
Abstract
Migraine is a largely inherited disorder of the brain characterized by a complex, but stereotypical, dysfunction of sensory processing. Often the most obvious clinical symptom is head pain, but non-headache symptoms such as photophobia, phonophobia and nausea are clearly part of the typical presentation. This review discusses the current pathophysiological concepts of migraine and migraine aura, such as a possible brainstem dysfunction and cortical spreading depression. Acute and preventive migraine treatment approaches are briefly covered with a focus on shortcomings of the currently available treatment options. A number of different receptors, such as calcitonin gene-related peptide (CGRP), TRPV1 and glutamate receptors, are currently being targeted by potential novel migraine therapeutics. The prospects of this research are exciting and are likely to improve patient care.
Similar content being viewed by others

Introduction
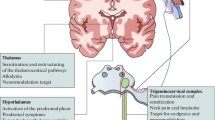
Migraine is a disorder characterized by a broad sensory processing dysfunction. Thereby, the perception of normal sensory stimuli, somatosensory, visual, auditory and olfactory, is thought to be centrally facilitated. This dysfunction has a strong genetic background [1] and leads to the perception of a combination of headache, photophobia, phonophobia, osmophobia and nausea. The disorder was considered to be vascular in origin for much of the 20th century [2], although it was considered to be a disorder of the nervous system by 19th century luminaries [3]. This was related to the pounding or throbbing nature of the pain and triggering by vasoactive substances that now seem more likely to be due to the prominent perception of pain in the context of the dense somatosensory innervation of intracranial vessels. This view is supported by data which include the fact that: vasoactive intestinal peptide - a strong intracranial vasodilator - does not trigger migraine [4]; intracranial vasodilatation also occurs secondary to experimental head pain stimulation [5], probably mediated by the trigeminal-parasympathetic reflex; and non-vasoconstrictor drugs, such as aspirin [6] and calcitonin gene-related peptide (CGRP) receptor antagonists can abort migraine attacks [7]. Most importantly, headache is only one of the neurological symptoms of migraine, where the dysfunction can really only be located in the brain itself.
This article provides an update on potential mechanisms of migraine and aura pathogenesis and reviews current and future medical strategies for the acute and preventive treatment of migraine.
Aura and cortical spreading depression
A subgroup of migraineurs experience aura, typically before the onset of head pain, with some of their attacks and there are several lines of evidence that cortical spreading depression is the pathophysiological substrate. In early clinical observations, it was noted that the progression of aura symptoms is consistent with a process transiently compromising cortical function at a speed of about 3 mm per minute [8]. Leao suggested that cortical spreading depression (CSD), advancing at the identical speed over the cortex, was the electrophysiological correlate of visual aura in humans. By now, the existence of CSD in humans has been proven using electrophysiological methods [9–11] and human imaging studies [12, 13]. A possible link between CSD and headache has been provided by the observation that CSD can activate trigeminal meningeal afferents [14], although contradictory data also exist [15]. Hence, CSD could not only induce aura symptoms, but also explain the head pain in patients with aura. This view is not supported by recent controlled trials which show that tonabersat, a possible gap-junction blocker and inhibitor of CSD [16], does not prevent migraine headache [17] but can prevent migraine aura [18]. It has been suggested that CSD also has a role in migraine without aura but the tonabersat studies suggest this is less likely. Silent aura, the occurrence of CSD confined to regions not clinically eloquent and still activating trigeminal afferents, is a tempting concept when looking for a unifying concept of migraine with and without aura. However, as noted above in the study by Hauge et al. [18], tonabersat was ineffective in migraine without aura and, as the drug reduced the frequency of aura attacks, this result clearly challenges the concept of the silent aura [18]. Moreover, in a recent case series, three patients were described who reported that their auras resolved when migraine preventives were started, while in parallel they experienced a worsening of the frequency of their migrainous headaches [19]. Finally, the sequence of a migraine attack has probably already been initiated long before the actual onset of CSD and aura. Migraine attacks often start with a typical premonitory phase when patients complain of tiredness, reduced concentration, irritability, yawning and other non-headache symptoms hours to days before the onset of aura and headache [20, 21]. At this point, many patients can predict the onset of a full-blown migraine attack and the start with non-headache symptoms underlines that migraine is much more than an isolated pain disorder.
