
Abstract
Background
The Hedgehog signaling pathway functions as an organizer in embryonic development. Recent studies have demonstrated constitutive activation of Hedgehog pathway in various types of malignancies. However, it remains unclear how Hedgehog pathway is involved in the pathogenesis of osteosarcoma. To explore the involvement of aberrant Hedgehog pathway in the pathogenesis of osteosarcoma, we investigated the expression and activation of Hedgehog pathway in osteosarcoma and examined the effect of SMOOTHENED (SMO) inhibition.
Results
To evaluate the expression of genes of Hedgehog pathway, we performed real-time PCR and immunohistochemistry using osteosarcoma cell lines and osteosarcoma biopsy specimens. To evaluate the effect of SMO inhibition, we did cell viability, colony formation, cell cycle in vitro and xenograft model in vivo. Real-time PCR revealed that osteosarcoma cell lines over-expressed Sonic hedgehog, Indian hedgehog, PTCH1, SMO, and GLI. Real-time PCR revealed over-expression of SMO, PTCH1, and GLI2 in osteosarcoma biopsy specimens. These findings showed that Hedgehog pathway is activated in osteosarcomas. Inhibition of SMO by cyclopamine, a specific inhibitor of SMO, slowed the growth of osteosarcoma in vitro. Cell cycle analysis revealed that cyclopamine promoted G1 arrest. Cyclopamine reduced the expression of accelerators of the cell cycle including cyclin D1, cyclin E1, SKP2, and pRb. On the other hand, p21cip1 wprotein was up-regulated by cyclopamine treatment. In addition, knockdown of SMO by SMO shRNA prevents osteosarcoma growth in vitro and in vivo.
Conclusions
These findings suggest that inactivation of SMO may be a useful approach to the treatment of patients with osteosarcoma.
Similar content being viewed by others

Background
Osteosarcoma is the most common primary bone malignant tumor occurring mainly in children [1]. After initial diagnosis is made by biopsy, treatment consists of preoperative chemotherapy, followed by definitive surgery and postoperative chemotherapy. Survival has improved over the past several decades. Indeed, patients with non-metastatic disease have a 70% chance of long-term survival. Unfortunately, patients with metastatic disease at diagnosis and those who have recurrent disease have a poor prognosis, with only 20% surviving at 5 years, indicating that new therapeutic options for them need to be actively explored. In cancer cells, dysregulation of cell division and apoptotic processes contribute to both drug resistance and metastatic potential [2, 3]. It has been reported that inactivation of the cell cycle regulatory pathway centered around the Rb gene is a critical step in the pathogenesis of osteosarcoma [4]. Although such dysregulation may constitute a potent source of new therapeutic targets, the molecular mechanisms of regulation of osteosarcoma cell proliferation are largely unknown.
Hedgehog (Hh) pathway has been implicated in different aspects of animal development, acting through several components, including the transmembrane proteins PATCHED (PTCH1) and SMOOTHENED (SMO), to activate the GLI zinc-finger transcription factors [5, 6]. Hh pathway is critical for many processes during embryonic and postnatal development, including proliferation, differentiation, specification of cell fate, left-right asymmetry, and morphogenesis [7]. Sporadic and familial mutations in the Hh pathway genes, PTCH1, suppressor-of-fused, and SMO, leading to elevated expression of downstream target genes including GLI, have been reported in basal cell carcinoma and the pediatric brain tumor medulloblastoma [8, 9]. In addition, the growth of many cancers has been suggested to depend on continuous Hh pathway even in the absence of activating mutations in the pathway (reviewed in ref. [10]).
To explore the involvement of Hh pathway in the pathogenesis of osteosarcoma, we investigated the expression and activation of the Hh pathway genes in osteosarcoma and examined the effect of inhibition of SMO by cyclopamine, a specific inhibitor of SMO [11] or SMO shRNA.
Results
Over-expression of Hh-GLI pathway molecules in osteosarcoma
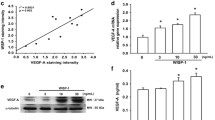
To examine the role of Hh�GLI pathway in osteosarcoma, we tested for the expression of Hh in osteosarcoma cell lines. Real-time PCR revealed that 4 of 5 human osteosarcoma cell lines increased Sonic Hedgehog (SHH) 2.1- to 18.8-fold (Fig. 1). In addition, 5 of 5 osteosarcoma cell lines increased Desert Hedgehog 1.3- to 24.4-fold (Fig. 1). To further examine Hh pathway molecules expression, we performed real-time PCR for Hh receptors and Hh target genes. PTCH1 was up-regulated 2.7-to 65.8-fold in 5 of 5 human osteosarcoma cell lines. SMO was up-regulated 2.1-to 5.8-fold in 4 of 5 human osteosarcoma cell lines. SMO was up-regulated 2.1-to 5.8-fold in 4 of 5 human osteosarcoma cell lines. GLI1 was up-regulated 2.5-to 8.9-fold in 5 of 5 human osteosarcoma cell lines. GLI2 was up-regulated 1.2-to 9.9-fold in 5 of 5 human osteosarcoma cell lines. To extend these findings, we performed immunocytochemistry for SMO and GLI2, and found that only osteosarcoma cells expressed detectable levels of SMO and GLI2. GLI2 was located in the nuclei of osteosarcoma cells (see additional file 1). We next examined SMO expression in osteosarcoma patient' biopsy specimens. Real-time PCR revealed that 9 of 9 human biopsy specimens of osteosarcoma increased SMO 1.44- to 55.5-fold (Fig. 2). In addition, real-time PCR revealed that expression of PTCH1 was increased in 8 of 9 patients' biopsy samples 2.44- to 29.4-fold (Fig. 2). GLI2 was up-regulated 2.5-to 58.4-fold in 9 of 9 human biopsy specimens of osteosarcoma (Fig. 2). Of most importance was the finding that markers of active Hh�GLI signaling, GLI2 and PTCH1 were consistently up-regulated in the examined osteosarcoma cells, demonstrating the aberrant Hh-GLI pathway activation [12–14]. Our findings suggest that Hh-GLI signaling is active in osteosarcomas.

