
Abstract
Background
The xanthophyll astaxanthin is a high-value compound with applications in the nutraceutical, cosmetic, food, and animal feed industries. Besides chemical synthesis and extraction from naturally producing organisms like Haematococcus pluvialis, heterologous biosynthesis in non-carotenogenic microorganisms like Escherichia coli, is a promising alternative for sustainable production of natural astaxanthin. Recent achievements in the metabolic engineering of E. coli strains have led to a significant increase in the productivity of carotenoids like lycopene or β-carotene by increasing the metabolic flux towards the isoprenoid precursors. For the heterologous biosynthesis of astaxanthin in E. coli, however, the conversion of β-carotene to astaxanthin is obviously the most critical step towards an efficient biosynthesis of astaxanthin.
Results
Here we report the construction of the first plasmid-free E. coli strain that produces astaxanthin as the sole carotenoid compound with a yield of 1.4 mg/g cdw (E. coli BW-ASTA). This engineered E. coli strain harbors xanthophyll biosynthetic genes from Pantoea ananatis and Nostoc punctiforme as individual expression cassettes on the chromosome and is based on a β-carotene-producing strain (E. coli BW-CARO) recently developed in our lab. E. coli BW-CARO has an enhanced biosynthesis of the isoprenoid precursor isopentenyl diphosphate (IPP) and produces β-carotene in a concentration of 6.2 mg/g cdw. The expression of crtEBIY along with the β-carotene-ketolase gene crtW148 (NpF4798) and the β-carotene-hydroxylase gene (crtZ) under controlled expression conditions in E. coli BW-ASTA directed the pathway exclusively towards the desired product astaxanthin (1.4 mg/g cdw).
Conclusions
By using the λ-Red recombineering technique, genes encoding for the astaxanthin biosynthesis pathway were stably integrated into the chromosome of E. coli. The expression levels of chromosomal integrated recombinant biosynthetic genes were varied and adjusted to improve the ratios of carotenoids produced by this E. coli strain. The strategy presented, which combines chromosomal integration of biosynthetic genes with the possibility of adjusting expression by using different promoters, might be useful as a general approach for the construction of stable heterologous production strains synthesizing natural products. This is the case especially for heterologous pathways where excessive protein overexpression is a hindrance.
Similar content being viewed by others
Background
Xanthophylls comprise the group of oxygenated carotenoids that are synthesized by many photosynthetic organisms and also by some non-photosynthetic yeasts, fungi, and bacteria via condensation of isoprenoid units and subsequent oxidation reactions. The xanthophyll astaxanthin (3,3'-dihydroxy-β,β-carotene-4,4'-dione) has gained considerable attention due to its beneficial effect on human health. It has been shown that astaxanthin bears a strong antioxidant and singlet oxygen-quenching activity [1] which can modulate biological functions ranging from lipid peroxidation to tissue protection against UV-light damage [2]. Preclinical studies have further shown that astaxanthin exhibits anti-inflammatory properties and reduces rethrombosis after thrombolysis [3]. Even more than the encouraging beneficial health properties, astaxanthin is used as a food colorant. The red-orange color of astaxanthin is closely connected with the quality of salmon or trout, for example. Therefore, the supplementation of astaxanthin or other carotenoids to their diets improves their value. Furthermore, the natural carotenoids in the diet of fish play an important role in reproduction [4].
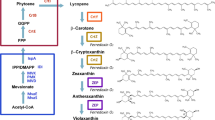
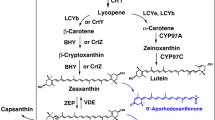
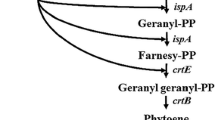
Besides chemical synthesis or extraction from naturally producing organisms like the green algae Haematococcus pluvialis or the yeast Xanthophyllomyces dendrorhous[5], the use of non-carotenogenic microorganisms like Escherichia coli as a heterologous biosynthesis host is a promising alternative for the sustainable production of natural astaxanthin. Recent achievements in metabolic engineering of E. coli and yeast strains have led to a significant increase in the productivity of isoprenoid compounds (in particular terpenoids and carotenoids), by increasing the metabolic flux towards the isoprenoid precursors [6–11]. For the heterologous biosynthesis of astaxanthin, however, the complete conversion of β-carotene to astaxanthin is obviously the most critical step towards an efficient biosynthesis of astaxanthin in E. coli[12–14]. The conversion of β-carotene to astaxanthin requires the introduction of keto-groups at 4 and 4' as well as hydroxyl-groups at 3 and 3'positions of the β-ionone rings. The addition of the keto groups is catalyzed by the β-carotene ketolase, which is encoded by crtO or by crtW genes [14, 15]. The introduction of the hydroxyl groups is catalyzed by the β-carotene hydroxylase. There are three known isoforms of this enzyme: CrtZ, CrtR, and cytochrome-P450 hydroxylase [16–19]. The isoforms of β-carotene ketolase and β-carotene hydroxylase as well as their variants from different organisms show differences in their substrate specificity concerning the 3-hydroxylation or 4-ketolation status of the β-ionone rings. As a result, the expression of ketolase and hydroxylase in naturally producing organisms as well as in a heterologous host leads to the formation of up to eight intermediates beside astaxanthin. See Figure 1 for details of astaxanthin biosynthesis in a non-carotinogenic strain like E. coli. E. coli produces farnesyl pyrophosphate (FPP) as an isoprenoid precursor that can be further converted to astaxanthin by heterologous biosynthetic genes. In the cyanobacterium Nostoc puntiforme PCC73102 two different genes of the crtW-type were found (NpF4798 (crtW148), NpF5919 (crtW38)). The recombinant enzymes CrtW38 and CrtW148 are both able to convert β-carotene into canthaxanthin, but only CrtW148 converts zeaxanthin into astaxanthin [20]. Similar observations have been made in the case of hydroxylase enzymes. CrtZ from Pantoea ananatis is able to introduce hydroxyl-groups into the β-ionone rings regardless of the presence of keto-groups in 4 and 4' positions [12]. The structurally different hydroxylases from cyanobacteria are not able to convert echinenone or canthaxanthin into astaxanthin [21].

Scheme of the putative astaxanthin biosynthesis pathway in recombinant E. coli , proceeding from the central intermediates pyruvate (Pyr) and glyceraldehyde-3-phosphate (GAP), highlighting the different possible routes of ketolase and hydroxylase reaction that can take place by the conversion of β-carotene (1) to astaxanthin (10). Dxs 1-deoxyxylulose-5-phosphate synthase, Idi isopentenyl diphosphate isomerise, IspA farnesyl diphosphate synthase, CrtE geranylgeranyl diphosphate synthase, CrtB phytoene synthase, CrtI phytoene desaturase, CrtY lycopene cyclase, W β-carotene ketolase (CrtW), Z β-carotene hydroxylase (CrtZ), IPP isopentenyl diphosphate, DMAPP dimethylallyl diphosphate, DXP 1-deoxyxylulose-5-phosphate, (2) β-cryptoxanthin, (3) echinenone, (4) 3'-hydroxyechinenone, (5) 3-hydroxyechinenone, (6) zeaxanthin, (7) canthaxanthin, (8) adonixanthin, (9) adonirubin.
