a preliminary titration) without washing and incubated again for 1 h at 37°C. Follow a final wash cycle, 50 μl of substrate and stopping solution were added and absorbance values were determined as previously described. Wells without the addition of unlabeled MAb were used as negative control and wells with the addition of unlabeled MAb (the same as the HRP-conjugated MAb) were used as a positive control. Results were expressed as percent binding of HRP-conjugated MAb. The amount of binding obtained in the absence of unlabeled antibody was set at 100% for each HRP-conjugated MAb.
Competitive ELISA with experimental sera
Competitive ELISAs were designed to analyze the capability of known experimental horse and avian sera to inhibit the binding of anti-WNV MAbs to the antigen. Fifty microliters of known sera, at sequential dilutions starting from 1/5, were incubated for 1 h at 37°C in WNV-coated immunoplates, together with 25 μl of HRP-conjugated MAb at the pre-determined optimal dilution. After washes, the colorimetric reaction was achieved as previously described and absorbance values at 492 nm determined. The optical density (OD) of each well was converted to the percent of inhibition (PI) of MAb binding by competition with serum antibodies using the following formula: PI = [1-(OD of serum-Mab mixture/OD of Mab alone)] × 100.
Antigen capture-ELISA (AC-ELISA)
ELISA plates were coated with purified MAbs at a concentration of 10 μg/ml and incubated overnight at 4 C in ELISA coating buffer. Fifty microliters of WNV-infected culture supernatants (listed above) at sequential dilutions were added to each well and plates were incubated for 1 h at 37°C. After washes, 50 μl of HRP-conjugated MAb at a pre-determined dilution was added and incubated again for 1 h at 37°C, followed by a final wash cycle. Diluting buffers, washing and colorimetric reactions were the same as for the ELISAs previously described. Several combinations of trapping and conjugated MAbs were tested. Uninfected cell culture supernatants were used as a negative control, and USUV and TBEV-infected culture supernatants were used to evaluate the specificity. The cut-off OD was calculated as the mean OD of negative samples plus two standard deviations.
Western blotting (WB)
Western blotting was performed using a standard protocol [34]. Briefly, SDS and b mercaptoethanol denatured WNV-antigen protein were separated on 12% SDS-PAGE [35] and transferred to nitrocellulose filters [36]. MAb tissue cultures were diluted 1/5 in a phosphate saline buffer at pH 7.2 containing 1% w/v of bovine albumin and 0.05% w/w of Tween 20. MAb binding was detected by incubation with alkaline phosphatase-labeled rabbit anti-mouse IgG, and the chromogenic substrate 5-bromo-4-chloro-3-indolylphosphate-Nitro Blue
Tetrazolium (BCIP/NBT). Horse sera were diluted 1/200 in the phosphate buffer described above. Antibody binding was detected by incubation with an HRP-labeled goat anti-horse IgG and the chromogenic substrate 4-Chloronaphthol (4CN).
Immunoperoxidase (IPMA)
IPMA was used to evaluate the reactivity profile of MAbs with all flaviviruses used in this study and previously described. Infected monolayers of Vero cells on 96-well microplates were prepared for each virus as described for IF. IPMA plates were filled with PBS with 20% of H2O2, dried and incubated with 100 μl of MAbs at 1:2 and 1.4 dilution in PBS for 1 h at 37°C. Cells were washed 3 times with PBS, after which 100 μl of HRP-conjugated goat anti-mouse IgG was added to each well and plates were incubated for 1 h at 37 C. After 3 further washings, substrate (5 mg/ml of 3-amino-9-ethylcarbazole in dimethyl sulfoxide) diluted in 0.05 M AEC buffer pH 5.2 (105 ml 0.2 M acetic acid, 395 ml 0.2 M sodium acetate, and 500 ml distilled water) was added. The colour reaction was allowed to develop for 20 minutes and the plates washed once with PBS and then examined under a light microscope. The uninfected cells were used as a negative control for each MAb dilution.
Virus-neutralization test (VNT)
The competence of MAbs to neutralize virus infectivity was investigated by VNT carried out in 96-well microplates using two different strains of WNV in parallel (Eg101, B956), belonging respectively to lineages I and II and Vero cells. Serial twofold dilutions of MAbs (2 wells/dilution) in 25 μl of serum-free culture medium were added to each well and incubated for 1 h at 37°C with an equal volume of tissue culture fluid containing 100 TCID50 of WNV. Virus back titration of the working dilution of virus is included, using six wells per tenfold dilution, to confirm the validity of the test results. A volume of 50 μl of Vero cells at log5 cells/ml in medium containing 10% fetal calf serum was added to each well. After incubation for 72-96 hours at 37°C with 5% CO2, wells were scored for cytopathic effect and neutralizing titers were expressed as the reciprocal of the final MAb dilution required to neutralize 100% of the inoculated cultures.