Neurogenic plasma protein extravasation
It has been suggested that some component of the migrainous pain is related to dural plasma protein extravasation with sterile neurogenic inflammation [22]. Electrical stimulation of the trigeminal ganglion induces plasma protein extravasation and this can be blocked by some of the substances which are active in acute migraine attacks (for example, sumatriptan) [23]. On the other hand, a blockade of neurogenic plasma protein extravasation is not completely predictive of anti-migraine efficacy, as other compounds which are effective in this experimental model of migraine, such as neurokinin-1 receptor antagonists, have failed in clinical migraine prevention trials [24, 25]. Hence, plasma protein extravasation seems to be an epiphenomenon rather than a pivotal mechanism of trigeminal activation and migraine generation [26].
Brainstem dysfunction in migraine pathogenesis?
In our view, the whole clinical picture of migraine can be better explained by a dysfunction of neuromodulatory structures in the brainstem, such as the locus coeruleus or periaqueductal grey matter. The locus coeruleus, the major noradrenergic nucleus, has a critical role in the regulation of cortical function and is known to modulate responses to afferent traffic [27]. Its connections are widely distributed throughout the neocortex and in positron emission tomography (PET) studies investigating acute migraine attacks, activation of an area of the dorsolateral brainstem that included the locus coeruleus has been shown [28, 29], although there is some remaining uncertainty regarding the exact anatomical location of this activation because of the limited spatial resolution of PET. Compared to other hypothesis on migraine neurobiology, dysfunction of such brainstem structures and networks could not only account for the somatosensory component of migraine (headache) but also for the auditory, olfactory and visual components. Moreover, a locus coeruleus dysfunction could also explain the distractibility and anxiety [27] which is often observed in migraineurs.
Pharmacological and interventional strategies of migraine prevention
Migraine prevention is an important component of therapy aimed at reducing the attack frequency and severity. Unfortunately, the mechanisms of action of current preventives are not well understood. A potential mechanism is the inhibition of cortical spreading depression but, as noted above, the efficacy against cortical spreading depression does not necessarily predict the efficacy in treating migraine without aura [18]. Substances that have proven beneficial in migraine, with and without aura, broadly comprise compounds from the following classes: beta-blockers (for example, propranolol), antidepressants (for example, amitriptyline), anticonvulsants (for example, valproate, topiramate), calcium channel blockers (for example, flunarizine) and serotonin antagonists (for example, methysergide) [30]. According to the pathophysiological concepts discussed above, these drugs most probably target the activity of modulatory circuits as well as the neuronal activity in afferent sensory pathways such as the trigeminal system [31]. Although many patients can be effectively managed using the available substances, side effects and contraindications because of co-morbidities can complicate treatment. A particular problem is the prediction of which patients will respond to which substance as treatment is often largely conducted by trial and error, which is frustrating for the patients and treating physicians alike. In order to obtain a better understanding of the mechanisms of the action of the available substances, phenotype-driven treatment approaches [32] as well as pharmacogenomic considerations [33], might help to overcome such issues. Moreover, newer treatment strategies, such as the use of Botulinum toxin in migraine prevention, are currently undergoing clinical trials (Table 1). Interventional neuromodulational approaches are also promising, including the stimulation of the occipital nerve, where functional imaging studies show that the central processing of pain signals in migraine in the thalamus may be modified by such therapies [34]. Studies, such as ONSTIM [35] and PRISM [36], are now complete with their implications being analysed (PRISM NCT00286078 and [37–39]). This exciting and developing area might be especially helpful in the treatment of medically refractory patients.
Pharmacological strategies for migraine attack treatment
Treatments for acute attacks can be divided into non-specific anti-pain compounds, such as simple analgesics and NSAIDS, and more migraine-specific treatment approaches, such as ergot-derivates and triptans, which are active at 5-HT1 receptors. Stratified care, the choice of an acute medication on the basis of attack characteristics, has been shown to be superior to stepped care [40] and, as triptans are most effective, a triptan is typically chosen for the more severe migraine attacks. Triptans, because of their better tolerability, have replaced ergotamine in most cases [41]. Sumatriptan and its six licensed successors are agonists at 5HT1B/D receptors. They reduce neuronal activity via these receptors at the trigeminocervical complex [42] and thalamic level [43] and these areas, instead of the blood vessels, are most probably also important sites for their therapeutic action in migraine. There are still situations where tolerability and contraindications to use are a problem. The main issue for triptans relates to their vasoconstrictor properties and related cardiovascular and cerebrovascular safety concerns. This necessitates that triptans are not used in patients with cerebrovascular or cardiovascular contraindications [44]. The new group of so-called Gepants, CGRP receptor antagonists, may soon offer an option to such patients. Two substances of this class, parenteral olcegepant [7] and oral telcagepant [45], have proved to be effective in the acute treatment of migraine without vascular liability [46]. Their effectiveness seems to be comparable to triptans [45].
