
Abstract
Background
Mycobacterium avium subspecies avium (M. avium) is frequently encountered in the environment, but also causes infections in animals and immunocompromised patients. In contrast, Mycobacterium avium subspecies paratuberculosis (M. paratuberculosis) is a slow-growing organism that is the causative agent of Johne's disease in cattle and chronic granulomatous infections in a variety of other ruminant hosts. Yet we show that despite their divergent phenotypes and the diseases they present, the genomes of M. avium and M. paratuberculosis share greater than 97% nucleotide identity over large (25 kb) genomic regions analyzed in this study.
Results
To characterize genome similarity between these two subspecies as well as attempt to understand their different growth rates, we designed oligonucleotide primers from M. avium sequence to amplify 15 minimally overlapping fragments of M. paratuberculosis genomic DNA encompassing the chromosomal origin of replication. These strategies resulted in the successful amplification and sequencing of a contiguous 11-kb fragment containing the putative Mycobacterium paratuberculosis origin of replication (oriC). This fragment contained 11 predicted open reading frames that showed a conserved gene order in the oriC locus when compared with several other Gram-positive bacteria. In addition, a GC skew analysis identified the origin of chromosomal replication which lies between the genes dnaA and dnaN. The presence of multiple DnaA boxes and the ATP-binding site in dnaA were also found in M. paratuberculosis. The strong nucleotide identity of M. avium and M. paratuberculosis in the region surrounding the origin of chromosomal replication led us to compare other areas of these genomes. A DNA homology matrix of 2 million nucleotides from each genome revealed strong synteny with only a few sequences present in one genome but absent in the other. Finally, the 16s rRNA gene from these two subspecies is 100% identical.
Conclusions
We present for the first time, a description of the oriC region in M. paratuberculosis. In addition, genomic comparisons between these two mycobacterial subspecies suggest that differences in the oriC region may not be significant enough to account for the diverse bacterial replication rates. Finally, the few genetic differences present outside the origin of chromosomal replication in each genome may be responsible for the diverse growth rates or phenotypes observed between the avium and paratuberculosis subspecies.
Similar content being viewed by others


Background
Mycobacteria are Gram-positive, acid-fast, pleomorphic, non-motile rods belonging to the order Actinomycetales. Mycobacterium avium complex organisms consist of the human and animal pathogens M. avium subsp. avium, M. avium subsp. paratuberculosis, and M. avium subsp. silvaticum [1]. DNA-DNA hybridization studies have long ago established a genetic similarity between M. avium subspecies avium (M. avium) and M. avium subspecies paratuberculosis (M. paratuberculosis) [2–4]. Now that whole genome sequencing technologies are available, investigators can begin to examine genetic relatedness in greater detail through direct nucleotide-nucleotide comparisons. These comparisons are particularly important in instances where two genetically similar bacteria have little or no specific diagnostic tests to distinguish each.
The literature reports genetic similarity between M. paratuberculosis and M. avium at between 72% and 95% [2, 4] depending on the region analyzed. However, despite the reported similarities, these mycobacteria are quite different phenotypically. M. paratuberculosis is an intracellular pathogen that infects ruminant animals, most notably cattle and sheep. The site of infection is the gastrointestinal tract, where it causes a chronic inflammatory ailment termed Johne's disease [5]. In contrast, M. avium is common in the environment, causes tuberculosis in birds, and disseminated infections in HIV patients [6]. Growth of M. paratuberculosis is characterized by its slow rate (doubling time of 22–26 hours, compared to 10–12 hours for M. avium) and requirement of mycobactin in culture media [5]. With the absence of a well-defined genetic system for M. paratuberculosis, a comparative genomic approach holds great potential in addressing the genetic basis for many of these phenotypic differences.
The genus Mycobacterium contains species that range from fast-growingsaprophytes such as M. smegmatis and M. fortuitum to slow-growing pathogens such asM. leprae, M. tuberculosis and M. paratuberculosis. Although the chromosomal origin of replication has been studied in some mycobacteria [7, 8], the genetic organization of the origin of replication in M. paratuberculosis has been previously unknown. Knowledge of the gene organization and sequence of this region is particularly important because chromosomal replication may be regulated by a common mechanism that could directly affect rate of growth.