Expression of activated Hh-GLI pathway molecules. Total RNA extracted from osteosarcoma cell lines were used for real-time PCR. Real-time PCR revealed that 4 of 5 human osteosarcoma cell lines increased Sonic Hedgehog (SHH) 2.1- to 18.8-fold (Fig. 1). In addition, 5 of 5 osteosarcoma cell lines increased Desert Hedgehog 1.3- to 24.4-fold (Fig. 1). To further examine Hh pathway molecules expression, we performed real-time PCR for Hh receptors and Hh target genes. PTCH1 was up-regulated 2.7-to 65.8-fold in 5 of 5 human osteosarcoma cell lines. SMO was up-regulated 2.1-to 5.8-fold in 4 of 5 human osteosarcoma cell lines. SMO was up-regulated 2.1-to 5.8-fold in 4 of 5 human osteosarcoma cell lines. GLI1 was up-regulated 2.5-to 8.9-fold in 5 of 5 human osteosarcoma cell lines. GLI2 was up-regulated 1.2-to 9.9-fold in 5 of 5 human osteosarcoma cell lines. The comparative Ct (ΔΔCt) method was used to determine fold change in expression using βII-microglobulin, GAPDH or ACTB. Each sample was run minimally at three concentrations in triplicate (error bar means S.D.). The experiment was triplicate with similar results.

Activation of Hh pathway in patient' biopsy samples. Real-time PCR revealed that 9 of 9 human biopsy specimens of osteosarcoma increased SMO 1.44- to 55.5-fold. Real-time PCR revealed that expression of PTCH1 was increased in 8 of 9 patients' biopsy samples 2.44- to 29.4-fold. GLI2 was up-regulated 2.5-to 58.4-fold in 9 of 9 human biopsy specimens of osteosarcoma. The comparative Ct (ΔΔCt) method was used to determine fold change in expression using βII-microglobulin, ACTB, and GAPDH. Each sample was run minimally at three concentrations in triplicate (error bar means S.D.). The experiment was triplicate with similar results.
Inhibition of SMO prevents osteosarcoma growth in vitro
To determine whether activation of Hh-GLI signaling is required for osteosarcoma cell growth, we used cyclopamine, a pharmacological agent known to effectively block Hh-GLI signaling by inhibiting SMO activation [11]. We performed real-time PCR to determine whether cyclopamine effectively inhibited the expression of the GLI target gene PTCH1 and GLI2[14]. Cyclopamine at 20 μM reduced mRNA levels of PTCH1 and GLI2 in osteosarcoma cells by more than 60%, consistent with the expected down-regulation of Hh-GLI signaling (Fig. 3A). As cyclopamine was used to prevent cancer cells growth at 10 to 20 μM [15–17] we decided 20 μM was appropriate concentration for osteosarcoma. MTT assay showed that cyclopamine slowed the growth of HOS and 143B in dose-dependent fashion (Fig. 3B). On the other hand, MTT assay showed that proliferation of osteosarcoma cells was enhanced by SHH. We next used a clonogenic assay to determine whether cells capable of forming anchorage-independent colonies were depleted by cyclopamine. This assay revealed cyclopamine reduced colony formation in soft agar (Fig. 3C). These findings suggest that inhibition of SMO inhibited osteosarcoma growth in vitro.

Inhibition of Hh pathway prevents osteosarcoma growth in vitro. A, We performed real-time PCR to determine which concentration of cyclopamine effectively inhibited Hh-GLI activity in osteosarcoma cells, and then measured the expression of the Hh-GLI pathway target PTCH1 and GLI2. Cyclopamine at 20 μM reduced mRNA levels of PTCH1 in 143B cell (error bar means S.D.). The comparative Ct (ΔΔCt) method was used to determine fold change in expression using ACTB. Each sample was run minimally at three concentrations in triplicate (error bar means S.D.). The experiment was triplicate with similar results. B, Growth of viable 143B and HOS cells over 3 days was slowed in dose-dependent fashion by cyclopamine treatment. The experiment was triplicate with similar results. C, Colony formation assay revealed cyclopamine reduced colony formation in soft agar. The experiment was triplicate with similar results. (*: P < 0.01) (error bar means S.D.)
Hh signaling regulates cell cycle of osteosarcoma
We examined cell cycle characteristics by flow cytometry. Of 143B cells cultured without cyclopamine, 39.8% of cells were in G1 phase, while 56.6% of cells were in G1 phase following treatment with cyclopamine. In the case of HOS cells were cultured without cyclopamine, 55.4% cells were in G1 phase. On the other hand, when cultured with cyclopamine, 72.3% of cells were in G1 phase (Fig. 4A). These findings suggested that cyclopamine promoted G1 arrest. We then examined the transcription of cell cycle-related genes. Real-time PCR revealed that cyclopamine prevented the transcription of accelerators of the cell cycle including cyclin D1, cyclin E1, SKP2, and NMYC (Fig. 4B). In mammalian cells, cyclin D, cyclin E, and p21cip1 are short-lived proteins that are controlled by ubiquitin-dependent proteolysis. We performed western blot analysis to determine protein levels, and found that cyclopamine reduced the levels of expression of cyclin D1 and cyclin E1 proteins. Cyclopamine also reduced the levels of expression of cyclin D1, cyclin E1, pRb, and SKP2 proteins (Fig. 4C). We next examined the expression of p21cip1, and found that p21cip1 protein was up-regulated by cyclopamine treatment (Fig. 4C). These findings suggested that cyclopamine promoted G1 arrest by inhibition of G1-S phase progression. These findings suggest that inhibition of SMO inhibited osteosarcoma growth via cell cycle regulation.