Selection of neutralization escape mutants
Neutralization escape mutants were selected by growing WNV in Vero cells in the presence of neutralizing MAbs using a modification of the method described by Holzmann et al., 1989 [37]. Essentially, serial 10-fold dilutions of WNV Eg101 (107 TCID50/ml) were incubated for 1 h at 37°C with an equal volume of ascites fluid containing between 1 and 8 mg/ml of neutralizing MAb. Aliquots of the virus-MAb mixture (100 μl) were adsorbed to Vero cell monolayer in 24-well plates for 1 h at 37°C, after which fresh medium was added. After incubation for 7 days at 37°C, samples of the supernatants showing CPE were screened for the presence of escape mutants by trapping ELISAs employing a non-neutralizing MAb (flavivirus-cross-reactive) as catcher and homologous neutralizing MAb in parallel with non-neutralizing MAb (flavivirus-cross-reactive) as conjugate. Mutants that were not recognized by homologous MAb while maintaining reactivity with the non-neutralizing MAb were selected and sub-cultured twice in the presence of ascites fluid. A fourth passage without MAb was performed and presumptive mutants were harvested when CPE was evident (48-72 h post-infection). Finally, each selected escape variant was cloned by limiting dilution, and amplified in Vero cells for the preparation of stock suspensions that were stored at -80°C and then titrated. The escape capability from MAb neutralization was investigated by virus neutralization tests performed using 100 TCDI50 of WNV selected variants and serial two-fold dilution of MAbs in Vero cells.
Sequence analysis
Viral RNA was extracted from wild-type WNV Eg101, and WNV escape mutants using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Regions of viral genome corresponding to genes for structural protein E were amplified for sequencing by reverse transcription-PCR [38]. One-step RT-PCR Kit (Qiagen) was used to run conventional RT-PCR assays, following the manufacturer's instructions. Amplification products were analyzed by electrophoresis on 2% agarose gels containing 0.5 μg/ml ethidium bromide. All fragments amplified by PCRs were sequenced by an automated fluorescence-based technique using an ABI-PRISM 3130 Genetic Analyzer (Applied Biosystems), following the manufacture's instruction. 3D macromolecular structure of WNV E protein was performed as described in Figure 3.
Abbreviations
- MAbs:
-
Monoclonal antibodies
- WNV:
-
West Nile virus
- USUV:
-
Usutu virus
- TBEV:
-
Tick-borne encephalits virus
- JEV:
-
Japanese encephalitis virus
- DENV1:
-
Dengue virus serotype 1
- DENV2:
-
Dengue virus serotype 2
- DENV3:
-
Dengue virus serotype 3
- DENV4:
-
Dengue virus serotype 4
- YFV:
-
Yellow fever virus
- DI:
-
Domain I of WNV E protein
- DII:
-
Domain II of WNV E protein
- DIII:
-
Domain III of WNV E protein
- IPMA:
-
Immunoperoxidase assay
- IF:
-
Immunofluorescence
- ELISA:
-
Enzyme linked immunosorbent assay
- AC-ELISA:
-
Antigen capture ELISA
- WB:
-
Western blotting
- VNT:
-
Virus-neutralization test
- CPE:
-
Cytopathic effect
- PBS:
-
Phosphate-buffered saline pH 7.4
- HRP:
-
Horseradish Peroxidase
- TCID50:
-
50% tissue culture infective dose
- OD:
-
Optical density.
References
Blitvich BJ: Transmission dynamics and changing epidemiology of West Nile virus. Anim Health Res Rev 2008, 9: 71-86. 10.1017/S1466252307001430
Brinton MA: The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol 2002, 56: 371-402. 10.1146/annurev.micro.56.012302.160654
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH: Structure of dengue virus: implicationsfor flavivirus organization, maturation, and fusion. Cell 2002, 108: 717-725. 10.1016/S0092-8674(02)00660-8
Modis Y, Ogata S, Clements D, Harrison SC: Structure of the dengue virus envelope protein after membrane fusion. Nature 2004,427(6972):313-319. 10.1038/nature02165
Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC: The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 1995, 375: 291-298. 10.1038/375291a0
Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, Fikrig E, Marasco WA, Koski RA, Modis Y: Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J Virol 2006, 80: 11000-11008. 10.1128/JVI.01735-06
Beasley DW, Barrett ADT: Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J Virol 2002, 76: 13097-13100. 10.1128/JVI.76.24.13097-13100.2002
Ludolfs D, Niedrig M, Paweska LT, Schmitz H: Reverse ELISA for the detection of anti West Nile virus IgG antibodies in humans. Eur J Clin Microbiol Infect Dis 2007, 26: 467-473. 10.1007/s10096-007-0309-1
Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH: Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 2005, 437: 764-769. 10.1038/nature03956
Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS: Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med 2005, 11: 522-530. 10.1038/nm1240
Sánchez MD, Pierson TC, McAllister D, Hanna SL, Puffer BA, Valentine LE, Murtadha MM, Hoxie JA, Doms RW: Characterization of neutralizing antibodies to West Nile virus. Virology 2005, 336: 70-82. 10.1016/j.virol.2005.02.020
Papa A, Danis K, Baka A, Bakas A, Dougas G, Lytras T, Theocharapoulos G, Chrysagis D, Vassiliadou E, Kamaria F, Liona A, Mellou K, Saroglou G, Panagiotopoulos T: Ongoing outbtreak of West Nile virus infections in humans in Greece. Euro Surveill 2010,15(34):Pii:19644.