At present, no recombinant xanthophyll-producing E. coli host has been reported that is able to accumulate astaxanthin as the sole carotenoid compound. However, significant improvements have been achieved by protein engineering of the β-carotene ketolase. By using random mutagenesis, crtW mutants form Paracoccus sp. and Sphingomonas sp., were generated that showed an up to 81% [22] and 90% [23] production of astaxanthin, respectively, when expressed in zeaxanthin-producing E. coli strains.
So far, all studies on the formation of xanthophyll compounds in E. coli use plasmids for the expression of the heterologous genes. To avoid the use of recombinant plasmids and to allow dispensing with selection makers like antibiotics, we present here a strain harboring the biosynthetic genes crtE, crtB, crtI, crtY, and crtZ from P. ananatis and crtW148 (NpF4798) from N. punctiforme PCC 73102 that are required for the formation of astaxanthin in E. coli stably inserted into the chromosome, along with an enhanced expression of the native E. coli genes idi and dxs. This strain is based on a β-carotene-producing basis strain (E. coli BW-CARO) recently developed in our lab [24]. Furthermore, by balancing the expression of crtZ and crtW148, which was determined by reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR), a plasmid-free E. coli strain was engineered that accumulated astaxanthin as the exclusive carotenoid.
Materials and methods
Bacterial strains, media, and chemicals
All strains and plasmids used or constructed in this study are listed in table 1. The bacterial strains used in this study were Escherichia coli BW-CARO [24] and E. coli DH5α. If not stated otherwise, strains of E. coli were grown at 37°C in LB medium. The minimal medium used had the following composition: KH2PO4 3 g L-1, K2HPO4 12 g L-1, (NH4)2SO4 5 g L-1, MgSO4 × 7H2O 0.3 g L-1, CaCl2 × 2H2O 0.015 g L-1, NaCl 0.1 g L-1, Glucose monohydrate 5 g L-1, FeSO4 × 7H2O/sodium citrate 15 ml L-1 (from the solution of 7.5 g L-1 FeSO4 × 7H2O and 100 g L-1 sodium citrate), thiamine, trace elements [25] 33 ml L-1. Antibiotics were used at the following final concentrations: ampicillin 100 μg ml-1, chloramphenicol 50 μg ml-1. Difco MacConkey agar base was purchased from Nordwald (Hamburg, Germany). Carotenoid standards were provided by the Institute of Technical Biochemistry, University of Stuttgart. All other chemicals and reagents are from Sigma-Aldrich or Roth (Germany) and were of the highest available purity.
Construction of plasmids
The crtW148 reading frame was obtained from plasmid pPQE32-148 [20]. After Nde I/Hind III digestion, the fragment was ligated into Nde I/Hind III digested pJF119ΔN [26] and transformed into E. coli DH5α. The resulting plasmid pJF119-crtW148 was further treated with Hind III and ligated with a Hind III digested FRT-cat-FRT fragment derived from plasmid pCAS30-FRT-cat-FRT [27].
The reading frame of crtZ was isolated from plasmid pJF119-crtZ [24] after Nde I/Bam HI treatment and cloned into pAW223 [Nde I/Bam HI]. The resulting plasmid pAW-crtZ was further treated with Hind III and ligated with a Hind III digested FRT-cat-FRT fragment. The DNA sequence of idi was amplified by polymerase chain reaction (PCR) from chromosomal DNA of E. coli LJ110 using the following oligonucleotides: a) GTGAGAACATATG CAAACGGAACACGTC b) CAAATGTCGGGATCC TTTTATTTAAGCTGGG. The PCR-product was treated with Nde I and Bam HI and was ligated into expression vector pJF119ΔN hydrolyzed with Nde I and Bam HI. The resulting plasmid pJF-idi was further treated with Hind III and ligated with a Hind III digested FRT-cat-FRT fragment to gain plasmid pJF119-idi- FRT-cat-FRT.
The dxs promoter replacement was conducted according to the method used by Yuan et al [28]. Thus, for the construction of pQE31-FRT-cat-FRT, the DNA sequence of FRT-cat-FRT was amplified by PCR from plasmid pCAS30-FRT-cat-FRT by using the following oligonucleotides: a) CACAGACTGAGGATCCTCGAG AGTCGACCTGCAGG and b) CTGTTTTATCAGACCGCTCGAG CGTTCTGATTTA. The PCR product was hydrolyzed with Xho I and ligated into the Xho I hydrolyzed plasmid pQE31.
Chromosomal integration
The individual expression cassettes of idi, crtW148, and crtZ from plasmid pJF-idi- FRT-cat-FRT, pJF-crtW-FRT-cat-FRT, and pAW-crtZ-FRT-cat-FRT, respectively, were amplified by PCR. The oligonucleotide primers for the Ptac-crtW expression cassette were: 1) CGGCGCATATTGCCCTGATGGACATTGACCCCACCCGCCTGGAAGAGTCGCATATTGTTCAAGGCGCACTCCCGTTCTGG and 2) AGCGCAACGATGGCTTTAAGTGTCAGATGGCTTCCTTCAGCAGACGGTTGATTGTCTGCAGGGTTATTGTCTCATGAGCG. Primers for the Prha-crtZ expression cassette were: ACCGTTCTTAATTCTGATATTTCATCGGTGATCTCCCGTCTGGGACATACCGATATGCATGCATCGATCACCACAATT and ATTCACGCTAGCCCATACACCACGACTTCCTAAAGTAATCAGTACAGTACGGATACC CAGGGTTATTGTCTCATGAGCGGATAC. Primers for the Ptac-idi expression cassette were: TGCAGGCATGAAACCGCGTCTTTTTTCAGATAAAAAGCGCAATCAGTCGCTCAA GGCGCACTCCCGTTCTGG and TAACATTACTCAGCAATAAACTGATATTCCGTCAGGCTGGAATAAGGATGGCCT TCTGCTTAATTTGATGCCTG.
For the integration of the expression cassettes, the purified PCR products were each transformed into E. coli BW-CARO and its variant strain carrying plasmid pKD46. Expression of the λ-Red enzymes and the preparation of competent cells were carried out as described previously [29]. Competent cells were electroporated with 0.2-0.4 μg of PCR product. After electroporation, the cells were resuspended in 1 ml LB-medium and incubated at 30°C for 12 h with shaking. Subsequently, the cell suspension was spread onto MacConkey-agar plates containing chloramphenicol and 1% of the respective sugar (D-galactose, D-ribose, or melibiose), corresponding to the targeted genes responsible for sugar degradation. MacConkey agar plates were incubated over-night at 37°C. 5-10 pale colonies per transformation were picked [24] and checked regarding correct recombination by control PCR. The chloramphenicol resistance cassette was eliminated using the plasmid pCP20 as previously described [30].