Several features of the oriC region are highly conserved among bacteria. The sequence immediately flanking the dnaA gene is considered the origin of chromosomal replication, or oriC region [9, 10]. This region contains several genes that encode proteins required for basic cellular functions, including the protein subunit of RNase P (RnpA), ribosomal protein L34 (RpmH), the replication initiator protein (DnaA), the beta subunit of DNA polymerase III (DnaN), the recombination repair protein RecF, and the DNA gyrase proteins GyrA and GyrB. The relative gene order in this region is also highly conserved in many bacteria, especially the Gram-positives [11]. Although intergenic sequences in this region are conserved only among closely related organisms, the DnaA box is found in the non-coding regions flanking dnaA in most bacteria studied [12]. DnaA boxes are conserved nucleotide sequences (TTGTCCACA) where the DnaA protein binds to DNA, triggering events that ultimately lead to replication initiation and DNA synthesis [9].
In an effort to understand the genetic basis for growth rate and other phenotypic differences between M. paratuberculosis and M. avium, we have analyzed the genetic similarity of these genomes using two strategies. First, the putative oriC region of M. paratuberculosis was amplified, sequenced and compared with M. avium and other bacteria. Second, we examined nucleotide identity outside the oriC region using DNA homology matrix analysis as well as using several hundred M. paratuberculosis sequences from a random shotgun library compared with M. avium sequences present in the unfinished microbial genomes database. Our results show that these subspecies not only have a conserved gene order surrounding the origin of chromosomal replication, but also have a high synteny and nucleotide identity throughout both genomes. In addition, this preliminary comparative survey of the genomes of M. avium and M. paratuberculosis show even greater similarity (97%) than the literature suggests (72% to 95%) [2].
Results
Identification of predicted ORFs encoding replication-related proteins
An ~11-kb contiguous genomic fragment from M. paratuberculosis was amplified and sequenced using 15 primer pairs designed from M. avium genomic sequence in the putative oriC region (Fig. 1). This strategy enabled the successful amplification of all 15 minimally overlapping fragments of ~800 bp in length for this region of the M. paratuberculosis chromosome. A putative replication origin was identified by GC skew analysis [14]. A strong inflection point in the GC plot marks this origin (Fig. 1). Eleven ORFs were identified using the gene prediction software Artemis [15] (release 3; The Sanger Centre http://www.sanger.ac.uk/Software/Artemis/). Similarity searches were conducted locally using the BLASTP algorithm through the Artemis interface. Seven of these ORFs have high identity to proteins essential for basic cellular processes, including replication, in other mycobacterial species (Table 1). The function of GidB is unknown, but it may have a role in cell division [11]. RNase P, which consists of the protein subunit RnpA and a catalytic RNA subunit, is essential for generating mature tRNAs by cleaving the 5'-terminal leader sequences of precursor tRNAs [16]. rpmH encodes ribosomal protein L34, and DnaA is the initiator protein for chromosome replication. The B-subunit of DNA polymerase is encoded by dnaN. The recF gene product is involved in recombination, DNA repair, and induction of the SOS response, and may also have a role in replication [17]. Bacterial DNA gyrase, a tetramer consisting of A and B subunits, catalyzes the ATP-dependent unwinding of covalently closed circular DNA [18]. The remaining predicted ORFs in this region have high similarity to hypothetical proteins from M. tuberculosis (Table 1).

Amplification strategy and organization of the M. paratuberculosis chromosomal origin of replication. The locations of primer pairs used for amplification and sequencing are marked with facing arrows above the kilobase (kb) scale. The GC skew is shown beneath the kb scale and has a window size of 500. OriC, right at the point of the GC inflection, designates the origin of replication. An open reading frame map of the ~11 kb fragment is represented by shaded boxes and the two divergent arrows immediately above identify the direction of transcription. The degree of substitution in comparison to the corresponding M. avium gene is indicated below the gene name. π (tau) is the overall substitution rate, ds is the synonymous substitution rate, and dn is the non-synonymous substitution rate. GidB, glucose inhibited division protein B. RnpA, RNAse protein component A. RpmH, ribosomal protein L34. DnaA, replication initiator. DnaN, DNA polymerase subunit III. GyrB, DNA gyrase subunit B.
