
Abstract
Background
Transmissible spongiform encephalopathies (TSEs), a group of neurodegenerative diseases, are thought to be caused by an abnormal isoform of a naturally occurring protein known as cellular prion protein, PrPC. The abnormal form of prion protein, PrPSc accumulates in the brain of affected individuals. Both isoforms are encoded by the same prion protein gene (PRNP), and the structural changes occur post-translationally. Certain mutations in the PRNP gene result in genetic TSEs or increased susceptibility to TSEs.
Case presentation
A 70 year old woman was admitted to the hospital with severe confusion and inability to walk. Relatives recognized memory loss, gait and behavioral disturbances over a six month period prior to hospitalization. Neurological examination revealed Creutzfeldt-Jakob disease (CJD) related symptoms such as incontinence, Babinski sign and myoclonus. EEG showed periodic sharp waves typical of sporadic CJD and cerebrospinal fluid analysis (CSF) was positive for the presence of the 14-3-3-protein. As the disease progressed the patient developed akinetic mutism and died in the tenth month after onset of the disease symptoms. Unfortunately, no autopsy material was available. PRNP sequencing showed the occurrence of a point mutation on one allele at codon 193, which is altered from ACC, coding for a threonine, to ATC, encoding an isoleucine (T193I).
Conclusion
Here we report a novel mutation of the PRNP gene found in an elderly female patient resulting in heterozygosity for isoleucine and threonine at codon 193, in which normally homozygosity for threonine is expected (T193). The patient presented typical clinical symptoms of CJD. EEG findings and the presence of the 14-3-3 protein in the CSF, contributed to CJD diagnosis, allowing the classification of this case as a probable CJD according to the World Health Organization (WHO) accepted criteria.
Similar content being viewed by others

Background
Transmissible spongiform encephalopathies (TSEs) are a group of neurodegenerative diseases of the central nervous system. These diseases share common features and can be found in both humans and animals. The infectious agent of TSEs is believed to be an abnormal isoform of a naturally occurring protein known as cellular prion protein, PrPC [1]. The abnormal prion protein isoform (PrPSc) accumulates in the brains of affected individuals. Both isoforms are encoded by the same gene PRNP, and the structural changes occur post-translationally. The lack of the endogenous PrP protein prevents the development of TSEs, as has been demonstrated in transgenic mice lacking the PRNP gene [2]. On the other hand, mutations that exist in the PRNP gene might facilitate the development of TSEs [3–5]. The most common TSE in humans is Creutzfeldt-Jakob disease (CJD), accounting for 85% of all TSE cases in humans [6]. There are three types of CJD, sporadic CJD (sCJD), infectious CJD (iCJD) and genetic CJD (gCJD). sCJD arises spontaneously, with unknown aetiology and affects mainly the elderly population at a frequency of one per million per year. iCJD is caused by the consumption of prion contaminated food or by medical treatments with prion contaminated biological materials and surgical instruments [7]. In gCJD, mutations in the PRNP gene predispose to disease [7, 8] by causing the expression of PrP protein with modified primary structure.
Several mutations have been identified in the prion protein, encoded by the PRNP gene. More than thirty have been associated or directly linked to TSEs [9–11]. Here we report a novel mutation in codon 193 (T193I) of the PRNP gene, in a patient with clinical features and EEG findings typical of CJD. Normally, codon 193, comprising nucleotides ACC, codes for a threonine, whereas in the case reported here it has been changed to ATC, thus encoding an isoleucine.
Case presentation
Patient
The patient was a 70 year old woman, with a history of diabetes mellitus, who was admitted to the hospital with confusion and inability to walk. Six months prior to admission, her relatives had started recognizing neurological symptoms, such as gait disturbance, memory loss and behavioral disturbances (irritability and aggression). Over the six month period that preceded the admission to the hospital, a rapid deterioration of her cognitive function was observed. She also displayed visual hallucinations and delusions requiring antipsychotic treatment.
On admission, the patient was bedridden, incontinent, severely confused with generalized hyperreflexia, bilateral Babinski sign, startle responses elicited by mild external stimuli, and multifocal generalized myoclonus which evolved to rhythmic. Two months later she developed akinetic mutism. The patient died three months after hospitalization.