Extraction and analysis of carotenoids
E. coli strains carrying carotenoid biosynthetic genes were cultivated in shake flasks in 50 ml LB medium or minimal medium. Cultivations were carried out at 30°C. At an optical density (OD600) of 0.3 - 0.4 the cultures were induced, if required, by addition of IPTG (0.5 mM, final conc.) and/or L-rhamnose (0.2% w/v, final conc.). Samples (1 ml) were withdrawn from cultures after different time points within a period of 48 h. The cells were harvested by centrifugation, washed with cold water, and subsequently extracted by vigorous shaking with acetone (500 μl) for 15 min at 50°C. Insoluble components of the extract were removed by centrifugation (20,000 × g). HPLC analysis was performed on Dionex HPLC Instrument (Idstein, Germany), installed with a Chromeleon Software, Gina autosampler, P580 pumps, and a diode array detector. Products were analyzed by loading 50 μl of the supernatant onto a C30-reverse-phase HPLC column (250 mm × 4.6 mm, 5 μm, YMC-Europa GmbH, Dinslaken, Germany) attached with a guard column containing matrix of the same material as the column. A solvent flow rate of 1.0 ml/min was used. The solvents used were Solvent A, consisting of MTBE (methyl-tert-butylether)/methanol/water (19/80/1) and Solvent B, consisting of MTBE/methanol/water (90/9/1). Gradient conditions: equilibration conditions at 2% B; 0 to 22 min linear gradient from 2% B to 100% B. The spectra of the eluted carotenoids were recorded online with a Dionex UVD340 diode array detector (200 - 600 nm).
Carotenoid compounds were identified by co-chromatography using authentic standard compounds and by analysis of its UV-Vis spectra.
For the quantification of the carotenoid compounds the integrated peak areas were compared to HPLC standard curves of authentic standards.
Detection and quantification of crtW148 and crtZ
Although isoprenoid products of CrtW148 and CrtZ were detected, neither enzyme activity tests nor SDS-PAGE were able to verify the presence of these proteins. Therefore, mRNA from crtW148 and crtZ was detected by means of absolute RT-qPCR using an internal standard.
(I) Generation of the internal standard
Internal hpd (hydroxyphenyl-pyruvate dioxygenase gene from Pseudomonas putida) RNA standard was generated. This was similar to a protocol published by Schuhmacher et al. [31] with the following exceptions: as template for the internal standard, pCAS1 was used [26].
(II) Sampling, total RNA isolation, and cDNA synthesis
E. coli BW-ASTA was cultivated in shaking flasks at 37°C (140 rpm) either in LB; LB plus L-rhamnose [12 mM]; LB plus L-rhamnose [12 mM] and IPTG [0.5 mM] or in minimal medium (MM); MM plus IPTG [0.5 mM]; or MM plus IPTG [0.5 mM] and L-rhamnose [12 mM]. For transcript level quantification, samples from the late exponential phase were taken. In total, two biological replicates were processed.
A cell number - OD correlation was generated by counting E. coli BW-ASTA using a Neubauer counting chamber. According to this correlation, 5 × 108 cells were pipetted into twice the amount RNA protect bacteria reagent (Qiagen) and processed according to manufacturer's protocol to prevent RNA degradation. Prior to RNA isolation, cell pellets were spiked with 10 μL internal hpd standard RNA [6.3 × 109 copies/μL]. RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). On-column DNAse digestion was performed using RNase-free DNase set from Qiagen. RNA concentration and quality were assessed photometrically (NanoDrop ND 1000) and analyzed using the Agilent Bioanalyzer 2100. In total, RNA concentrations ranged from 396 ng/μL to 710 ng/μL. Only RNA with 260/280 nm ratios of 1.8 to 2.0 and 260/230 nm ratios greater than 1.8 were used for reverse transcription. Reverse transcription (RT) of 1 μg total RNA was performed using QuantiTect Rev. Transcription Kit (Qiagen, Hilden, Germany) in a total volume of 20 μL according to manufacturer's protocol. cDNA was stored at -80°C until further use.
(III) Quantitative real-time PCR
Quantitative real-time PCR (qPCR) primers were designed using Roche's online Universal ProbeLibrary (UPL) Assay Design Center (https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp Roche Applied Science, Mannheim, Germany)). The specificity of primer pairs was checked using Basic Local Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov/); for primer sequences, hydrolysis probes and PCR efficiency using plasmid or cDNA as template see Table 2. Primers were purchased from Metabion, Martinsried, Germany (purification HPLC); hydrolysis probes were ordered from Roche Applied Science, Mannheim, Germany.
qPCR was performed in a LightCycler® 480 System using LightCycler® 480 multiwell plates 96 (white) and the standard run protocol UPL (software: LightCycler® 480 SW 1.5.0 - version 1.5.0.39). An absolute quantification (method: Abs Quant/2nd Derivative Max) was performed using plasmids pCAS1 (6718 bp, harboring hpd), pJF119-crtZ (5905 bp, harboring crtZ,) and pJF119-crtW148 (6038 bp, harboring crtW148) as standard. A dilution series of Hind III hydrolyzed plasmid DNA (109 - 104 copies/μL) in triplicate was used for standard curve generation. Copy number was calculated according to Equation 1:

with N: copies × μL-1; NA: copies/mol (Avogadro constant with 6.022 × 1023 × mol-1), Conc.hP: concentration hydrolyzed plasmid (g × μL-1); MW: molecular weight (g × mol-1) and MWhP: molecular weight hydrolyzed plasmid.
(IV) Calculation of internal standard recovery and absolute copy numbers
The recovery (r) of the internal RNA standard (hpd) was calculated by dividing the experimentally determined copy number (Nhpd,quant) of the internal standard by the copy number of the internal standard (Nhpd,applied) originally used for spiking the samples multiplied with dilution factor D (Equation 2):

where D is a dilution factor caused by the process of RNA isolation, RT, and quantification by qPCR (see Equation 3).

with VRNAα [L]: volume used for RNA isolation; VRNAω [L]): sample volume after RNA isolation; VRNA->cDNA [L]: RNA volume used for cDNA synthesis; VcDNA->qPCR [L]: cDNA volume used for qPCR reaction and VcDNA [L]: total volume of cDNA synthesis reaction. Absolute copy numbers were calculated using Equation 4:

with NmRNA, cell being the absolute copy number per cell and NmRNAα being the copy number of the mRNA of interest in total volume used for RNA isolation (50 μL).