Sequence homology and conserved gene order in the oriC region of mycobacteria and other gram-positive bacteria
Alignment of the region surrounding oriC for several mycobacteria and other gram-positive bacteria provides some interesting comparisons (Fig. 2). The M. paratuberculosis oriC region conforms to the conserved gene order that is present in other mycobacteria as well as the closely related Streptomyces coelicolor. Even the more distantly related Bacillus subtilis shows some degree of synteny in this region. The fast growing M. smegmatis species contains a gnd sequence between dnaN and recF, which is absent in the slow-growing mycobacteria (Fig. 2). However, there appear to be no notable differences between M. avium and M. paratuberculosis at this level. The M. smegmatis coding sequence, gnd, has similarity to the 6-phophogluconate dehydrogenase genes in E. coli, but the mycobacterial protein is predicted to be about 200 amino acids shorter than the E. coli homolog. The length of non-coding intergenic regions between rpmH – dnaA and dnaA – dnaN is well conserved among the bacteria shown in figure 2. In many bacteria where a functional oriC has been identified, this gene order is conserved and oriC is adjacent to the dnaA gene [9, 10, 19].

Comparative gene order in the oriC region of mycobacteria and other Gram-positive bacteria. The relative gene order in this region of M. paratuberculosis conforms to the highly conserved order found in other gram-positive bacteria. Numbers indicate the length of the ORF or intergenic region. Arrows show the direction of transcription.
The amino acid sequence of each gene product was compared with the corresponding sequence in M. paratuberculosis for all species in this study (Table 2). The data show that while gene order is conserved, the percent identity declines in comparisons with mycobacteria other than M. avium. This percent identity declines even further in comparisons with non-mycobacterial sequences such as S. coelicolor and Corynebacteria glutamicum (Table 2).
Conserved functional motifs in the M. paratuberculosis putative oriC
Fuzznuc (EMBOSS; http://www.hgmp.mrc.ac.uk/Software/EMBOSS/index.html) was used to identify potential DnaA boxes in the M. paratuberculosis oriC region. The Gram-positive organisms in this study harbor 10 – 30 DnaA boxes (with 1 – 3 mismatches from the consensus sequence TTGTCCACA) flanking the dnaA sequence [8, 20–23] and 35 were found surrounding the M. paratuberculosis dnaA gene (Fig. 3). In addition, a hexameric sequence thought to be recognized by ATP-DnaA (AGATCT) was found in the 3' non-coding sequence adjacent to dnaA (Fig. 3b). The significance of additional dnaA boxes in M. paratuberculosis is likely necessary to open the DNA helix of this GC rich organism (69% GC content).

Non-coding sequences flanking M. paratuberculosis dnaA harbor 35 DnaA boxes. Nucleotide sequence of the rpmH-dnaA intergenic region (A) and dnaA-dnaN intergenic region (B) are shown. Sequences matching the DnaA box consensus(TTGTCCACA) with 1 – 3 mismatches are marked with an arrow. In (B), an A/T-rich region is underlined and the potential ATP-DnaA recognition site is boxed.
The dnaA gene is divided into four functional domains based on analysis of several dnaA mutants [24]. These domains consist of (1) an area near the N-terminus thought be involved in ability of the DnaA protein to aggregate, (2) ATP binding, (3) a domain that maps to a region near the C-terminus and is involved in DNA binding, (4) and a final domain of unknown function, but may bind DnaB. The conserved ATP-binding site that is found in domain III in other bacteria was also located in M. paratuberculosis (Fig. 3b). An AT-rich stretch of 19 nucleotides (74% A+T), which in other bacteria serves as the site of local unwinding of DNA after DnaA-DNA interaction, was located in non-coding sequence adjacent to dnaA (Fig. 3b). The non-coding sequences flanking dnaA are slightly AT-rich in general, relative to the rest of the genome sequence, consistent with findings in other gram-positive bacteria (38% – 40% A/T, vs. ~33% in the entire sequence).