Cyclopamine treatment promotes G1 arrest. A, HOS and143B cells were treated with 10 μM cyclopamine. After 48-hour treatment cells were collected and subjected to cell cycle analysis. When 143B cells were cultured without cyclopamine, 39.8% of cells were in G1 phase. On the other hand, when cultured with cyclopamine, 56.6% of cells were in G1 phase. In the case of HOS cells cultured without GSI, 55.4% of cells were in G1 phase, while 72.3% of cells were in G1 phase when treated with cyclopamine (error bar means S.D.). B, Real-time PCR was performed to quantify mRNAs of cell cycle related genes. Twenty-four-hour treatment with cyclopamine reduced levels of cyclin D1, Cyclin E1, SKP2, and NMYC transcription (error bar means S.D.). The comparative Ct (ΔΔCt) method was used to determine fold change in expression using βII-microglobulin and GAPDH. Each sample was run minimally at three concentrations in triplicate (error bar means S.D.). The experiment was triplicate with similar results. C, Western blot analysis of levels of cell cycle-related genes. Forty-eight-hour treatment with cyclopamine reduced levels of expression of cyclin D1, cyclin E1, SKP2, and phosphprylated RB (pRb) proteins. Expression of P21cip1 protein was upregulated by cyclopamine treatment. The experiment was triplicate with similar results (cyclopamine: 10 μM).
Knock down of SMO prevents osteosarcoma growth in vivo
To confirm the effect of SMO suppression, we examined the effect of SMO shRNA. 143B was transfected with control shRNA or SMO shRNA. SMO shRNA reduced the expression of SMO mRNA (Fig. 5A). MTT assay revealed that knock-down of SMO prevented osteosarcoma growth in vitro (Fig. 5A). We next used a clonogenic assay to determine whether cells capable of forming anchorage-independent colonies were depleted by SMO shRNA. This assay revealed SMO shRNA reduced colony formation in soft agar (Fig. 5B). These findings show that suppression of SMO prevents osteosarcoma growth in vitro. We then examined the transcription of cell cycle-related genes. Real-time PCR revealed that SMO shRNA prevented the transcription of accelerators of the cell cycle including cyclin D1, cyclin E1, SKP2, and E2F1 (see additional file 2). To examine the in vivo effect of SMO shRNA, nude mice were inoculated with control shRNA or SMO shRNA transfected 143B osteosarcoma cells intradermally. Results demonstrated significant inhibition of tumor growth SMO shRNA versus control shRNA (Fig. 6A, B). Kaplan-Meier analysis showed that SMO shRNA conferred a significant survival benefit (Fig. 6B). Next, we performed real-time PCR using formed tumors. Real-time PCR revealed that transcription of GLI1, GLI2, and PTCH1 was decreased in tumors formed by SMO shRNA-transfected 143B. These findings showed that SMO shRNA prevented the transcription of Hh target genes in vivo. In addition, SMO shRNA prevented the transcription of accelerators of the cell cycle including cyclin E1, SKP2, and E2F1 (see additional file 3). Histological analysis indicated that SMO shRNA prevented cell proliferation. The control tumors exhibited a number of cells positive for Ki67, a marker of cell proliferation. In contrast, SMO shRNA transfected tumors exhibited little evidence of proliferation, as evidenced by lack of Ki67 positivity. The number of Ki67-positive cells was decreased to 30% of control revel by SMO shRNA (Fig. 6C). These findings suggest that inhibition of SMO prevents osteosarcoma growth by cell cycle regulation in vivo.

Knock down of SMO by SMO shRNA prevents osteosarcoma growth in vitro. A, Real-time PCR revealed that SMO shRNA effectively knock down SMO mRNA. (error bar means S.D.). The comparative Ct (ΔΔCt) method was used to determine fold change in expression using ACTB. Each sample was run minimally at three concentrations in triplicate (error bar means S.D.). The experiment was triplicate with similar results. B, Growth of viable 143B cells over 4 days was slowed by SMO shRNA. The experiment was triplicate with similar results. C, Colony formation assay revealed that SMO shRNA reduced colony formation in soft agar. The experiment was triplicate with similar results. (*: P < 0.01) (Error bar means S.D.)

SMO shRNA prevents osteosarcoma xenograft growth in vivo and prolongs survival. A, SMO shRNA or control shRNA transfected 143B cells (1 × 106) were inoculated subcutaneously. Established 143B tumors were measured. The tumor volume at day 7 was set at 1, and tumor volumes at subsequent time points were calculated. SMO shRNA inhibited tumor growth at 8 weeks compared to control shRNA. B, Kaplan-Meier survival curves from SMO shRNA groups and control shRNA. Kaplan-Meier analysis showed that SMO shRNA conferred a significant survival benefit (n = 6, p < 0.05). C, Immunohistochemical examination of ki67 was performed in xenograft tumors. SMO shRNA decreased SMO RNA in vivo. Ki67 staining revealed that proliferation of osteosarcoma cells was decreased by GSI treatment. The number of Ki67-positive cells was decreased to 30% of control revel by SMO shRNA (error bar means S.D.) (*: P < 0.01).
Discussion
Although the role of Hh signaling in various cancers [18–21], it's role in the pathogenesis of osteosarcoma has not been reported. In the present study, we found that Shh, Dhh, PTCH1, SMO, GLI1 and GLI2 transcripts were over-expressed in osteosarcoma cell line. In addition, SMO, PTCH1, and GLI2 were over-expressed in osteosarcoma biopsy specimens'. In general, it is accepted that enhanced Hh pathway activation leads to downstream expression of target genes including PTCH1 and GLI, and hence, the levels of these transcripts are often used as surrogate markers of Hh pathway activity [22]. In addition, SHH promoted osteosarcoma cells proliferation. Our findings suggest that Hh pathway is activated in osteosarcomas. On the other hand, GLI1 was down-regulated in human osteosarcoma biopsy specimens (data not shown). The reason for GLI1 down-regulation could not be determined. One possibility is that the GLI1 promoter is inactivated in human osteosarcomas by epigenetic modification. We found that GLI1 promoter contains a CG-rich region. Wong et al. reported that Hh pathway activity down-stream of SMO is mediated by GLI2 [23]. These data suggest that Hh activity down-stream of SMO is mediated by GLI2 instead of GLI1 in osteosarcoma.