NmRNAα is calculated by Equation 5:

with NmRNA,quant: copy number estimated by qPCR.
Results
Construction of a β-carotene-producing strain
Recently our group developed a method for the integration of heterologous expression cassettes into the chromosome of E. coli and the subsequent screening [24]. From this work resulted a plasmid-free strain (E. coli BW-CARO) carrying the four genes crtEBIY from P. ananatis in individual expression cassettes under the control of IPTG inducible tac-promoters. To enhance the expression level of the native 1-deoxyxylulose-5-phosphate synthase gene (dxs), a rate-limiting enzyme of the isoprenoid pathway [32], the native dxs promoter was exchanged by the strong bacteriophage promoter T5 [28]. First, a chloramphenicol resistance gene, flanked by FRT sites, was cloned upstream of the T5-promoter into pQE31. The resistance gene and promoter region of the plasmid gained (pQE31-FRT-cat-FRT) was PCR amplified and subsequently inserted upstream of the native dxs in E. coli BW-CARO using the λ-Red recombineering technique. After elimination of the resistance gene, the resulting strain E. coli BW-CARO-dxs was further modified by the insertion of an additional copy of the native isopentenyl diphosphate isomerase gene (idi), a further rate-limiting enzyme of the isoprenoid pathway [33]. The E. coli idi gene was cloned into the expression vector pJF119ΔN and to allow transient selection for chromosomal integration, the cloned gene was hooked up to a downstream chloramphenicol resistance cassette. The expression cassette of the resulting plasmid pJF119-FRT-cat-FRT, including resistance gene, was PCR amplified and transformed into E. coli BW-CARO-dxs harboring the λ-Red recombinase expression plasmid pKD46. The selected PCR primers led to the homologous recombination of the expression cassette into the galactose locus (galETKM). The engineered E. coli strains gained (E. coli BW-CARO, E. coli BW-CARO-dxs, and E. coli BW-CARO-dxs-idi) were cultivated in complex medium and the carotenoid content was analyzed. All strains produce β-carotene as the sole carotenoid (Figure 2A). As expected, the amount of β-carotene increased due to the enhanced expression of dxs and idi. The exchange of the native dxs promoter by the T5-promoter in E. coli BW-CARO increased the amount of β-carotene per cell by a factor of 2.6. After insertion of the additional idi copy into E. coli BW-CARO-dxs the amount of β-carotene per cell increased by the factor of 1.9 (β-carotene content in E. coli BW-CARO 1.24 ± 0.17 mg/g cellular dry weight (cdw); E. coli BW-CARO-dxs 3.254 ± 0.24 mg/g cdw; E. coli BW-CARO-dxs-idi 6.18 ± 0.78 mg/g cdw).

HPLC analysis of the carotenoids accumulated during cultivation of plasmid-free E. coli strains: A BW-CARO-dxs-idi in LB medium (48 h), B BW-ASTA in LB medium (48 h), C BW-CANT in LB medium (48 h), D BW-ASTA in LB medium plus L-rhamnose (48 h), E BW-ASTA in MM medium plus IPTG (24 h), F BW-ASTA in MM medium plus IPTG and L-rhamnose (48 h), G BW-ASTA in MM medium plus IPTG (48 h), H astaxanthin standard. Detection wavelength 470 nm. (1) β-carotene, (3) echinenone, (6) zeaxanthin, (7) canthaxanthin, (8) adonixanthin, (9) adonirubin, (10) astaxanthin, (10') cis-astaxanthin.
Chromosomal integration of crtW and crtZ
The reading frame of the β-carotene ketolase gene (crtW148) from N. punctiforme PCC73102 was amplified from plasmid pQE32-148 and cloned into the expression vector pJF119ΔN, resulting in plasmid pJF-crtW148. In transformants of β-carotene-producing E. coli BW-CARO-dxs-idi with pJF-crtW148 the di-keto carotenoid canthaxanthin represented 85% (0.896 ± 0.12 mg/g cdw) of the total carotenoid content with about 10% β-carotene and small amounts of echinenone (Figure 2C). The ratio of the three carotenoids did not vary during the 48 h cultivation. The addition of the inducer IPTG had neither influence on the carotenoid formation nor on growth (data not shown).
After cloning of a FRT-cat-FRT cassette into pJF-crtW148, chromosomal integration of the Ptac-crtW148 expression cassette into the melibiose locus (melAB) of E. coli BW-CARO-dxs-idi, and elimination of the cat resistance cassette, the engineered strain E. coli BW-CANT showed an about 20% higher formation of canthaxanthin (1.085 ± 0.15 mg/g cdw) but the percentage of canthaxanthin of all carotenoids was 85% as in the plasmid carrying strain E. coli BW-CARO-dxs-idi pJF-crtW148. The addition of IPTG up to 1 mM into LB-medium cultures of E. coli BW-CARO-dxs-idi pJF-crtW148 and E. coli BW-CANT, respectively, did not change the ratio of canthaxanthin, β-carotene, and echinenone as well as the total carotenoid yield, which we take as evidence of the leakiness of the tac-promoter in LB-medium.
In order to engineer an astaxanthin-producing strain, the β-carotene hydroxylase gene (crtZ) was inserted into the chromosome of E. coli BW-CANT. To allow a variable expression of crtZ compared to the tac-promoter controlled biosynthetic genes, crtZ was expressed under control of the rhamnose-promoter (PrhaBAD). For this purpose crtZ was cloned from plasmid pJF119-crtZ into pAW223. To verify the functional expression of this construct, pAW-crtZ was introduced into β-carotene-producing E. coli. Cultivation of E. coli BW-CARO-dxs-idi pAW-crtZ and subsequent carotenoid analysis showed that zeaxanthin was produced as the only carotenoid product (1.48 mg/g cdw).
Cloning of a FRT-cat-FRT cassette into pAW-crtZ and chromosomal integration of the PrhaBAD-crtZ expression cassette into the ribose locus (rbsDABCK) of E. coli BW-CANT and elimination of the resistance cassette resulted in the strain E. coli BW-ASTA, containing all required biosynthetic genes for the formation of astaxanthin as single expression units, respectively, on the chromosome. Cultivation of E. coli BW-ASTA in LB-medium showed that astaxanthin is the predominant carotenoid product (71%). Besides astaxanthin, zeaxanthin (25%) was the only other detectable carotenoid (Figure 2B, Figure 3). The addition of the inducer L-rhamnose to E. coli BW-ASTA cultures led to an increase of zeaxanthin (69%) compared to astaxanthin (20%) (Figure 2D, Figure 3). The enhanced expression of crtZ by L-rhamnose induction therefore led to a significant increase of zeaxanthin, obviously due to a low activity of CrtW148 for the substrate zeaxanthin. The induction of E. coli BW-ASTA LB medium cultures with IPTG in the presence of 0.2% L-rhamnose or without had neither effect on the amount of carotenoid nor on the ratio of astaxanthin and zeaxanthin.