Routine hematological and biochemical examinations produced normal results. CSF examination (protein, glucose and cell count) was normal, while EEG showed generalized findings, periodic triphasic sharp waves, recurring at intervals of 0.5–1 sec (Fig 1). Magnetic resonance imaging of the brain showed no significant abnormalities, except for a minor brain atrophy which could be related to the patient's age.

Electroencephalogram of the suspected CJD patient showing typical periodic bursts of triphasic sharp waves.
There was a lack of family history concerning neurological disorders. Although the clinical course followed the typical rapid progression of CJD, the relatives did not consent to having a neuropathological examination carried out, neither did they agree to further genetic investigation of the immediate family members, and thus it has not been possible to determine if the mutation has been passed to the patient's offspring.
14-3-3 protein detection in the CSF
Cerebrospinal fluid (CSF) was collected by lumbar puncture. Both the lumbar puncture and the EEG, were performed three and eight days after the patient's admission to the hospital (about six months after the occurrence of the reported symptoms observed by family members).
The proteins contained in 30 μl of CSF were separated on a 12% polyacrylamide gel and then transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was stained as described [12] with a polyclonal anti-14-3-3β antibody (Santa Cruz, sc-629) and an alkaline phosphatase conjugated secondary antibody (Pierce, 31340). CDP-Phototope Star (New England Biolabs, N701) was used to develop the membrane.
Detection of 14-3-3 protein in CSF, is considered to be a sensitive and specific marker for sporadic CJD. CSF tested positive twice for the presence of 14-3-3 protein (Fig. 2), supporting a probable CJD diagnosis [13, 14].

Immunoblot detection of 14-3-3 in the CSF. The patient was tested positive for the presence of 14-3-3 in the CSF. Lane 1: healthy control, lane 2: patient sample, lane 3: positive control (CSF) and lane 4: positive control (brain tissue homogenate)
DNA purification and sequencing
Genomic DNA was isolated from whole blood using Promega's Wizard genomic DNA purification kit according to the manufacturer's instructions. The proper quality and quantity of the genomic DNA for polymerase chain reaction (PCR) amplification was confirmed by spectrophotometry. The PRNP open reading frame was amplified by PCR with the following primers designed as cloning primers; HumoS: 5'- GCT CTA GAG CAT GGC GAA CCT T and HumoAS: 5'- GCT CTA GA T CAT CCC ACG ATC AGG AA. The bases of the primers corresponding to the open reading frame of PRNP are shown in bold. Amplification was confirmed by agarose gel electrophoresis. Sequencing was performed first at MWG-Biotech (Ebersberg, Germany) and then the sequencing results were confirmed by two other independent laboratories (Department of Neurology, University of Crete, MRC Prion Unit and Department of Neurodegenerative Diseases, University College, London).
PRNP sequence analysis revealed the existence of a causative mutation at codon 193. Upon comparison with the 762 bp sequence of the human PrP cDNA record (Genebank accession number M13899), it was found that the sequenced PRNP gene exhibits a point mutation at codon 193, comprising nucleotides 577–579. The point mutation occurs as a C to T transition at position 578 which causes the switch of codon 193 from ACC, encoding threonine, to ATC, encoding isoleucine. As shown on the electropherogram (Fig. 3), the patient is heterozygous for threonine and isoleucine at codon 193. The patient was found to be homozygous for methionine at codon 129 (M129).

Electropherogram of the DNA sequencing showing the position 193 were the mutation was detected. Codon ACC codes for a Threonine and codon ATC for an Isoleucine. The arrow indicates the position on the electropherogram, where both C and T are present.
Conclusion and discussion
The genotype of the host seems to play an important role in prion disease pathogenesis. Susceptibility to prion diseases is enhanced if the individual possesses certain genotypes. The human genetic TSEs account for 5–15% of human TSE cases and have been linked to specific mutations of the PRNP gene [9, 10, 15]. Susceptibility to sCJD has been associated with homozygosity for methionine at the polymorphic codon 129 [16, 17]. In the Greek population 50% homozygosity for M129 has been reported [18].
Here we report the case of a 70 year old female patient carrying the T193I mutation, who presented typical clinical symptoms of CJD. The presence of a mutation in the PRNP gene along with the presentation of symptoms typical for CJD supports the diagnosis of probable CJD according to the WHO accepted criteria [13]. The observed mutation was associated with homozygosity for methionine at codon 129. The patient's age at onset of the symptoms and the homozygosity M129 is consistent with the average age at onset observed in other gCJD cases [9].