SMO is a central transducer of the Hh signal and important anticancer drug target [11, 14, 19, 22, 24–33]. Warzecha et al reported that cyclopamine is able to inhibit proliferation of osteosarcoma cell lines [34]. In agreement with their findings, our results showed that inhibition of SMO by cyclopamine or SMO shRNA is efficient in suppressing tumourigenic properties of osteosarcoma cells both in vitro and in vivo. We used cyclopamine to inhibit SMO in xenograft model at first. We performed that treatment with 25 mg/kg cyclopamine reduced numbers of ki67-positive cells (see additional file 4). These findings suggest that inhibition of SMO prevents osteosarcoma growth by cell cycle regulation in vivo. Although it appeared that osteosarcoma growth was prevented by cyclopamine, all mice died for undetermined reasons by 1 month after cyclopamine treatment (data not shown). We next performed 10 mg/kg cyclopamine treatment, and found no difference in osteosarcoma growth between cyclopamine treatment and the control group (data not shown). Unfortunately, a therapeutic dose of this agent in the 143B xenograft model could not be obtained. It has been reported that cyclopamine might not be a good candidate for a drug in the treatment of malignant tumors because it had several serious side effects in young mice, including weight loss and dehydration, suggesting that it may not be possible to achieve a therapeutic dose in our xenograft model system [28, 35]. In efforts to solve these problems, we used SMO shRNA. SMO shRNA inhibited osteosarcoma growth. Kaplan-Meier analysis showed that SMO shRNA conferred a significant survival benefit. It was reported that administration of RNAi resulted in silencing of the target genes in vivo [36–41]. These findings demonstrate the therapeutic potential of SMO shRNA for the treatment of osteosarcoma. Although SMO is the major signal transducer of the Hh pathway, SMO inhibition suppresses tumorigenesis by down-regulation of β-catenin mediated Wnt signaling [42]. It was reported that deregulation of β-catenin signaling is implicated in the pathogenesis of osteosarcoma [43, 44]. Further examination might be needed the relationship between SMO inhibition and Wnt-β-catenin signaling in osteosarcoma.
Cyclopamine promoted G1 arrest in osteosarcoma in vitro. We also found that cyclopamine treatment regulated the expression of cell cycle regulators. Quantitative real-time PCR and western blot analysis revealed that cyclin D1, E1, SKP2, and pRB were down-regulated upon SMO inhibition with cyclopamine. Cyclin D1, cyclin E1, SKP2, and pRb have been reported to promote G1-S phase progression [45–48]. Our findings suggest that cyclopamine promoted cell cycle arrest via down-regulation of cyclins and pRb. It has been reported that cyclin D1 and cyclin E1 are direct targets of Hh signaling [49, 50]. GLI2 mediated the mitogenic effects of Shh by transcriptional activation of cyclin D1 and cyclin D2 in developing hair follicles [51]. Our findings are consistent with the results of these previous studies. We showed that cyclopamine decreased the transcription of SKP2. The relationship between Hh signaling and SKP2 have not been reported. We attempted to find a GLI binding site (GACCACCCA) in the -1000 to +20 region of the 5' flanking sequence of SKP2, but found no GLI binding consensus sequence. These findings suggest that transcription of SKP2 might not be regulated by GLI. It has been reported that the SKP2 gene contains a functional E2F response element and is transcribed by E2F1 [52]. E2F1 is an early transcriptional target of GLI2 [53]. In addition, E2F1 transcription is activated by Rb phosphorylation. Our findings suggest that down-regulation of E2F1 and pRb indirectly reduced the transcription of SKP2. In addition, we showed that cyclopamine treatment promoted p21Cip1 up-regulation. p21cip1 can bind to various cyclin dependent kinases and that it inhibits their kinase activity. Our findings suggest that inhibition of the Hh pathway reduces the expression of the SKP2 subunit of the ubiquitin-ligase complex SCFSKP2, which in turn inhibit proteasome�mediated degradation of p21Cip1 and promote cell cycle arrest.
It has been reported that cyclopamine treatment induced apoptosis in tumor cells [20, 32, 54]. We performed apoptosis assay, but could not detect apoptosis of 143B osteosarcoma cell line (data not shown). This finding may be the result of differences in cell viability between osteosarcoma and other cancer cell lines.
Several key signalling pathways, such as Hedgehog, Notch, Wnt and BMP-TGFbeta-Activin (bone morphogenetic protein-transforming growth factor-beta-Activin), are involved in most processes essential to the proper development of an embryo. It is also becoming increasingly clear that these pathways can have a crucial role in tumorigenesis (reviewed in [19]). We previously reported that activation of Notch signaling promote the progression of human osteosarcoma [55]. Additionally, some recent reports have provided evidence for direct interaction or cross-talk between these pathways (reviewed in [56]). Further examination should be performed to elucidate these pathways interaction in osteosarcoma pathogenesis.
Several recent papers have demonstrated that anti-tumor effect by SMO inhibitors are mostly due to their effect on stromal cells [57, 58]. On the other hand some papers have reported that Hh signaling pathway is activated in cancer cells [14, 17, 21, 23, 59]. Although, there is a possibility that anti-osteosarcoma effect by cyclopamine was partially dependent to the effect on bone marrow stromal cell, anti-tumor effect of SMO shRNA revealed that inactivation of SMO directly inhibits osteosarcoma proliferation in vitro and in vivo.
The hypothesis that malignant tumours are generated by rare populations of Tumour-initiating cells (TIC), also called cancer stem cells, that are more tumourigenic than other cancer cells has gained increasing credence [31, 60]. Some reports have shown the existence of TICs in bone and soft tissue sarcomas [61–65]. Magali et al. reported that loss of Smo causes depletion of TICs whereas constitutively active Smo augments TICs number and accelerates disease [20, 66]. These data suggest that inhibition of Hh pathway might affect the proliferation of TICs of osteosarcoma.
In conclusion, our findings demonstrate that the Hh pathway is functionally activated in osteosarcoma. This novel finding improves understanding of osteosarcoma and may be important in understanding the proliferation of osteosarcoma cells. Our findings suggest that inactivation of SMO may be an attractive target for the treatment of patients with osteosarcoma.