A vast majority of all M. paratuberculosis K-10 genomic sequence have considerable nucleotide similarity to sequences from the human pathogenic isolate M. avium 104
As a basis for all nucleotide comparisons between M. avium and M. paratuberculosis in this study, an alignment of the 16s rRNA gene was performed. That analysis revealed a 100% nucleotide identity over the entire 1,472-bp gene (data not shown). Likewise, the oriC region in M. paratuberculosis was found to share a high level of nucleotide identity (~98%) with M. avium. Calculation of the rates of total nucleotide diversity (3) and synonymous substitution per synonymous site (ds) and non-synonymous substitution per non-synonymous site (dn) revealed patterns of variation within the range observed from sequence data outside the oriC region. These calculations showed a high degree of similarity between the two sequences and a predominance of synonymous over non-synonymous substitutions (Fig. 1). The patterns of nucleotide substitution varied considerably between genes in this region of the genome. For instance, there was complete nucleotide identity in the rpmH and recF genes and only 94% identity in the gene rnpA. To verify that these observed differences were real and not as a result of sequencing errors in the yet unfinished M. avium genome, we confirmed the data by resequencing the entire 11 kb region from an isolate clone of M. avium and obtained identical results (not shown).
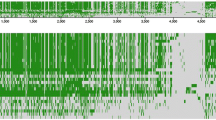
We next determined if the nucleotide identities would remain consistently high when M. paratuberculosis sequences outside the oriC region were compared with M. avium. Sequencing of the M. paratuberculosis K-10 cattle isolate is nearing completion in our laboratories and TIGR http://www.tigr.org is in the finishing stages of M. avium isolate 104. Beginning with nucleotide number 1 in the dnaA coding region of each genome, a comparison of 2 million bases of M. paratuberculosis with 2 million bases from M. avium by Pustell DNA matrix analysis [25], indicates that genomic similarity continues outside the surrounding oriC region (Fig. 4). When evaluating similarities between two sequences of this size, a matrix comparison is the method of first choice. In addition, the matrix method displays matching regions in the context of the sequence as a whole, making it easy to determine if the regions are repeated or inverted. For example, figure 4 shows a large 56.6 kb genomic inversion of the region surrounding nucleotide 350,000. The DNA identity matrix also identified sequences that were present in one genome, but absent in the other as shown by the broken diagonal lines (Fig. 4). These data show remarkable similarity over large regions in both mycobacterial genomes.

DNA matrix analysis of a contiguous 2 million nucleotide section of the M. avium (y-axis) and M. paratuberculosis (x-axis) genomes. Four 500,000 nucleotide matrices are shown with the nucleotide segments indicated above each plot. A long unbroken diagonal line from the upper left corner to the lower right corner indicates that the sequences are collinear. The diagonal line (in blue) that runs from the lower left to the upper right at the 350,000 nucleotide region indicates that one sequence is the reverse complement of the other. The arrows (in red) show sequences present in M. avium but absent in M. paratuberculosis and the arrowhead (in green) shows a sequence represented only in M. paratuberculosis. The initial nucleotide in the dnaA coding sequence was defined as number one in both genomes for this analysis. The parameters for this DNA identity matrix include: a window size of 30, a minimum percent score of 80, and a hash value of 4.
Finally, we analyzed 548 recombinant clones from a randomly sheared M. paratuberculosis small insert library in order to obtain specific rates of nucleotide substitutions. Sequences from these clones represented over 350,000 bp of unique (non-overlapping) M. paratuberculosis genomic DNA and comprised 7% of the estimated 5 Mb genome sequence. From this analysis, we estimated the rates of total synonymous and non-synonymous substitutions for 200 fragments that were aligned in-frame and then analyzed with the program NAGV2 [26] using the methods of Nei and Gojobori [27]. The results of these analyses show that the average nucleotide diversity between the two species is 2.59% ± 0.06% (range 0% to 18.8%; median, 1.85% ± 0.05%). The results also show that the average rates of synonymous substitution per synonymous site are 3.38% ± 1.32% (range, 0% to 19.5%; median, 3.5% ± 1.5%). In contrast, the rates of non-synonymous substitution per non-synonymous site were 1.89% ± 0.05% (range, 0% to 12.9%; median 1.3% ± 0.05%). These results not only indicate that the two subspecies have a high degree of nucleotide identity (>97%), but also suggest that the patterns of substitution have favored synonymous substitutions as can be expected from positive selection.
Discussion
With the genome sequencing projects of M. paratuberculosis and M. avium nearing completion, we have been able to compare large amounts of sequence data for the first time. Our results show substantial nucleotide identity above even that reported previously in the literature [2–4]. Paradoxically, the overall nucleotide identity between these phenotypically distinct mycobacteria appears similar to that observed with two phenotypically identical Helicobacter pylori isolates at ≥98% nucleotide identity [28].