The announcement of the success of a phase II dose-ranging proof-of-concept study with the 5-HT1F receptor agonist CO-144 offers another prospect for a non-vasoconstrictor acute anti-migraine therapy [47]. The success of CGRP receptor antagonists and 5-HT1F receptor agonists reinforce a neurally-based approach to migraine and emphasize that migraine is a brain disorder. Other strategies, such as nitric oxide synthase inhibitors [48], vanilloid TRPV1 receptor antagonists [NCT00269022, [49]] and glutamate, AMPA/kainate receptor [50] as well as pure kainate receptor antagonists, will hopefully follow providing an even broader repertoire of migraine-specific drugs (Table 1, [16, 51]).
Conclusion
Migraine is a disorder of the brain characterized by a complex sensory dysfunction. Hence, therapeutic approaches with neural targets are most promising. Currently, a number of such pharmacological and interventional therapies are being investigated and it is likely that patients can benefit from CGRP receptor antagonists and occipital nerve stimulation in the near future. In addition to these new treatment strategies, defining patient populations who are more likely to respond to the already available therapeutic options also has great potential to improve patient care in daily practice.
Abbreviations
- CGRP:
-
calcitonin gene-related peptide
- CSD:
-
cortical spreading depression
- PET:
-
position emission tomography.
References
Russell MB: Is migraine a genetic illness? The various forms of migraine share a common genetic cause. Neurol Sci. 2008, 29 (Suppl 1): S52-54. 10.1007/s10072-008-0887-4.
Moskowitz MA, Buzzi MG, Sakas DE, Linnik MD: Pain mechanisms underlying vascular headaches: Progress Report 1989. Rev Neurol (Paris). 1989, 145: 181-193.
Gowers WR: A Manual of Diseases of the Nervous System. 1989, Philadelphia: P. Blakiston, Son & Co
Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M: Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008, 28: 226-236. 10.1111/j.1468-2982.2007.01497.x.
May A, Bahra A, Buchel C, Frackowiak RS, Goadsby PJ: PET and MRA findings in cluster headache and MRA in experimental pain. Neurology. 2000, 55: 1328-1335.
Lampl C, Voelker M, Diener HC: Efficacy and safety of 1,000 mg effervescent aspirin: individual patient data meta-analysis of three trials in migraine headache and migraine accompanying symptoms. J Neurol. 2007, 254: 705-712. 10.1007/s00415-007-0547-2.
Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM: Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004, 350: 1104-1110. 10.1056/NEJMoa030505.
Lashley K: Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiat. 1941, 46: 331-339.
Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M: Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006, 129 (Pt 3): 778-790. 10.1093/brain/awh716.
Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, Parkin MC, Lauritzen M: Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002, 33: 2738-2743. 10.1161/01.STR.0000043073.69602.09.
Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE: Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res. 1996, 740: 268-274. 10.1016/S0006-8993(96)00874-8.
Olesen J, Larsen B, Lauritzen M: Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981, 9: 344-352. 10.1002/ana.410090406.
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, et al: Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA. 2001, 98: 4687-4692. 10.1073/pnas.071582498.
Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA: Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002, 8: 136-142. 10.1038/nm0202-136.
Ebersberger A, Schaible HG, Averbeck B, Richter F: Is there a correlation between spreading depression, neurogenic inflammation, and nociception that might cause migraine headache?. Ann Neurol. 2001, 49: 7-13. 10.1002/1531-8249(200101)49:1<7::AID-ANA4>3.0.CO;2-K.
Goadsby PJ: Emerging therapies for migraine. Nat Clin Pract Neurol. 2007, 3: 610-619. 10.1038/ncpneuro0639.
Goadsby PJ, Ferrari MD, Csanyi A, Olesen J, Mills JG: Randomized, double-blind, placebo-controlled, proof-of-concept study of the cortical spreading depression inhibiting agent tonabersat in migraine prophylaxis. Cephalalgia. 2009, 29: 742-750. 10.1111/j.1468-2982.2008.01804.x.
Hauge AW, Asghar MS, Schytz HW, Christensen K, Olesen J: Effects of tonabersat on migraine with aura: a randomised, double-blind, placebo-controlled crossover study. Lancet Neurol. 2009, 8: 718-723. 10.1016/S1474-4422(09)70135-8.