Methods
Cell culture
HOS, 143B, MG63, and Saos-2 cells were purchased from the American Type Culture Collection (ATCC, Manassas, USA). NOS-1 was purchased from RIKEN cell bank (Tsukuba, Japan) [67]. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). Human osteoblast cells (NHOst) were purchased from Sanko Junyaku (Tokyo, Japan). Cells were cultured with OBM™ (Cambrex, East Rutherford, NJ, USA) or DMEM supplemented with 10% FBS. All cells were grown in a humidified atmosphere containing 5% CO2 at 37°C.
Patient' specimens
All human osteosarcoma biopsy specimens were obtained from primary lesions. Biopsy was performed before chemotherapy or radio therapy for diagnostic purpose. Normal bone tissue was obtained from femur during total hip arthroplasty. The study protocol was approved by the institutional review board of the Kagoshima University. All patients and controls gave written informed consent.
MTT assay
Cells were incubated with substrate for MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) for 4 h, and washed with PBS and lysed to release formazan from cells. Then cells were analyzed in a Safire microplate reader (BIO-RAD, Hercules, CA, USA) at 562 nm. Cyclopamine and tomatidine was purchased from Funakoshi (Tokyo, Japan). 143B cell were serum starved for 12 h, and then cultured with recombinant human sonic hedgehog (R&D Systems, Minneapolis, Japan).
SMO shRNA was purchased from SABiosciences (Maryland, USA). SMO and control shRNAs were cloned into pGeneClip™ Neomycin Vector, which express shRNA under the control of the U1 promoter. Lipofection of shRNA was performed every other day as recommended in the supplier's protocol using FuGENE 6 (Roche, Basel, Switzerland).
Colony formation assay
Colony formation assay was performed as previously described [68]. Briefly, cells were suspended in DMEM containing 0.33% agar and 10% fetal bovine serum and plated onto the bottom layer containing 0.5% agar. The cells were plated at a density of 5 × 103 per well in a 24-well plate, and colonies were counted 14 days later. Each condition was analyzed in triplicate, and all experiments were repeated three times.
Real-time PCR
All primer sets amplified 100- to 200-bp fragments. Total RNA was extracted using the miR-Vana RNA isolation system or TRIzol (Invitrogen, Carlsbad, CA, USA). Reactions were run using SYBR Green (BIO-RAD, Hercules, CA, USA) on a MiniOpticon™ machine (BIO-RAD, Hercules, CA, USA). The comparative Ct (ΔΔCt) method was used to determine fold change in expression using βII-microglobulin, or GAPDH, or ACTB. Each sample was run at three concentrations in triplicate. The following primers were used. Desert hedgehog: 5-TGATGACCGAGCGTTGTAAG-3, 5-GCCAGCAACCCATACTTGTT-3; Indian Hedgehog: 5-ACTTCTGCCTGGTCCTGTTG-3, 5-AGCGATCTTGCCTTCATAGC-3; Sonic hedgehog: 5-ACCGAGGGCTGGGACGAAGA-3, 5-ATTTGGCCGCCACCGAGTT-3; PATCHED: 5-TAACGCTGCAACAACTCAGG-3, 5-GAAGGCTGTGACATTGCTGA-3; SMOOTHENED: 5-GGGAGGCTACTTCCTCATCC-3, 5-GGCAGCTGAAGGTAATGAGC-3; GLI1: 5-GTGCAAGTCAAGCCAGAACA-3, 5-ATAGGGGCCTGACTGGAGAT-3, GLI2: 5-CGACACCAGGAAGGAAGGTA-3, 5-AGAACGGAGGTAGTGCTCCA-3; cyclin D1: 5-ACAAACAGATCATCCGCAAACAC-3, 5-TGTTGGGGCTCCTCAGGTTC-3; cyclin E1: 5-CCACACCTGACAAAGAAGATGATGAC-3, 5-GAGCCTCTGGATGGTGCAATAAT-3; SKP2: 5-TGGGAATCTTTTCCTGTCTG-3, 5-GAACACTGAGACAGTATGCC-3; NMYC: 5-CTTCGGTCCAGCTTTCTCAC-3, 5-GTCCGAGCGTGTTCAATTTT-3; βII-microgloblin: 5-TCAATGTCGGATGGATGAAA-3, 5-GTGCTCGCGCTACTC TCTCT-3; GAPDH: 5-GAAGGTGAAGGTCGGAGTC-3, 5-GAAGATGGTGATGGGATTTC-3; ACTB: 5-AGAAAATCTGGCACCACACC-3, 5-AGAGGCGTACAGGGATAGCA-3.
Immunohistochemistry
The following primary antibodies were used; anti-SMO (diluted 1:200, Santa Cruz, CA. U.S.A), anti-GLI2 (diluted 1:200, Abcam, Cambridge, UK), and anti-ki67 (Zymed laboratories, San Francisco, USA). The following secondary antibodies were used:; fluorescein-conjugated goat anti-mouse IgG antibody (diluted 1:200; Jackson ImmunoResearch, PA, USA) and rhodamine-conjugated donkey anti-rabbit IgG antibody (diluted 1:200; Chemicon, Temecula, CA). The cells were counterstained with Hoechst 33258 (Molecular Probes, Carlsbad, CA, USA) to identify nuclei. Immunohistochemistry with each second antibody alone without primary antibody was performed as a control.
Western blot
Cells were lysed using NP40 lysis buffer (0.5% NP40, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 3 mM pAPMSF (Wako Chemicals, Kanagawa, Japan), 5 mg/ml aprotinin (Sigma, StLouis, USA), 2 mM sodium orthovanadate (Wako Chemicals, Kanagawa, Japan), and 5 mM EDTA). Lysates were subjected to SDS-PAGE and subsequent immunoblotting with antibodies to cyclin D1, E1, p21, SKP2, and pRb (Santa Cruz, CA. U.S.A). Detection was performed using the ECL detection system (Amersham, Giles, UK).