Calisher CH, Karabatsos N, Dalrymple JM, Shope RE, Porterfield JS, Westaway EG, Brandt WE: Antigenic relationships between flaviviruses as determined by cross-neutralization tests with polyclonal antisera. J Gen Virol 1989, 70: 37-43. 10.1099/0022-1317-70-1-37
Calzolari M, Bonilauri P, Bellini R, Albieri A, Defilippo F, Maioli G, Galletti G, Gelati A, Barbieri I, Tamba M, Lelli D, Carra E, Cordioli P, Angelini P, Dottori M: Evidence of simultaneous circulation of West Nile and Usutu viruses in mosquitoes sampled in Emilia-Romagna region (Italy) in 2009. PLoS One 2010,5(12):e14324. 10.1371/journal.pone.0014324
Tamba M, Bonilauri P, Bellini R, Calzolari M, Albieri A, Sambri V, Dottori M, Angelini P: Detection of Usutu virus within a West Nile virus surveillance program in Northern Italy. Vector Borne Zoonotic Dis 2010, 11: 551-557.
Chambers TJ, Halevy M, Nestorowicz A, Rice CM, Lustig S: West Nile virus envelope proteins: nucleotide sequence analysis of strains differing in mouse neuroinvasiveness. J Gen Virol 1998, 79: 2375-2380.
Throsby M, Geuijen C, Goudsmit J, Bakker AQ, Korimbocus J, Kramer RA, Clijsters-Van der Horst C, De Jong M, Jongeneelen M, Thijsse S, Smit R, Visser TJ, Bijl N, Marissen WE, Loeb M, Kelvin DJ, Preiser W, Ter Meulen J, DE Kruif J: Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J Virol 2006, 80: 6982-6992. 10.1128/JVI.00551-06
Chávez JH, Silva JR, Amarilla AR: Domain III peptides from flavivirus envelope protein are useful antigens for serologic diagnosis and targets for immunization. Biologicals 2010,38(6):613-618. 10.1016/j.biologicals.2010.07.004
Beasley D, Holbrook M, Travassos Da Rosa A, Coffey L, Carrara A, Bohm RP, Phillippi-Falkenstein K, Ratterree MS, Lillibridge KM, Ludwig GV, Estrada-Franco J, Weaver SC, Tesh RB, Shope RE, Barrett ADT: Use of a recombinant envelope protein subunit antigen for specific serological diagnosis of West Nile virus infection. J Clin Microbiol 2004, 42: 2759-2765. 10.1128/JCM.42.6.2759-2765.2004
Holbrook M, Shope R, Barrett A: Use of recombinant E protein domain IIIbased enzyme-linked immunosorbent assays for differentiation of tick-borne encephalitis serocomplex flaviviruses from mosquito-borne flaviviruses. J Clin Microbiol 2004, 42: 4101-4110. 10.1128/JCM.42.9.4101-4110.2004
Chu J, Chiang C, Ng M: Immunization of flavivirus West Nile recombinant envelope domain III protein induced specific immune response and protection against West Nile virus infection. J Immunol 2007, 178: 2699-26705.
Alka , Bharati K, Malik Y, Vrati S: Immunogenicity and protective efficacy of the E. coli-expressed domain III of Japanese encephalitis virus envelope protein in mice. Med Microbiol Immunol 2007, 196: 227-231. 10.1007/s00430-007-0043-4
Leng C, Liu S, Tsai J, Li Y, Chen M, Liu H, Lien S, Yueh A, Hsiao K, Lai L, Liu F, Chong P, Chen H: A novel dengue vaccine candidate that induces cross-neutralizing antibodies and memory immunity. Microbes Infect 2009, 11: 288e95.
