
Abstract
Background
Inherited prion diseases are rare autosomal dominant disorders associated with diverse clinical presentations. All are associated with mutation of the gene that encodes prion protein (PRNP). Homozygous mutations with atypical clinical phenotypes have been described but are extremely rare.
Case presentation
A Chinese patient presented with a rapidly progressive cognitive and motor disorder in the clinical spectrum of sCJD. Investigations strongly suggested a diagnosis of CJD. He was found to carry a homozygous mutation at PRNP codon 200 (E200D), but there was no known family history of the disorder. The estimated allele frequency of E200D in East Asian populations is incompatible with it being a highly penetrant mutation in the heterozygous state.
Conclusion
In our view the homozygous PRNP E200D genotype is likely to be causal of CJD in this patient. Homotypic PrP interactions are well known to favour the development of prion disease. The case is compatible with recessively inherited prion disease.
Similar content being viewed by others
Background
Prion diseases are rare and transmissible neurodegenerative disorders associated with accumulation of assemblies of misfolded prion protein (PrP). Over 85% of the annual incidence of human prion disease takes the form of sCJD, typically presenting as a rapidly progressive dementia with ataxia, myoclonus and motor problems. Inherited prion diseases (IPD) comprise the large majority of the remainder of cases, which have a wide variety of clinical presentations, sometimes indistinguishable from sCJD, but often with much slower clinical change [1]. All IPD is associated with coding mutation of the gene that encodes prion protein (PRNP) and a large number of variants have been described. IPD segregates in families as an autosomal dominant trait, although some variants have relatively late clinical onset and carriers may die from other conditions before the disease manifests. The most common mutation that causes IPD, E200K, has a clinical presentation that is hard to distinguish from sCJD. E200K is frequent in particular regions of the world, and a small number of patients with a homozygous E200K mutation have been seen, having a slightly earlier onset of disease compared with heterozygous patients, and some altered clinical features [2]. A patient with a homozygous Q212P mutation has been reported. Here we present and discuss the case of a patient with CJD and a homozygous E200D mutation, not previously described.
Case presentation
A 61 year old man of Chinese ethnicity with no significant past medical history presented with a 6 week history of behavioural change including aggression, poor memory, praxis problems, slurred speech and loss of balance. There was a recent history of weight loss and vomiting. On examination he appeared to understand and speak English but the content of his speech was disorganised and he appeared confused. Examination revealed normal eye movements. He was dysarthric and demonstrated ataxia in all four limbs with truncal ataxia also. There was no myoclonus, or pyramidal or extrapyramidal signs at this stage.
The patient’s father died aged 61 of a non-neurological condition, the status of the patient’s mother is not known. There were no prior diagnoses of CJD or dementia in the wider family known to the patient’s children.
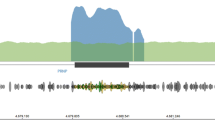
Initial work-up showed normal full blood count, standard biochemical profile, B12 and folate. Paraneoplastic, VGKC and anti-NMDA antibodies were negative as were HIV, syphilis and hepatitis serology. EEG was reported as mild slowing only, with no periodic sharp wave complexes. A brain MRI showed abnormal symmetric restricted diffusion in the corpus striatum bilaterally and cortical diffusion restriction in the frontal lobes and around the parieto-occipital fissure bilaterally (Fig. 1). CSF was acellular, with normal protein and glucose. CSF Viral PCR was negative but 14–3-3 and S-100b were abnormal. A diagnosis of CJD was confirmed by a positive reaction when CSF was analysed in quadruplicate by RT-QuIC (real-time quaking induced conversion, Fig. 2). The E200D mutation was detected by bidirectional sequencing and was linked to a methionine homozygous genotype at polymorphic codon 129.