Animal experiments
143B cells (1 × 106) were mixed with a collagen gel in a 1:1 volume, and were inoculated subcutaneously in 5-week-old nude mice. The mice were randomly assigned to receive either cyclopamine (25 mg/kg-10 mg/kg) or an equal volume of DMSO as control. Cyclopamine and saline solution were administered by intraperitoneal injection. The treatment with cyclopamine was initiated 1 week after tumor inoculation when the tumors had grown to visible size. The injections were repeated every other day. Tumor size was measured with calipers weekly, and tumor volume was calculated using a formula of LW2/2 (L and W representing the length and width of tumors). SMO shRNA (SABiosciences, Maryland, USA) transfected 143B cells (1 × 106) or control shRNA (1 × 106) cells were mixed with a collagen gel in a 1:1 volume, and were inoculated subcutaneously in 5-week-old nude mice. Tumor size was measured with calipers weekly, and tumor volume was calculated using a formula of LW2/2 (L and W representing the length and width of tumors). All experimental procedures were performed in compliance with the guiding principles for the Care and Use of Animals described in the American Journal of Physiology and with the Guidelines established by the Institute of Laboratory Animal Sciences, Faculty of Medicine, Kagoshima University. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize possible alternatives to in vivo techniques.
Cell cycle analysis
Cell cycle analysis was performed by Reprocell (Tokyo, Japan). At 48 h after cyclopamine treatment, cells were collected by trypsinization and washed with DPBS. Cells were fixed in 70% (v/v) ethanol at 4°C, washed with PBS, and resuspended with 500 μl of staining solution [PBS pH 7.4, 100 μg/ml DNase-free RNase, 1 mg/ml propidium iodide]. Cells were then analyzed by flow cytometry using a FACS Vantage SE (Becton Dickinson, Franklin Lakes, NJ). Data were gated using pulse width and pulse area to exclude doublets, and the percentage of cells present in each phase of the cell cycle was calculated using FlowJo software (Tree Star, Ashland, OR, USA).
Statistics and supplemental data
Each sample was analyzed in triplicate, and experiments were repeated three times. In all figures, error bars are standard divisions. All statistical analyses were performed using Microsoft Office Excel (Microsoft, Albuquerque, New Mexico, USA) and STASTISCA (StatSoft, Tulsa, OK, USA). Differences between mean values were evaluated by the unpaired t-test, and differences in frequencies by Fisher's exact test. Differences were considered significant at P < 0.05.
Abbreviations
- (Hh):
-
Hedgehog
- (SMO):
-
SMOOTHENED
- (PTCH1):
-
PATCHED
- (SHH):
-
Sonic hedgehog
- (DHH):
-
Desert hedgehog.
References
Gibbs CP, Weber K, Scarborough MT: Malignant bone tumors. Instr Course Lect. 2002, 51: 413-428.
Igney FH, Krammer PH: Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002, 2: 277-288. 10.1038/nrc776
Lafleur EA, Koshkina NV, Stewart J, Jia SF, Worth LL, Duan X, Kleinerman ES: Increased Fas expression reduces the metastatic potential of human osteosarcoma cells. Clin Cancer Res. 2004, 10: 8114-8119. 10.1158/1078-0432.CCR-04-0353
Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, Minna JD, Dryja TP, Weinberg RA: Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci USA. 1990, 87: 2775-2779. 10.1073/pnas.87.7.2775
Ingham PW, McMahon AP: Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001, 15: 3059-3087. 10.1101/gad.938601
Ruiz i Altaba A, Sanchez P, Dahmane N: Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. 2002, 2: 361-372. 10.1038/nrc796
McMahon AP, Ingham PW, Tabin CJ: Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003, 53: 1-114. full_text
Fogarty MP, Kessler JD, Wechsler-Reya RJ: Morphing into cancer: the role of developmental signaling pathways in brain tumor formation. J Neurobiol. 2005, 64: 458-475. 10.1002/neu.20166
Gorlin RJ: Nevoid basal cell carcinoma syndrome. Dermatol Clin. 1995, 13: 113-125.
Ruiz i Altaba A, Mas C, Stecca B: The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17: 438-447. 10.1016/j.tcb.2007.06.007
Chen JK, Taipale J, Cooper MK, Beachy PA: Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16: 2743-2748. 10.1101/gad.1025302
Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP: Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996, 10: 301-312. 10.1101/gad.10.3.301
Hynes M, Stone DM, Dowd M, Pitts-Meek S, Goddard A, Gurney A, Rosenthal A: Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron. 1997, 19: 15-26. 10.1016/S0896-6273(00)80344-X
Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A: Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA. 2004, 101: 12561-12566. 10.1073/pnas.0404956101
Chung MK, Kim HJ, Lee YS, Han ME, Yoon S, Baek SY, Kim BS, Kim JB, Oh SO: Hedgehog signaling regulates proliferation of prostate cancer cells via stathmin1. Clin Exp Med. 2009
Mao L, Xia YP, Zhou YN, Dai RL, Yang X, Duan SJ, Qiao X, Mei YW, Hu B, Cui H: A critical role of Sonic Hedgehog signaling in maintaining the tumorigenicity of neuroblastoma cells. Cancer Sci. 2009, 100: 1848-1855. 10.1111/j.1349-7006.2009.01262.x
Singh RR, Cho-Vega JH, Davuluri Y, Ma S, Kasbidi F, Milito C, Lennon PA, Drakos E, Medeiros LJ, Luthra R, Vega F: Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009, 69: 2550-2558. 10.1158/0008-5472.CAN-08-1808
Kasper M, Regl G, Frischauf AM, Aberger F: GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006, 42: 437-445. 10.1016/j.ejca.2005.08.039
Rubin LL, de Sauvage FJ: Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006, 5: 1026-1033. 10.1038/nrd2086
Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF: Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007, 13: 944-951. 10.1038/nm1614
Lindemann RK: Stroma-initiated hedgehog signaling takes center stage in B-cell lymphoma. Cancer Res. 2008, 68: 961-964. 10.1158/0008-5472.CAN-07-5500
Watkins DN, Peacock CD: Hedgehog signalling in foregut malignancy. Biochem Pharmacol. 2004, 68: 1055-1060. 10.1016/j.bcp.2004.04.025
Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH, Dlugosz AA, Reiter JF: Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med. 2009, 15: 1055-1061. 10.1038/nm.2011
Sasai K, Romer JT, Kimura H, Eberhart DE, Rice DS, Curran T: Medulloblastomas derived from Cxcr6 mutant mice respond to treatment with a smoothened inhibitor. Cancer Res. 2007, 67: 3871-3877. 10.1158/0008-5472.CAN-07-0493
Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, Kon C, Gatchalian C, Porter JA, Rubin LL, Wang FY: Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc Natl Acad Sci USA. 2003, 100: 4616-4621. 10.1073/pnas.0732813100
Romer J, Curran T: Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005, 65: 4975-4978. 10.1158/0008-5472.CAN-05-0481
Sanchez P, Clement V, Ruiz i Altaba A: Therapeutic targeting of the Hedgehog-GLI pathway in prostate cancer. Cancer Res. 2005, 65: 2990-2992.
Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T: Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell. 2004, 6: 229-240. 10.1016/j.ccr.2004.08.019
Zhang X, Harrington N, Moraes RC, Wu MF, Hilsenbeck SG, Lewis MT: Cyclopamine inhibition of human breast cancer cell growth independent of Smoothened (Smo). Breast Cancer Res Treat. 2009, 115: 505-521. 10.1007/s10549-008-0093-3
Hegde GV, Munger CM, Emanuel K, Joshi AD, Greiner TC, Weisenburger DD, Vose JM, Joshi SS: Targeting of sonic hedgehog-GLI signaling: a potential strategy to improve therapy for mantle cell lymphoma. Mol Cancer Ther. 2008, 7: 1450-1460. 10.1158/1535-7163.MCT-07-2118
Lauth M, Bergstrom A, Shimokawa T, Toftgard R: Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA. 2007, 104: 8455-8460. 10.1073/pnas.0609699104
Chen X, Horiuchi A, Kikuchi N, Osada R, Yoshida J, Shiozawa T, Konishi I: Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: it's inhibition leads to growth suppression and apoptosis. Cancer Sci. 2007, 98: 68-76. 10.1111/j.1349-7006.2006.00353.x
Tabs S, Avci O: Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur J Dermatol. 2004, 14: 96-102.
Warzecha J, Gottig S, Chow KU, Bruning C, Percic D, Boehrer S, Brude E, Kurth A: Inhibition of osteosarcoma cell proliferation by the Hedgehog-inhibitor cyclopamine. J Chemother. 2007, 19: 554-561.
Kimura H, Ng JM, Curran T: Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008, 13: 249-260. 10.1016/j.ccr.2008.01.027
Makimura H, Mizuno TM, Mastaitis JW, Agami R, Mobbs CV: Reducing hypothalamic AGRP by RNA interference increases metabolic rate and decreases body weight without influencing food intake. BMC Neurosci. 2002, 3: 18- 10.1186/1471-2202-3-18
Matsui Y, Kobayashi N, Nishikawa M, Takakura Y: Sequence-specific suppression of mdr1a/1b expression in mice via RNA interference. Pharm Res. 2005, 22: 2091-2098. 10.1007/s11095-005-8178-8
Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J: Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004, 432: 173-178. 10.1038/nature03121
Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J: RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003, 9: 347-351. 10.1038/nm828
Zender L, Hutker S, Liedtke C, Tillmann HL, Zender S, Mundt B, Waltemathe M, Gosling T, Flemming P, Malek NP: Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci USA. 2003, 100: 7797-7802. 10.1073/pnas.1330920100
Yano J, Hirabayashi K, Nakagawa S, Yamaguchi T, Nogawa M, Kashimori I, Naito H, Kitagawa H, Ishiyama K, Ohgi T, Irimura T: Antitumor activity of small interfering RNA/cationic liposome complex in mouse models of cancer. Clin Cancer Res. 2004, 10: 7721-7726. 10.1158/1078-0432.CCR-04-1049
Arimura S, Matsunaga A, Kitamura T, Aoki K, Aoki M, Taketo MM: Reduced level of smoothened suppresses intestinal tumorigenesis by down-regulation of Wnt signaling. Gastroenterology. 2009, 137: 629-638. 10.1053/j.gastro.2009.04.059
Haydon RC, Deyrup A, Ishikawa A, Heck R, Jiang W, Zhou L, Feng T, King D, Cheng H, Breyer B: Cytoplasmic and/or nuclear accumulation of the beta-catenin protein is a frequent event in human osteosarcoma. Int J Cancer. 2002, 102: 338-342. 10.1002/ijc.10719
Iwaya K, Ogawa H, Kuroda M, Izumi M, Ishida T, Mukai K: Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clin Exp Metastasis. 2003, 20: 525-529. 10.1023/A:1025821229013
Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI: Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004, 6: 1229-1235. 10.1038/ncb1194
Hara T, Kamura T, Kotoshiba S, Takahashi H, Fujiwara K, Onoyama I, Shirakawa M, Mizushima N, Nakayama KI: Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol. 2005, 25: 9292-9303. 10.1128/MCB.25.21.9292-9303.2005
Hwang HC, Clurman BE: Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005, 24: 2776-2786. 10.1038/sj.onc.1208613
Swanton C: Cell-cycle targeted therapies. Lancet Oncol. 2004, 5: 27-36. 10.1016/S1470-2045(03)01321-4
Duman-Scheel M, Weng L, Xin S, Du W: Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002, 417: 299-304. 10.1038/417299a
Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P: Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002, 277: 5548-5555. 10.1074/jbc.M105708200
Mill P, Mo R, Fu H, Grachtchouk M, Kim PC, Dlugosz AA, Hui CC: Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003, 17: 282-294. 10.1101/gad.1038103
Zhang L, Wang C: F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2006, 25: 2615-2627. 10.1038/sj.onc.1209286
Regl G, Kasper M, Schnidar H, Eichberger T, Neill GW, Ikram MS, Quinn AG, Philpott MP, Frischauf AM, Aberger F: The zinc-finger transcription factor GLI2 antagonizes contact inhibition and differentiation of human epidermal cells. Oncogene. 2004, 23: 1263-1274. 10.1038/sj.onc.1207240
Tsuda N, Ishiyama S, Li Y, Ioannides CG, Abbruzzese JL, Chang DZ: Synthetic microRNA designed to target glioma-associated antigen 1 transcription factor inhibits division and induces late apoptosis in pancreatic tumor cells. Clin Cancer Res. 2006, 12: 6557-6564. 10.1158/1078-0432.CCR-06-0588
Tanaka M, Setoguchi T, Hirotsu M, Gao H, Sasaki H, Matsunoshita Y, Komiya S: Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br J Cancer. 2009, 100: 1957-1965. 10.1038/sj.bjc.6605060