The high nucleotide identity shared between M. paratuberculosis and M. avium directly conflicts with their divergent phenotypic characteristics. Because of strong similarity in the oriC region, alternative hypotheses should be tested to explain the growth rate differences between M. avium and M. paratuberculosis. Genomic rearrangements and the presence of unique genes identified by matrix analysis in this study are two such possibilities that could account for some of the phenotypic differences. We have recently reported on M. paratuberculosis coding sequences that are absent in M. avium [29]. From an analysis of 48% of the M. paratuberculosis genome, only 27 predicted coding sequences were found to be absent in M. avium. Therefore, an estimated total of 50–60 M. paratuberculosis coding sequences might be absent in M. avium following a whole genome analysis. This extremely low number of unique M. paratuberculosis genes is in stark contrast to E. coli where the MG1655 isolate contains 528 genes not found in the EDL933 isolate [30]. Further analysis of this limited number of unique coding sequences will be critical in developing specific diagnostic reagents. Finally, a detailed analysis of coding sequences unique to each respective mycobacterial genome and their genetic regulatory networks will be necessary to understand the molecular basis for growth rate and other phenotypic differences.
Other potential explanations include the presence of global regulators, insertion sequences, transcription-translation rates, genomic rearrangements and ribosomal RNA operons. Each respective genome possesses insertion elements (IS900, IS1311) at unique loci that could distinctly affect growth difference or other phenotype by insertional mutation. Foley-Thomas et al. [31] compared the expression of the luciferase gene in M. paratuberculosis with the fast-growing M. smegmatis and concluded that the rates of transcription and translation may not account for the slow growth of M. paratuberculosis.
We present evidence for at least one large-scale genomic rearrangement between these two subspecies. This rearrangement consists of a 56.6 kb inversion that contains approximately 61 predicted coding sequences (Bannantine and Kapur, unpublished). Genomic rearrangements such as that described could have a profound effect on phenotype. The presence of multiple copies of ribosomal RNA operons within a genome can be directly attributed to faster growth rate. The increased gene dosage results in more ribosomes and therefore increased protein translational capacity. However, only one rRNA operon is present in each subspecies and this is also true for the fast growers Mycobacterium abscessus and Mycobacterium chelonae [32]. These fast growing mycobacteria have multiple promoters that increase the transcriptional rate of the rRNA operon to overcome gene dosage limitations [32]. The rRNA operon promoter structures have not been mapped by primer extension for either M. paratuberculosis or M. avium, but if M. avium had multiple functional rRNA operon promoters, that may account for the growth rate differences.
The genetic organization of the origin of replication has been characterized in several Gram-positive pathogens including B. subtilis, S. coelicolor, M. tuberculosis, M. avium, M. leprae, and M. smegmatis [8]. The results of our investigation on the oriC region of M. paratuberculosis show that each of the 15 primer pairs, designed from M. avium sequence data, resulted in the successful amplification and subsequent sequencing of an ~11 kb region of the M. paratuberculosis genome. The sequenced region encodes 11 putative proteins, several of which show a high level of identity to proteins that are known or predicted to be involved in DNA replication. However, we found a cluster of substitutions in a region of rnpA (data not shown). It is noteworthy that in this region of the gene, each of the nucleotide substitutions results in an amino acid replacement. While mutations in this region of the gene are known to result in dramatic differences in ability of bacteria to respond to environmental stresses [33], the functional significance of these differences between M. avium and M. paratuberculosis are at present unknown. While these sequencing efforts have revealed a conserved gene order in the oriC of Gram-positive bacteria [11], the nucleotide and amino acid identity between M. paratuberculosis and M. avium in this region is much stronger when compared to other mycobacteria and other Gram-positive bacteria (see Table 2). It is well recognized that the characterization of gene organization in the oriC region as well as the complete genome sequence will provide a springboard for addressing questions such as the nature of the slow growth rate of M. paratuberculosis as compared to the genetically related rapidly-growing mycobacteria. Progress on these research fronts will improve our chances of understanding and controlling infections caused by M. paratuberculosis and related pathogens.