The PrP protein primary structure is well conserved over a variety of mammals [19]. Interestingly, T193 of the human PrP protein can be found in the homologue proteins at the same position in all thirty six species listed in the cited publication, except in shrew, in which it is replaced by serine. Threonine and serine, are both hydrophilic polar amino acids with uncharged side chains, while isoleucine is a hydrophobic amino acid. The T193I mutation reported here, may affect the three dimensional structure of the protein. Since TSEs are linked to conformational changes of the prion protein, this mutation may be associated with the occurrence of the disease.
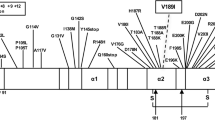
The mutation reported here results in an amino acid change within α-helix 2 (H2) of the PrP protein, which comprises amino acids 173–194 [20]. H2 is characterized by the occurrence of different causative mutations, most of which are linked to the appearance of a CJD phenotype [10].
Of particular interest is the T188A mutation, which is the most closely located mutation to codon 193 on H2 that has been reported [21]. Similarly to T193I, in the T188A case, threonine is replaced by a hydrophobic residue and both cases emerged with homozygosity for methionine at codon 129. Both patients were women of similar age i.e. 70 and 69 years old, respectively, and the brain MRI scan revealed no typical CJD findings, but a minor brain atrophy. EEG and the CSF analysis for the 14-3-3 protein were typical for sCJD in both cases. Clinical symptoms like the failure of cognitive abilities, myoclonus and visual disturbances were observed in both patients. Additionally, both patients share the absence of family history for dementia, which is probably caused by the reduced penetrance of the mutation [9, 22].
Codon 196 is very closely located to codon 193 although it does not belong to H2. The E196K mutation associated with M129 homozygosity [22] affected a 69 year old woman, who in contrast to the patient reported here, had a family history of dementia. The onset symptoms in both patients included behavioral abnormalities, albeit of a different type (the patient carrying the E196K mutation displayed emotional lability, inappropriate giggling, anorexia versus irritability and aggression). Gait disturbances were observed in both patients and both became bedridden as the disease progressed. Furthermore, both patients tested positive for the presence of 14-3-3 protein in the CSF. However, the EEG of the patient carrying the E196K mutation showed no periodic activity and there was a slight difference in the period from the onset of symptoms to death (10 versus 12 months).
During the CJD surveillance program supported by the Greek Ministry of Health, a total of twenty five patients suspected of having CJD, and ten of their relatives, were genotypically examined with respect to the PRNP gene. Full sequencing of the above samples revealed no abnormalities in the PRNP sequence and in all samples homozygosity for threonine at codon 193 was found. These data indicate that the T193I alteration of the PrP protein reported here is a mutation rather than a rare polymorphism.
Such mutations may either be inherited by the individual from the parents following the rules of Mendelian inheritance, or they may occur spontaneously. In the second case, a lack of family history should be expected. Lack of family history in gCJD cases is often reported [9] and it is suggested to occur due to the partial penetrance of the mutation.
References
Prusiner SB: Novel proteinaceous infectious particles cause scrapie. Science. 1982, 216 (4542): 136-144. 10.1126/science.6801762.
Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C: Mice devoid of PrP are resistant to scrapie. Cell. 1993, 73 (7): 1339-1347. 10.1016/0092-8674(93)90360-3.
Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG: Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A. 2000, 97 (18): 10168-10172. 10.1073/pnas.97.18.10168.
Peoc'h K, Guerin C, Brandel JP, Launay JM, Laplanche JL: First report of polymorphisms in the prion-like protein gene (PRND): implications for human prion diseases. Neurosci Lett. 2000, 286 (2): 144-148. 10.1016/S0304-3940(00)01100-9.
Tranchant C, Geranton L, Guiraud-Chaumeil C, Mohr M, Warter JM: Basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Neurology. 1999, 52 (6): 1244-1249.
Brandel JP: Clinical aspects of human spongiform encephalopathies, with the exception of iatrogenic forms. Biomed Pharmacother. 1999, 53 (1): 14-18. 10.1016/S0753-3322(99)80055-9.
Prusiner SB, Telling G, Cohen FE, DeArmond SJ: Prion diseases of humans and animals. Seminars in Virology. 1996, 7 (15): 159-173. 10.1006/smvy.1996.0021.