Production of zeaxanthin (A) and astaxanthin (B) by E. coli BW-ASTA (in mg per liter of culture (rhomboid)) and in mg per gram cellular dry weight (cdw; (square)) after cultivation for 48 h under different conditions. Values represent the mean (n = 4) ± standard deviation. * indicates significant changes between un-induced (LB or MM) and induced state (LB Rha, LB IPTG or MM IPTG, MM Rha IPTG, respectively). p ≤ 0.05, unpaired t-test assuming unequal variances.
Controlled gene expression in E. coli BW-ASTA using glucose-containing medium
In order to avoid the formation of zeaxanthin by E. coli BW-ASTA, an increase of the ketolase activity or a decrease of the hydroxylase activity would be necessary. To better control the expression by the tac- and, in particular, by the rha-promoter that are both not tightly controlled in LB-medium (see Figure 4), minimal medium with glucose as C- and energy source was used. The cultivation of E. coli BW-ASTA in minimal medium without the addition of inducer molecules resulted in the formation of only small amounts (0.11 mg/g cdw) of astaxanthin, which we interpret as the result of the higher repression of the tac-promoter in minimal medium in contrast to LB-medium. On the other hand, the addition of IPTG to the cultures, which induces the Ptac controlled gene expression of dxs, idi, crtE, crtB, crtI, crtY, and crtW, resulted after 48 h in a 12-fold increase in the formation of carotenoids. After 24 h of incubation in minimal medium, E. coli BW-ASTA synthesized 0.96 ± 0.14 mg/g cdw astaxanthin and the by-products adonirubin (13%), canthaxanthin (12%), and β-carotene (1%) (Figure 2E). In contrast to the cultivation in LB medium no zeaxanthin formation was observed. After an incubation time of 48 h astaxanthin was produced as exclusive carotenoid (>95%) in a concentration of (2.07 ± 0.15 mg/l; 1.41 ± 0.11 mg/g cdw) (Figure 3). This result shows that during the cultivation of E. coli BW-ASTA in minimal medium with glucose and IPTG the β-carotene produced is predominantly converted by the ketolase (CrtW148) into cantaxanthin, which is subsequently slowly hydroxylated by CrtZ. The concurrent addition of both inducers, IPTG and L-rhamnose, also led after 24 h to the formation of astaxanthin (68%), adonirubin (14%), cantaxanthin (12%), and traces of β-carotene (1%). In the late stationary phase (48 h) of the culture astaxanthin was still the predominant carotenoid with >90%, however, the cells also contained about 5% of zeaxanthin as a by-product (Figure 2F).

Transcript copy numbers/cell quantified by absolute RT-qPCR. Copy numbers ± STABW of crtW148 (grey) and crtZ (black) from E. coli BW-ASTA cultivated in LB or minimal medium (MM) and harvested in late exponential phase are shown. Rha: Induction with L-rhamnose [12 mM]. IPTG: Induction with IPTG [0.5 mM]. * indicates significant transcript number changes between un-induced (LB or MM) and induced state (LB Rha, LB Rha IPTG or MM IPTG, MM Rha IPTG, respectively). p ≤ 0.05, n = 4, unpaired t-test assuming unequal variances.
Transcriptional analysis of crtW148 and crtZ
In order to ascertain whether the assumed expression levels of CrtW148 and CrtZ based on carotenoid formation by E. coli BW-ASTA were in accordance with the mRNA levels of crtW148 and crtZ, we performed absolute RT-qPCR studies. A method previously described by Schuhmacher et al. [31] that uses an external standard for absolute transcript number quantification, was adapted to the Roche UPL system allowing for absolute transcript number determination per cell.
From the cultivation of E. coli BW-ASTA in LB medium and in minimal medium, with or without induction of the tac- and rha-promoter, respectively, cells were withdrawn in the late exponential phase and were analyzed for the crtZ and crtW148 transcript level by RT-qPCR. The number of transcripts of crtW148 and crtZ under the different conditions are shown in Figure 4. In LB medium, comparable transcript numbers per cell could be detected for crtW148 with or without IPTG induction (between 22 ± 4 and 25 ± 1 copies/cell). A 15% higher copy number was detected for crtZ (41 ± 3 copies/cell compared to 48 ± 5 to 53 ± 3 copies/cell) after induction with L-rhamnose. The transcript level of crtZ reflects the formation of zeaxanthin when cultivated in complex medium. An increased expression of crtZ correlated with an increased formation of zeaxanthin by E. coli BW-ASTA. Under all cultivation conditions in LB medium, zeaxanthin and astaxanthin had been detected. By using glucose-containing minimal medium the transcription of the heterologous genes was more tightly regulated than in LB medium. Transcript numbers per cell of crtW148 without the addition of IPTG were 12 ± 1. For crtZ in average, less than one copy number per cell was detected in minimal medium without induction. This reflects the tight regulation of the rhamnose-promoter under these conditions. These results are in good accordance with the product formation; in minimal medium without induction, only small amounts of astaxanthin as the sole carotenoid were detected (Figure 3). The induction by IPTG led to a 2.3 to 2.6 fold increase in mRNA level of crtW148, and resulted in a 20-fold increase of the carotenoid concentration. Here, astaxanthin was the only carotenoid that was accumulated by E. coli BW-ASTA after 48 h (Figure 2). This shows that the low mRNA level of crtZ yielded in a sufficient amount of hydroxylase activity converting all the produced precursors into astaxanthin. The addition of L-rhamnose led to a significant increase in transcript levels of crtZ (8.6 fold). In these cases, both astaxanthin (90%) and zeaxanthin (5%) had been detected as products after 48 h.
Discussion
The in vivo biosynthesis of a complex natural product in a heterologous host like E. coli first requires the introduction and the functional expression of biosynthetic genes that enable the conversion of available cell intermediates towards the desired product. The heterologous biosynthesis of astaxanthin by E. coli has been achieved in numerous studies by the expression of respective carotenoid biosynthetic genes using recombinant plasmids [20, 23, 33–36]. In this study, we constructed a plasmid-free E. coli strain that carries each of the astaxanthin biosynthetic genes (crtE,B,I,Y,Z,W) as individual expression units on the chromosome. For the integration of the expression cassettes we used a method, recently developed in our lab, that allows fast and reliable integration and screening [24]. It is based on the λ-Red mediated recombination technique developed for the directed knock-out [29]. This method utilizes the replacement of E. coli s' rare sugar degradation genes which are dispensable for most biotechnological applications. The replacement of these genes can easily be visualized by the use of MacConkey differential agar medium carrying the corresponding sugar compound.