Ross J, Li L: Recent advances in understanding extrinsic control of hematopoietic stem cell fate. Curr Opin Hematol. 2006, 13: 237-242. 10.1097/01.moh.0000231420.92782.8f
Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, de Sauvage FJ: Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci USA. 2009, 106: 4254-4259. 10.1073/pnas.0813203106
Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D: A paracrine requirement for hedgehog signalling in cancer. Nature. 2008, 455: 406-410. 10.1038/nature07275
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, Warmuth M: Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008, 14: 238-249. 10.1016/j.ccr.2008.08.003
Clarke MF, Fuller M: Stem cells and cancer: two faces of eve. Cell. 2006, 124: 1111-1115. 10.1016/j.cell.2006.03.011
Murase M, Kano M, Tsukahara T, Takahashi A, Torigoe T, Kawaguchi S, Kimura S, Wada T, Uchihashi Y, Kondo T: Side population cells have the characteristics of cancer stem-like cells/cancer-initiating cells in bone sarcomas. Br J Cancer. 2009, 101: 1425-1432. 10.1038/sj.bjc.6605330
Levings PP, McGarry SV, Currie TP, Nickerson DM, McClellan S, Ghivizzani SC, Steindler DA, Gibbs CP: Expression of an exogenous human Oct-4 promoter identifies tumor-initiating cells in osteosarcoma. Cancer Res. 2009, 69: 5648-5655. 10.1158/0008-5472.CAN-08-3580
Fujii H, Honoki K, Tsujiuchi T, Kido A, Yoshitani K, Takakura Y: Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int J Oncol. 2009, 34: 1381-1386.
Wang L, Park P, Lin CY: Characterization of stem cell attributes in human osteosarcoma cell lines. Cancer Biol Ther. 2009, 8 (6): 10.1158/1535-7163.MCT-09-0089.
Hirotsu M, Setoguchi T, Matsunoshita Y, Sasaki H, Nagao H, Gao H, Sugimura K, Komiya S: Tumour formation by single fibroblast growth factor receptor 3-positive rhabdomyosarcoma-initiating cells. Br J Cancer. 2009, 101: 2030-2037. 10.1038/sj.bjc.6605407
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D: Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009, 458: 776-779. 10.1038/nature07737
Hotta T, Motoyama T, Watanabe H: Three human osteosarcoma cell lines exhibiting different phenotypic expressions. Acta Pathol Jpn. 1992, 42: 595-603.
Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D, Brat DJ, Perry A, Eberhart CG: Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004, 64: 7787-7793. 10.1158/0008-5472.CAN-04-1446
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) (B) 18390419, (C) 19591725, (C) 21591919, (C) 21591920, and Grant-in-Aid from the Ministry of Health, Labour and Welfare for the Third Term Comprehensive Control Research for Cancer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TS was involved in the design and execution of the experiments, drafted the manuscript and contributed to the overall experimental design. MH conducted most of the experiments. HS was conducted a most of experiments. HG was conducted a part of experiments. YM was conducted a part of experiments. HN was conducted a part of experiments. OK was conducted a part of experiments. SK contributed to the overall experimental design. All authors read and approved the final manuscript.
Masataka Hirotsu, Takao Setoguchi, Hiromi Sasaki contributed equally to this work.
Electronic supplementary material
12943_2009_520_MOESM1_ESM.TIFF
Additional file 1: A, Immunohistochemical examination revealed that SMO was expressed on cytoplasm of 143B and GLI2 was localized in the nucleus of 143B. B, MTT assay showed that Sonic hedgehog promote proliferation of osteosarcoma cells. The experiment was triplicate with similar results. (TIFF 261 KB)
12943_2009_520_MOESM2_ESM.TIFF
Additional file 2: Real-time PCR was performed to quantify mRNAs of cell cycle related genes. SMO shRNA reduced levels of cyclin D1, cyclin E1, SKP2, and E2F1 transcription (error bar means S.D.). The comparative Ct (ΔΔCt) method was used to determine fold change in expression using ACTB. The experiment was triplicate with similar results. (TIFF 111 KB)
12943_2009_520_MOESM3_ESM.TIFF
Additional file 3: We performed real-time PCR using formed tumors. Real-time PCR revealed that transcription of GLI1, GLI2, and PTCH1 was decreased in tumors formed by SMO shRNA-transfected 143B. In addition, SMO shRNA reduced levels of Cyclin E1, SKP2, and E2F1 transcription. The comparative Ct (ΔΔCt) method was used to determine fold change in expression using ACTB. The experiment was triplicate with similar results. (TIFF 138 KB)
12943_2009_520_MOESM4_ESM.TIFF
Additional file 4: Cyclopamine prevents proliferation of osteosarcoma in vivo. Immunohistochemical examination of ki67 was performed in xenograft tumors. Ki67 staining revealed that proliferation of osteosarcoma cells was decreased by cyclopamine treatment. The numbers of Ki67-positive cells was decreased to 50% of control revel by cyclopamine administration at day 14 (error bar means S.D.). (TIFF 335 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hirotsu, M., Setoguchi, T., Sasaki, H. et al. Smoothened as a new therapeutic target for human osteosarcoma. Mol Cancer 9, 5 (2010). https://doi.org/10.1186/1476-4598-9-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-9-5