Prusiner SB, Scott MR: Genetics of prions. Annu Rev Genet. 1997, 31: 139-175. 10.1146/annurev.genet.31.1.139.
Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E: Genetic prion disease: the EUROCJD experience. Hum Genet. 2005, 118 (2): 166-174. 10.1007/s00439-005-0020-1.
Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H: Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002, 249 (11): 1567-1582. 10.1007/s00415-002-0896-9.
Ladogana A, Puopolo M, Poleggi A, Almonti S, Mellina V, Equestre M, Pocchiari M: High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005, 64 (9): 1592-1597. 10.1212/01.WNL.0000160118.26865.11.
Wiltfang J, Otto M, Baxter HC, Bodemer M, Steinacker P, Bahn E, Zerr I, Kornhuber J, Kretzschmar HA, Poser S, Ruther E, Aitken A: Isoform pattern of 14-3-3 proteins in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. J Neurochem. 1999, 73 (6): 2485-2490. 10.1046/j.1471-4159.1999.0732485.x.
WHO: Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation. 1998
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, Bernheimer H, Cardone F, Delasnerie-Laupretre N, Cuadrado Corrales N, Ladogana A, Bodemer M, Fletcher A, Awan T, Ruiz Bremon A, Budka H, Laplanche JL, Will RG, Poser S: Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000, 55 (6): 811-815.
Sanchez-Valle R, Nos C, Yague J, Graus F, Dominguez A, Saiz A: Clinical and genetic features of human prion diseases in Catalonia: 1993-2002. Eur J Neurol. 2004, 11 (10): 649-655. 10.1111/j.1468-1331.2004.00967.x.
Palmer MS, Dryden AJ, Hughes JT, Collinge J: Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991, 352 (6333): 340-342. 10.1038/352340a0.
Plaitakis A, Viskadouraki AK, Tzagournissakis M, Zaganas I, Verghese-Nikolakaki S, Karagiorgis V, Panagiotides I, Kilindireas C, Patsouris E, Haberler C, Budka H, Sklaviadis T: Increased incidence of sporadic Creutzfeldt-Jakob disease on the island of Crete associated with a high rate of PRNP 129-methionine homozygosity in the local population. Ann Neurol. 2001, 50 (2): 227-233. 10.1002/ana.1285.
Saetta AA, Michalopoulos NV, Malamis G, Papanastasiou PI, Mazmanian N, Karlou M, Kouzoupis A, Korkolopoulou P, Patsouris E: Analysis of PRNP gene codon 129 polymorphism in the Greek population. Eur J Epidemiol. 2006, 21 (3): 211-215. 10.1007/s10654-006-0012-z.
van Rheede T, Smolenaars MM, Madsen O, de Jong WW: Molecular evolution of the mammalian prion protein. Mol Biol Evol. 2003, 20 (1): 111-121. 10.1093/molbev/msg014.
Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K: NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000, 97 (1): 145-150. 10.1073/pnas.97.1.145.
Collins S, Boyd A, Fletcher A, Byron K, Harper C, McLean CA, Masters CL: Novel prion protein gene mutation in an octogenarian with Creutzfeldt-Jakob disease. Arch Neurol. 2000, 57 (7): 1058-1063. 10.1001/archneur.57.7.1058.
Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Laupretre N, Laplanche JL: Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000, 15 (5): 482-10.1002/(SICI)1098-1004(200005)15:5<482::AID-HUMU16>3.0.CO;2-1.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/6/169/prepub
Acknowledgements
The study was supported by the Greek Ministry of Health through the Center for Control of Infectious Diseases KEEL. Oral consent was obtained from the patient's relatives for publication of the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
KK conducted the immunoblot analysis and drafted the manuscript. IP conducted the immunoblot analysis, the PCR amplification and drafted the manuscript. SB and DK performed the clinical observations and reviewed the manuscript. HL and AP performed the sequencing reaction and reviewed the manuscript. JC repeated and confirm the sequencing analysis and TS overviewed the work and reviewed the manuscript. All authors read and approved the final manuscript.
Konstantia Kotta, Ioannis Paspaltsis contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kotta, K., Paspaltsis, I., Bostantjopoulou, S. et al. Novel mutation of the PRNPgene of a clinical CJD case. BMC Infect Dis 6, 169 (2006). https://doi.org/10.1186/1471-2334-6-169
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-6-169