The chromosomal insertion of heterologous biosynthetic genes has for obvious reasons some advantages compared to the use of heterologous plasmids. Thus, plasmids may be the best choice for the cloning and short-term expression of recombinant genes, in particular for the maximum overproduction of a given protein. Especially in metabolic engineering applications, however, a too strong gene expression may be unfavorable for long-term productivity [37]. Yoon et al. [38] observed that a high expression of lycopene biosynthetic genes in E. coli leads to a decrease in growth and lycopene production. Therefore, the chromosomal integration of heterologous expression cassettes can be favorably compared to multi-copy plasmids in terms of metabolic burden effects, structural instability, and most important segregational instability [39]. Furthermore, a stable chromosomal insertion obviates the use of selection-markers (e.g. antibiotics) that are commonly used for the maintenance of plasmids during cultivation. Especially antibiotics are both costly and can hamper the product purification in food and pharmaceutical production processes. On the other hand, a low enzyme activity of a heterologous downstream pathway can result in a reduced product yield or in an accumulation of pathway intermediates [40]. Thus, the in vivo biosynthesis of carotenoids in E. coli requires an appropriate heterologous gene expression, adapted to the supply of isoprenoid precursors to avoid the effect of metabolic burden and to avoid the accumulation of pathway intermediates. The increased biosynthesis of β-carotene (up to 6.2 mg/g cdw) by the enhanced expression of idi and dxs in E. coli BW-CARO demonstrates that the expression of the heterologous biosynthetic genes and accordingly the enzyme activity of the corresponding proteins in this strain do not limit the formation of β-carotene. Similar observations were made by Chiang et al. [41] for a lycopene-producing strain that carries a single copy of a lycopene biosynthetic gene cluster on the chromosome. Surprisingly, the additional expression of crtW148 and crtZ in the β-carotene-producing strain reduced the overall formation of carotenoids about three times compared to E. coli BW-CARO-dxs-idi. This leads to the suggestion that the recombinant proteins (CrtW148 or CrtZ) or a product of their enzymatic reaction effect the formation of the carotenoid precursors upstream of phytoene, because no other carotenoid accumulated in the cell. This stands in contrast to the study by Scaife et al. [36] who found that expression of a β-carotene-ketolase and -hydroxylase within a β-carotene-producing strain significantly increases the total carotenoid yield. The maximum astaxanthin yield of almost 2 mg/g in this study and ours is in the same range and represents the highest astaxanthin content in E. coli that has been achieved so far.
Besides the chromosomal integration, another aim of our work was to find conditions by which E. coli synthesizes astaxanthin as the sole carotenoid. For the conversion of β-carotene to astaxanthin, the β-carotene ketolase gene from Nostoc punctiforme (crtW148) and the β-carotene hydroxylase gene from Pantoea ananatis (crtZ) were chosen, because the corresponding proteins (CrtW148, CrtZ; see Figure 1) are known to be bifunctional and therefore accept β-carotene as well as hydroxylated or ketolated products, respectively, as substrate [12, 20]. In order to vary the expression level of crtW148 and crtZ, the hydroxylase gene was expressed under control of the rha-promoter in contrast to the other heterologous genes that were controlled by a tac-promoter, respectively. The cultivation of E. coli BW-ASTA in LB medium showed the formation of astaxanthin as the predominant product but also a significant amount of zeaxanthin that increased upon the additional induction of the rha-promoter. In contrast, the additional IPTG induction of the crtW148 controlling tac-promoter had no significant influence on the product formation. The carotenoid formation is in concordance with the results of mRNA quantification. The qPCR measurement showed that both, tac-promoter (crtW148) and rha-promoter (crtZ), are only marginally repressed under the given conditions (Figure 4).
It is supposed that hydroxylase and ketolase compete for their substrate and that only a balanced expression of these two enzymes might lead to a complete conversion of β-carotene to astaxanthin [12, 42–44]. This hypothesis is supported by our results. We suppose that in the astaxanthin biosynthesis by E. coli BW-ASTA the hydroxylation reaction occurs fast with β-carotene as well as with the ketolated intermediates as substrates. No intermediates were detected under these conditions which are mostly ketolated (Figure 2). The CrtW148 ketolase does utilize β-carotene and to a minor extent hydroxylated intermediates. But during the course of cultivation the ketolase was not able to convert the accumulated zeaxanthin into adonixanthin or astaxanthin, completely. Although the bifunctionality of CrtW148 from N. punctiforme was proven [20], the conversion of zeaxanthin to astaxanthin by CrtW148 is obviously the most limiting step towards the efficient biosynthesis of astaxanthin in our system. To improve the conversion of zeaxanthin to astaxanthin, protein engineering of a CrtW-type ketolase had been used successfully, but a complete transformation to astaxanthin was not achieved [22, 23]. In contrast to this research, our working hypothesis was to avoid the accumulation of zeaxanthin by increasing the activity ratio of CrtW148 to CrtZ not by enhanced crtW148 expression or enzyme activity/specificity but instead by lowering the crtZ expression level. This was achieved by the use of D-glucose containing minimal medium that led to a better balanced regulation of both heterologous promoters. Especially the rhamnose-promoter, that regulates the expression of crtZ, is more tightly controlled by the catabolite repression [45, 46]. Cultivation of E. coli BW-ASTA under this condition with induction of the IPTG-controlled heterologous promoters led in the early phase of the cultivation to the formation of astaxanthin and the intermediates adonirubin and canthaxanthin (Figure 2) that vanished, presumably due to the transformation into astaxanthin during the course of cultivation. We take this as evidence that the adjustment of the expression level can direct the pathway towards the desired product, astaxanthin. The low expression of crtZ and the, in contrast, high expression of crtW148 (Figure 4) make it obvious that the synthesized β-carotene is due to the kinetics of the reactions, preferentially converted into canthaxanthin, which is secondly completely transformed by the slower hydroxylation reaction via adonirubin into astaxanthin. Similar observations concerning the competition between hydroxylase and ketolase have been made for transgenic maize where plants harboring only an endogenous hydroxylase but an exogenous ketolase gene under an endospecific promoter accumulated astaxanthin. Astaxanthin, however, was not accumulated by plants harboring both exogenous hydroxylase and ketolase genes [44]. The authors indicated that the avoidance of the adonixanthin accumulation is crucial for astaxanthin production in transgenic maize endosperm [44]. In contrast, we find the avoidance of zeaxanthin accumulation to be the crucial step for sole astaxanthin synthesis in the bacterium E. coli BW-ASTA.