Axial diffusion weighted image showing bilateral high signal in the striatum and cortex

CSF RT-QuIC trace of reaction seeded with 30 μl of CSF (blue) and 15 μl (orange). Results expressed as the mean of the two highest of four replicates
Over the course of his admission his neurological state rapidly worsened. He began to exhibit startle responses and multifocal myoclonus. Later he became increasingly agitated with distressing visual hallucinations which were managed with lorazepam. In the final days of his illness he became mute and bed bound with generalised spasticity. Following discussion with his family palliative care was commenced. Death was certified within 4 weeks of admission (total duration 10 weeks).
Discussion and conclusions
Although no post-mortem examination was done the diagnosis of this patient is almost certain. The MRI findings and RT-QuIC test are both highly specific in isolation. We are not aware of a single report in the literature of a false positive diagnosis when both of these tests are abnormal. The E200D variant has been found in the East Asian population with an allele frequency of approximately 1 in 10,000 (based on 2 occurrences in 18,394 East Asian exomes reported on gnomAD) [3]. As the variant has not been reported in the heterozygous state in any CJD patient, it is highly likely to be benign as this genotype.
The homozygous E200D genotype has never been observed in population databases. It should be extremely rare and found predominantly in families with some degree of consanguinity, which was possible but not certain in this case. Family history is a good guide to help decision making about the penetrance of novel PRNP variants, however in this case we would not expect a family history as the parents are expected to be heterozygous carriers of a benign variant. In our opinion, it seems likely that the homozygous E200D genotype was causal in this patient, the alternative possibility is of a coincidental occurrence of sCJD with a genotype of PRNP never before observed [4]. The situation seems similar to the Q212P variant of PRNP which appears to have low penetrance in the heterozygous state, but has caused an atypical early onset prion disease in the homozygous state [5].
Availability of data and materials
All data is available on reasonable request to the corresponding author.
Abbreviations
- CJD:
-
Creutzfeldt-Jakob disease
- PRNP:
-
Prion protein gene
- VGKC:
-
Voltage-gated potassium channel
- NMDA:
-
N-Methyl-D-aspartic acid
- RT-QuIC:
-
Real-time Quaking Induced Conversion Assay
- PCR:
-
Polymerase chain reaction
References
Mead S, Lloyd S, Collinge J. Genetic factors in mammalian prion diseases. Annu Rev Genet. 2019;53(1):117–47. https://doi.org/10.1146/annurev-genet-120213-092352.
Nitsan Z, Cohen OS, Chapman J, Kahana E, Korczyn AD, Appel S, et al. Familial Creutzfeldt-Jakob disease homozygous to the E200K mutation: clinical characteristics and disease course. J Neurol. 2020;267(8):2455–8. https://doi.org/10.1007/s00415-020-09826-z.
Minikel EV, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra329. https://doi.org/10.1126/scitranslmed.aad5169.
Mok TH, et al. Evaluating the causality of novel sequence variants in the prion protein gene by example. Neurobiol Aging. 2018;71:265 e261–7. https://doi.org/10.1016/j.neurobiolaging.2018.05.011.
Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC prion unit. Hum Mutat. 2010;31(7):E1551–63. https://doi.org/10.1002/humu.21281.
Acknowledgements
We would like to thank the patient and his children for contributing information for this letter.
Funding
Clinical research studies at the National Prion Clinic and MRC Prion Unit at UCL are funded by the National Institute of Health Research’s Biomedical Research Centre at University College London NHS Foundation Trust. Genetic analysis was funded by the Medical Research Council (UK). These funders had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AH, HO, SM assessed the patient and/or family; TC and LD did genetic analysis; JC, SM provided supervision. AG did the RT-QuIC analysis. All authors were involved in drafting the case report. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The patient was a participant in the National Prion Monitoring Cohort (UK) a prospective research study approved by NHS Research Ethics Committee.
Consent for publication
Written consent for the publication, including clinical details of the deceased and their family, was obtained from the next of kin.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hassan, A., Campbell, T., Darwent, L. et al. Case report of homozygous E200D mutation of PRNP in apparently sporadic Creutzfeldt-Jakob disease. BMC Neurol 21, 248 (2021). https://doi.org/10.1186/s12883-021-02274-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-021-02274-w