Goncalvez AP, Purcell RH, Lai CJ: Epitope determinants of a chimpanzee Fab antibody that efficiently cross-neutralizes Dengue type 1 and type 2 viruses map to inside and in close proximity to fusion loop of the Dengue type 2 virus envelope glycoprotein. J Virol 2004, 78: 12919-129128. 10.1128/JVI.78.23.12919-12928.2004
Gould LH, Foellmer JH, Oliphant T, Wang T, Ledizet M, Murakami A, Noonan K, Lambeth K, Kar K, Anderson JF, De Silva AM, Diamond MS, Koski RA, Marasco WA, Fikrig E: Protective and Therapeutic Capacity of Human Single-Chain Fv-Fc Fusion Proteins against West Nile Virus. J Virol 2005, 79: 14606-14613. 10.1128/JVI.79.23.14606-14613.2005
Razumov IA, Kazachinskaia EI, Ternovoi VA, Protopopova EV, Galkina IV, Gromashevskii VL, Prilipov AG, Kachko AV, Ivanova AV, L'vov DK, Loktev VB: Neutralizing monoclonal antibodies against Russian strain of the West Nile virus. Viral Immunol 2005, 18: 558-568. 10.1089/vim.2005.18.558
Oliphant T, Nybakken GE, Engle M, Xu Q, Nelson CA, Sukupolvi-Petty S, Marri A, Lachmi BE, Olshevsky U, Fremont DH, Pierson TC, Diamond MS: Antibody recognition and neutralization determinants on domains I and II of West Nile virus envelope protein. J Virol 2006, 80: 12149-12159. 10.1128/JVI.01732-06
Mcminn PC, Lee E, Hartley S, Roehrig JT, Dalgarno L, Weir C: Murray valley encephalitis virus envelope protein antigenic variants with altered Hemagglutination properties and reduced Neuroinvasiveness in Mice. Virol 1995, 211: 10-20. 10.1006/viro.1995.1374
Sánchez MD, Pierson TC, Degrace MM, Mattei LM, Hanna SL, Del Piero F, Doms RW: The neutralizing antibody response against West Nile virus in naturally infected horses. Virology 2007, 59: 336-348.
Liu J, Liu B, Cao Z, Inoue S, Morita K, Tian K, Zhu Q, Gao GF: Characterization and application of monoclonal antibodies specific to West Nile virus envelope protein. J Virol Methods 2008, 154: 20-26. 10.1016/j.jviromet.2008.09.019
Galfre G, Milstein C: Preparation of monoclonal antibodies, strategies and procedure. Methods Enzymol 1981, 73: 3-5.
Harlow E, Lane D: Antibodies A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1988:469.
Tjissen P, Kurstak E: Highly efficient and simple methods for the preparation of peroxidase and active peroxidase-antibodies conjugates for enzyme immunoassays. Analyt Biochem 1984, 136: 451-457. 10.1016/0003-2697(84)90243-4
Capucci L, Fusi , Lavazza A, Pacciarini ML, Rossi C: Detection and preliminary characterization of a new rabbit calicivirus related to hemorrhagic disease virus but nonpathogenic. J Virol 1996, 70: 8614-8623.
Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227: 680-685. 10.1038/227680a0
Towbin H, Staehelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotchnology 1992, 24: 145-149.
Holzmann H, Mandl CW, Guirakhoo F, Heinz FX, Kunz C: Characterization of antigenic variants of tick-borne encephalitis virus selected with neutralizing monoclonal antibodies. J Gen Virol 1989, 70: 219-222. 10.1099/0022-1317-70-1-219
Jourdain E: Oiseaux savages et virus West Nile: étude éco-épidémiologique en Camarge PhD thesis. Grenoble: Universidad de Joseph Fourier; 1995.
Acknowledgements
Many thanks to Daniela Gamba, Giuliana Botti and all technicians of the IZSLER Virology Section for their precious technical work. The Authors would like to thank Michael John of the Vita-Salute San Raffaele University for the English language editing of this manuscript. This study was partially founded by EuroWestNile, European Union Research Project supported by the Seventh Framework Programme for Research, FP7-HEALTH-2010-single-stage.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DL carried out most of the experiments, interpreted the results and wrote the manuscript. AM, ES, EC, IB, HZ were involved in virological and molecular analysis. EB, LC were involved in monoclonal antibodies production and in Western blotting analysis. PC designed the experiment. All of the authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lelli, D., Moreno, A., Brocchi, E. et al. West Nile virus: characterization and diagnostic applications of monoclonal antibodies. Virol J 9, 81 (2012). https://doi.org/10.1186/1743-422X-9-81
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-81