Conclusions
In this study, we engineered a plasmid-free E. coli strain that carries biosynthetic genes for the in vivo biosynthesis of astaxanthin. The stable chromosomal insertion of the heterologous genes enables dispensing with selection makers that are required for the maintenance of recombinant plasmid. The biosynthetic genes were each integrated as single expression units into the chromosome of E. coli. This approach allows the control of individual gene expression levels, which is complicated to achieve if the astaxanthin biosynthetic genes are organized within one operon. This E. coli engineering strategy might also be useful as a general approach for the construction of stable production strains for the heterologous biosynthesis of natural products for which excessive protein overexpression is a hindrance.
The adjustment of the crtZ expression level in the astaxanthin-producing strain was applied successfully for the complete in vivo conversion of β-carotene into astaxanthin by recombinant E. coli cells.
References
Tatsuzawa H, Maruyama T, Misawa N, Fujimori K, Nakano M: Quenching of singlet oxygen by carotenoids produced in Escherichia coli - attenuation of singlet oxygen-mediated bacterial killing by carotenoids. FEBS Lett. 2000, 484: 280-284. 10.1016/S0014-5793(00)02149-9.
Guerin M, Huntley ME, Olaizola M: Haematococcus astaxanthin: applications for human health and nutrition. Trends Biotechnol. 2003, 21: 210-216. 10.1016/S0167-7799(03)00078-7.
Lauver DA, Driscoll EM, Lucchesi BR: Disodium disuccinate astaxanthin prevents carotid artery rethrombosis and ex vivo platelet activation. Pharmacology. 2008, 82: 67-73. 10.1159/000132085.
Chen H-M, Meyers SP: Ensilage Treatment of Crawfish Waste for Improvement of Astaxanthin Pigment Extraction. Journal of Food Science. 1983, 48: 1516-1520. 10.1111/j.1365-2621.1983.tb03528.x. 10.1111/j.1365-2621.1983.tb03528.x.
Rodriguez-Saiz M, de la Fuente JL, Barredo JL: Xanthophyllomyces dendrorhous for the industrial production of astaxanthin. Appl Microbiol Biotechnol. 2010, 88: 645-658. 10.1007/s00253-010-2814-x.
Alper H, Miyaoku K, Stephanopoulos G: Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005, 23: 612-616. 10.1038/nbt1083.
Farmer WR, Liao JC: Precursor balancing for metabolic engineering of lycopene production in Escherichia coli. Biotechnol Prog. 2001, 17: 57-61. 10.1021/bp000137t.
Ignea C, Cvetkovic I, Loupassaki S, Kefalas P, Johnson CB, Kampranis SC, Makris AM: Improving yeast strains using recyclable integration cassettes, for the production of plant terpenoids. Microb Cell Fact. 2011, 10: 4- 10.1186/1475-2859-10-4.
Jin YS, Stephanopoulos G: Multi-dimensional gene target search for improving lycopene biosynthesis in Escherichia coli. Metab Eng. 2007, 9: 337-347. 10.1016/j.ymben.2007.03.003.
Lee PC, Mijts BN, Schmidt-Dannert C: Investigation of factors influencing production of the monocyclic carotenoid torulene in metabolically engineered Escherichia coli. Appl Microbiol Biotechnol. 2004, 65: 538-546.
Maury J, Asadollahi MA, Moller K, Clark A, Nielsen J: Microbial isoprenoid production: an example of green chemistry through metabolic engineering. Adv Biochem Eng Biotechnol. 2005, 100: 19-51.
Fraser PD, Miura Y, Misawa N: In vitro characterization of astaxanthin biosynthetic enzymes. J Biol Chem. 1997, 272: 6128-6135. 10.1074/jbc.272.10.6128.
Hasunuma T, Miyazawa S, Yoshimura S, Shinzaki Y, Tomizawa K, Shindo K, Choi SK, Misawa N, Miyake C: Biosynthesis of astaxanthin in tobacco leaves by transplastomic engineering. Plant J. 2008, 55: 857-868. 10.1111/j.1365-313X.2008.03559.x.
Misawa N, Kajiwara S, Kondo K, Yokoyama A, Satomi Y, Saito T, Miki W, Ohtani T: Canthaxanthin biosynthesis by the conversion of methylene to keto groups in a hydrocarbon beta-carotene by a single gene. Biochem Biophys Res Commun. 1995, 209: 867-876. 10.1006/bbrc.1995.1579.
Tao L, Cheng Q: Novel beta-carotene ketolases from non-photosynthetic bacteria for canthaxanthin synthesis. Mol Genet Genomics. 2004, 272: 530-537. 10.1007/s00438-004-1083-8.
Blasco F, Kauffmann I, Schmid RD: CYP175A1 from Thermus thermophilus HB27, the first beta-carotene hydroxylase of the P450 superfamily. Appl Microbiol Biotechnol. 2004, 64: 671-674. 10.1007/s00253-003-1529-7.
Masamoto K, Misawa N, Kaneko T, Kikuno R, Toh H: Beta-carotene hydroxylase gene from the cyanobacterium Synechocystis sp. PCC6803. Plant Cell Physiol. 1998, 39: 560-564.
Misawa N, Nakagawa M, Kobayashi K, Yamano S, Izawa Y, Nakamura K, Harashima K: Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J Bacteriol. 1990, 172: 6704-6712.
Alvarez V, Rodriguez-Saiz M, de la Fuente JL, Gudina EJ, Godio RP, Martin JF, Barredo JL: The crtS gene of Xanthophyllomyces dendrorhous encodes a novel cytochrome-P450 hydroxylase involved in the conversion of beta-carotene into astaxanthin and other xanthophylls. Fungal Genet Biol. 2006, 43: 261-272. 10.1016/j.fgb.2005.12.004.
Steiger S, Sandmann G: Cloning of two carotenoid ketolase genes from Nostoc punctiforme for the heterologous production of canthaxanthin and astaxanthin. Biotechnol Lett. 2004, 26: 813-817.
Makino T, Harada H, Ikenaga H, Matsuda S, Takaichi S, Shindo K, Sandmann G, Ogata T, Misawa N: Characterization of cyanobacterial carotenoid ketolase CrtW and hydroxylase CrtR by complementation analysis in Escherichia coli. Plant Cell Physiol. 2008, 49: 1867-1878. 10.1093/pcp/pcn169.
Ye RW, Stead KJ, Yao H, He H: Mutational and functional analysis of the beta-carotene ketolase involved in the production of canthaxanthin and astaxanthin. Appl Environ Microbiol. 2006, 72: 5829-5837. 10.1128/AEM.00918-06.
Tao L, Wilczek J, Odom JM, Cheng Q: Engineering a beta-carotene ketolase for astaxanthin production. Metab Eng. 2006, 8: 523-531. 10.1016/j.ymben.2006.06.001.
Albermann C, Trachtmann N, Sprenger GA: A simple and reliable method to conduct and monitor expression cassette integration into the Escherichia coli chromosome. Biotechnol J. 2010, 5: 32-38. 10.1002/biot.200900193.