Wolthausen J, Sternberg S, Gerloff C, May A: Are cortical spreading depression and headache in migraine causally linked?. Cephalalgia. 2009, 29: 244-249. 10.1111/j.1468-2982.2008.01713.x.
Kelman L: The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache. 2004, 44: 865-872. 10.1111/j.1526-4610.2004.04168.x.
Giffin NJ, Kowacs F, Libri V, Williams P, Goadsby PJ, Kaube H: ffect of the adenosine A1 receptor agonist GR79236 on trigeminal nociception with blink reflex recordings in healthy human subjects. Cephalalgia. 2003, 23: 287-292. 10.1046/j.1468-2982.2003.00511.x.
Moskowitz MA: Basic mechanisms in vascular headache. Neurol Clin. 1990, 8: 801-815.
Buzzi MG, Moskowitz MA: The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br J Pharmacol. 1990, 99: 202-206.
May A, Goadsby PJ: Substance P receptor antagonists in the therapy of migraine. Expert Opin Investig Drugs. 2001, 10: 673-678. 10.1517/13543784.10.4.673.
Goldstein DJ, Offen WW, Klein EG, Phebus LA, Hipskind P, Johnson KW, Ryan RE: Lanepitant, an NK-1 antagonist, in migraine prevention. Cephalalgia. 2001, 21: 102-106. 10.1046/j.1468-2982.2001.00161.x.
Peroutka SJ: Neurogenic inflammation and migraine: implications for the therapeutics. Mol Interv. 2005, 5: 304-311. 10.1124/mi.5.5.10.
Aston-Jones G, Cohen JD: An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005, 28: 403-450. 10.1146/annurev.neuro.28.061604.135709.
Weiller C, May A, Limmroth V, Juptner M, Kaube H, Schayck RV, Coenen HH, Diener HC: Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995, 1: 658-660. 10.1038/nm0795-658.
Afridi SK, Matharu MS, Lee L, Kaube H, Friston KJ, Frackowiak RS, Goadsby PJ: A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005, 128 (Pt 4): 932-939. 10.1093/brain/awh416.
Lance JW, Goadsby PJ: Mechanism and Management of Headache. 2005, Philadelphia: Elsevier Butterworth Heinemann, 7
Shields KG, Goadsby PJ: Propranolol modulates trigeminovascular responses in thalamic ventroposteromedial nucleus: a role in migraine?. Brain. 2005, 128 (Pt 1): 86-97.
Wheeler SD: Phenotype-driven preventive strategies for migraine and other headaches. Neurologist. 2009, 15: 59-70. 10.1097/NRL.0b013e318165eb94.
Johnson MP, Fernandez F, Colson NJ, Griffiths LR: A pharmacogenomic evaluation of migraine therapy. Expert Opin Pharmacother. 2007, 8: 1821-1835. 10.1517/14656566.8.12.1821.
Matharu MS, Bartsch T, Ward N, Frackowiak RSJ, Weiner RL, Goadsby PJ: Central neuromodulation in chronic migraine patients with suboccipital stimulators: a PET study. Brain. 2004, 127: 220-230. 10.1093/brain/awh022.
Saper J, Goadsby PJ, Silberstein S, Dodick DW: Occipital nerve stimulation (ONS) for treatment of intractable chronic migraine (ICM): 3-Month Results from the ONSTIM Feasibility Study. Neurology. 2009, 72 (Suppl 3): A252.
Lipton RB, Goadsby PJ, Cady RK, Aurora SK, Grosberg BM, Freitag F, et al: PRISM study: occiptal nerve stimulation for treatment-refractory migraine. Cephalalgia. 2009, 29 (Suppl 1): 30.
Goadsby PJ, Dodick D, Mitsias P, Khan K, Khan A, Brewer AR, Saper J, Silberstein SD: ONSTIM: occipital nerve stimulation for the treatment of chronic migraine. European Journal of Neurology. 2005, 12 (Suppl 2): 198.
Burns B, Watkins L, Goadsby PJ: Successful treatment of medically intractable cluster headache using occipital nerve stimulation (ONS). Lancet. 2007, 369: 1099-1106. 10.1016/S0140-6736(07)60328-6.
Magis D, Allena M, Bolla M, De Pasqua V, Remacle JM, Schoenen J: Occipital nerve stimulation for drug-resistant chronic cluster headache: a prospective pilot study. Lancet Neurology. 2007, 6: 314-321. 10.1016/S1474-4422(07)70058-3.
Lipton RB, Stewart WF, Stone AM, Lainez MJ, Sawyer JP: Stratified care vs step care strategies for migraine: the Disability in Strategies of Care (DISC) Study: A randomized trial. JAMA. 2000, 284: 2599-2605. 10.1001/jama.284.20.2599.