Pan JG, Rhee JS, Lebeault JM: Physiological constraints in increasing biomass concentration of Escherichia coli B in fed-batch culture. Biotechnology Letters. 1987, 9: 89-94. 10.1007/BF01032744. 10.1007/BF01032744.
Albermann C, Ghanegaonkar S, Lemuth K, Vallon T, Reuss M, Armbruster W, Sprenger GA: Biosynthesis of the vitamin E compound delta-tocotrienol in recombinant Escherichia coli cells. Chembiochem. 2008, 9: 2524-2533. 10.1002/cbic.200800242.
Vallon T, Ghanegaonkar S, Vielhauer O, Muller A, Albermann C, Sprenger G, Reuss M, Lemuth K: Quantitative analysis of isoprenoid diphosphate intermediates in recombinant and wild-type Escherichia coli strains. Appl Microbiol Biotechnol. 2008, 81: 175-182. 10.1007/s00253-008-1707-8.
Yuan LZ, Rouviere PE, Larossa RA, Suh W: Chromosomal promoter replacement of the isoprenoid pathway for enhancing carotenoid production in E. coli. Metab Eng. 2006, 8: 79-90. 10.1016/j.ymben.2005.08.005.
Datsenko KA, Wanner BL: One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000, 97: 6640-6645. 10.1073/pnas.120163297.
Cherepanov PP, Wackernagel W: Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995, 158: 9-14. 10.1016/0378-1119(95)00193-A.
Schuhmacher T, Lemuth K, Hardiman T, Vacun G, Reuss M, Siemann-Herzberg M: Quantifying cytosolic messenger RNA concentrations in Escherichia coli using real-time polymerase chain reaction for a systems biology approach. Anal Biochem. 2010, 398: 212-217. 10.1016/j.ab.2009.11.025.
Harker M, Bramley PM: Expression of prokaryotic 1-deoxy-D-xylulose-5-phosphatases in Escherichia coli increases carotenoid and ubiquinone biosynthesis. FEBS Lett. 1999, 448: 115-119. 10.1016/S0014-5793(99)00360-9.
Wang CW, Oh MK, Liao JC: Engineered isoprenoid pathway enhances astaxanthin production in Escherichia coli. Biotechnol Bioeng. 1999, 62: 235-241. 10.1002/(SICI)1097-0290(19990120)62:2<235::AID-BIT14>3.0.CO;2-U.
Choi SK, Harada H, Matsuda S, Misawa N: Characterization of two beta-carotene ketolases, CrtO and CrtW, by complementation analysis in Escherichia coli. Appl Microbiol Biotechnol. 2007, 75: 1335-1341. 10.1007/s00253-007-0967-z.
Kajiwara S, Kakizono T, Saito T, Kondo K, Ohtani T, Nishio N, Nagai S, Misawa N: Isolation and functional identification of a novel cDNA for astaxanthin biosynthesis from Haematococcus pluvialis, and astaxanthin synthesis in Escherichia coli. Plant Mol Biol. 1995, 29: 343-352. 10.1007/BF00043657.
Scaife MA, Burja AM, Wright PC: Characterization of cyanobacterial beta-carotene ketolase and hydroxylase genes in Escherichia coli, and their application for astaxanthin biosynthesis. Biotechnol Bioeng. 2009, 103: 944-955. 10.1002/bit.22330.
Jones KL, Kim SW, Keasling JD: Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab Eng. 2000, 2: 328-338. 10.1006/mben.2000.0161.
Yoon SH, Lee YM, Kim JE, Lee SH, Lee JH, Kim JY, Jung KH, Shin YC, Keasling JD, Kim SW: Enhanced lycopene production in Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate. Biotechnol Bioeng. 2006, 94: 1025-1032. 10.1002/bit.20912.
Friehs K: Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004, 86: 47-82.
Leonard E, Ajikumar PK, Thayer K, Xiao WH, Mo JD, Tidor B, Stephanopoulos G, Prather KL: Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc Natl Acad Sci USA. 2010, 107: 13654-13659. 10.1073/pnas.1006138107.
Chiang CJ, Chen PT, Chao YP: Replicon-free and markerless methods for genomic insertion of DNAs in phage attachment sites and controlled expression of chromosomal genes in Escherichia coli. Biotechnol Bioeng. 2008, 101: 985-995. 10.1002/bit.21976.
Fraser PD, Shimada H, Misawa N: Enzymic confirmation of reactions involved in routes to astaxanthin formation, elucidated using a direct substrate in vitro assay. Eur J Biochem. 1998, 252: 229-236. 10.1046/j.1432-1327.1998.2520229.x.
Misawa N, Yamano S, Linden H, de Felipe MR, Lucas M, Ikenaga H, Sandmann G: Functional expression of the Erwinia uredovora carotenoid biosynthesis gene crtl in transgenic plants showing an increase of beta-carotene biosynthesis activity and resistance to the bleaching herbicide norflurazon. Plant J. 1993, 4: 833-840. 10.1046/j.1365-313X.1993.04050833.x.
Zhu C, Naqvi S, Breitenbach J, Sandmann G, Christou P, Capell T: Combinatorial genetic transformation generates a library of metabolic phenotypes for the carotenoid pathway in maize. Proc Natl Acad Sci USA. 2008, 105: 18232-18237. 10.1073/pnas.0809737105.
Holcroft CC, Egan SM: Interdependence of activation at rhaSR by cyclic AMP receptor protein, the RNA polymerase alpha subunit C-terminal domain, and rhaR. J Bacteriol. 2000, 182: 6774-6782. 10.1128/JB.182.23.6774-6782.2000.
Tobin JF, Schleif RF: Positive regulation of the Escherichia coli L-rhamnose operon is mediated by the products of tandemly repeated regulatory genes. J Mol Biol. 1987, 196: 789-799. 10.1016/0022-2836(87)90405-0.
Acknowledgements
The authors are grateful to Prof. Gerhard Sandmann (Universität Frankfurt) for providing us with plasmid pPQE32-148, to Holger Beuttler (Institute of Technical Biochemistry, Universität Stuttgart) for providing carotenoid standards, to Cristina Prada for her help with the construction of E. coli BW-CARO-dxs-idi, and particularly to Prof. Georg Sprenger for helpful discussions, continuous support, and critical reading of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 706/TP B3).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CA initiated and coordinated the project. KL and KS performed the experiments. KL was responsible for RT-qPCR. KL and CA wrote the paper. All authors approved the final version of the manuscript.
Karin Lemuth, Kristin Steuer contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lemuth, K., Steuer, K. & Albermann, C. Engineering of a plasmid-free Escherichia coli strain for improved in vivo biosynthesis of astaxanthin. Microb Cell Fact 10, 29 (2011). https://doi.org/10.1186/1475-2859-10-29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-10-29