Tfelt-Hansen P, Saxena PR, Dahlof C, Pascual J, Lainez M, Henry P, Diener H, Schoenen J, Ferrari MD, Goadsby PJ: Ergotamine in the acute treatment of migraine: a review and European consensus. Brain. 2000, 123 (Pt 1): 9-18. 10.1093/brain/123.1.9.
Hoskin KL, Kaube H, Goadsby PJ: Sumatriptan can inhibit trigeminal afferents by an exclusively neural mechanism. Brain. 1996, 119 (Pt 5): 1419-1428. 10.1093/brain/119.5.1419.
Shields KG, Goadsby PJ: Serotonin receptors modulate trigeminovascular responses in ventroposteromedial nucleus of thalamus: a migraine target?. Neurobiol Dis. 2006, 23: 491-501. 10.1016/j.nbd.2006.04.003.
Dodick D, Lipton RB, Martin V, Papademetriou V, Rosamond W, MaassenVanDenBrink A, Loutfi H, Welch KM, Goadsby PJ, Hahn S, et al: Consensus Statement: Cardiovascular Safety Profile of Triptans (5-HT1B/1D Agonists) in the Acute Treatment of Migraine. Headache. 2004, 44: 414-425. 10.1111/j.1526-4610.2004.04078.x.
Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, Leibensperger H, Froman S, Assaid C, Lines C, et al: Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet. 2008, 372: 2115-2123. 10.1016/S0140-6736(08)61626-8.
Petersen KA, Birk S, Lassen LH, Kruuse C, Jonassen O, Lesko L, Olesen J: The CGRP-antagonist, BIBN4096BS does not affect cerebral or systemic haemodynamics in healthy volunteers. Cephalalgia. 2005, 25: 139-147. 10.1111/j.1468-2982.2004.00830.x.
Reuter U, Pilgrim AJ, Diener HC, Farkkila M, Ferrari MD: COL-144, a selective 5-HT1F agonist, for the treatment of migraine attacks. Cephalalgia. 2009, 29: 122.
Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J: Nitric oxide synthesis inhibition in migraine. Lancet. 1997, 349: 401-402. 10.1016/S0140-6736(97)80021-9.
Rami HK, Thompson M, Stemp G, Fell S, Jerman JC, Stevens AJ, Smart D, Sargent B, Sanderson D, Randall AD, et al: Discovery of SB-705498: A potent, selective and orally bioavailable TRPV1 antagonist suitable for clinical development. Bioorg Med Chem Lett. 2006, 16: 3287-3291. 10.1016/j.bmcl.2006.03.030.
Sang CN, Ramadan NM, Wallihan RG, Chappell AS, Freitag FG, Smith TR, Silberstein SD, Johnson KW, Phebus LA, Bleakman D, et al: LY293558, a novel AMPA/GluR5 antagonist, is efficacious and well-tolerated in acute migraine. Cephalalgia. 2004, 24: 596-602. 10.1111/j.1468-2982.2004.00723.x.
Weiss B, Alt A, Ogden AM, Gates M, Dieckman DK, Clemens-Smith A, Ho KH, Jarvie K, Rizkalla G, Wright RA, et al: Pharmacological characterization of the competitive GLUK5 receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J Pharmacol Exp Ther. 2006, 318: 772-781. 10.1124/jpet.106.101428.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/7/71/prepub
Acknowledgements
TS is supported by the Deutsche Forschungsgemeinschaft (Projekt SP 1215/1-1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
TS declares no competing interests. PJG has consulted for, advised or collaborated with Advanced Bionics, Allergan, Almirall, ATI, AstraZeneca, Belgian Research Council, Boehringer-Ingelheim, BMS, Boston Scientific, Colucid, Eli-Lilly, Fidelity Foundation, GlaxoSmithKline, Johnson & Johnson, Kalypsys, Medtronic, MAP, Migraine Research Foundation, Migraine Trust, Minster, Medical Research Council-UK, MSD, NINDS, Netherlands Research Council, Neuralieve, Neuraxon, NTP, Organisation for Understanding Cluster Headache-UK and Pfizer.
Authors' contributions
TS and PJG each participated in the writing and editing of this minireview. Both authors have read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sprenger, T., Goadsby, P.J. Migraine pathogenesis and state of pharmacological treatment options. BMC Med 7, 71 (2009). https://doi.org/10.1186/1741-7015-7-71
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-7-71