The conservation of functional sequence motifs in the oriC of other Gram-positive organisms has provided clues to the mechanism of bacterial replication. For instance, DnaA monomers bind to specific, non-palindromic 9-nucleotide sequences called DnaA boxes, and this interaction is thought to initiate replication. The oriC of Gram-positive bacteria typically contains 10 – 30 of these DnaA boxes, often found in non-coding regions flanking the dnaA gene. The interaction of DnaA with DnaA boxes promotes the local unwinding of a nearby AT-rich region, providing an entry site for the DnaB/DnaC helicase complex. The dnaA gene itself is divided into four domains that differ in the extent of sequence homology [34]. Domain IV is responsible for DnaA box recognition and domain III is a highly conserved region containing the ATP-binding site [13, 35]. Domain I participates in cooperative DnaA protein-DNA interactions [36].
The genetic relatedness of M. paratuberculosis with other mycobacterial subspecies has been the root cause of the lack of development of M. paratuberculosis-specific diagnostic tests. By comparing the genome sequences of both M. paratuberculosis and M. avium, specific diagnostic tests may be developed and a better understanding of the molecular differences that contribute to unique phenotypes will be obtained. Finally, knowledge of the complete genome sequence of M. paratuberculosis is expected to facilitate the identification of diagnostic sequences in this economically significant veterinary pathogen.
Conclusion
With the genomes of M. paratuberculosis and M. avium nearly completed, investigators will be able to analyze the similarities and differences between these genomes with amazing detail. Through a comparative genomic analysis of over 2 million nucleotides, we have shown that the two subspecies, avium and paratuberculosis, are highly similar at the gene and nucleotide level. This is in stark contrast to the phenotypic differences that each displays.
Methods
Strains and growth media
A cattle isolate (K-10) of M. paratuberculosis [31] has been chosen for genome sequencing studies. The organism was grown in Middlebrook 7H9 broth supplemented with OADC (Difco Laboratories, Detroit, MI), Tween 80, and mycobactin J (Allied Monitor, Fayette, MO) as described by Bannantine et al. [37]. M. avium strain 104 was grown in Middlebrook 7H9 broth. DNA was extracted using the Qiagen QIAamp Tissue Kit (Chatsworth, CA).
Primer design and amplifications
A web-interfaced program, Primer3 http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi, was used. Primers were designed based on available M. avium strain 104 genomic sequence data http://www.tigr.org for the amplification of 11 genes in a contiguous ~11 kb M. paratuberculosis fragment surrounding the putative origin of replication (oriC). By this strategy, a total of 15 primer pairs were constructed for the amplification of 15 minimally overlapping fragments of ~800 bp in length for this region of the M. paratuberculosis genome. Amplification reactions included the high fidelity DNA polymerase, Pfu (Stratagene, La Jolla, CA) and an annealing temperature of 58°C.
Library construction
A random 2.2-kb insert library of M. paratuberculosis K-10 has been constructed as follows. Total M. paratuberculosis genomic DNA was isolated and randomly sheared using a nebulizer and compressed nitrogen according to protocols developed by Bruce Roe's laboratory http://www.genome.ou.edu. The resulting DNA fragments were separated by gel electrophoresis and fragments in the range of 2.1–2.2 kb were purified. After polishing the ends of the fragments using Klenow (New England Biolabs, Beverly, MA), they were cloned into SmaI-restricted/CIAP pUC18 vector. The resulting library was >90% recombinant and contained more than 50,000 independent recombinant clones.
DNA Sequencing and Analysis
The DMSO protocol (ABI Automated DNA Sequencing Chemistry Guide, ABI, Foster City, CA) was implemented for carrying out the sequencing reactions and data were collected using ABI 377 automated DNA sequencers at the Advanced Genetic Analysis Center at the University of Minnesota. The data was analyzed using the DNAStar (Madison, WI) package and Artemis [15]. Rates of synonymous and non-synonymous substitution were calculated by the un-weighted method of Nei and Gojobori [27]. Pustell DNA matrix analysis [25] was performed using MacVector version 7.1 software.
Nucleotide Sequence Accession Number
The GenBank accession number for the M. paratuberculosis 11-kb oriC region is AF222789. The M. paratuberculosis random sequences can be accessed via the M. paratuberculosis genome project website: http://www.cbc.umn.edu/ResearchProjects/AGAC/Mptb/Mptbhome.html.
References
Thorel MF, Krichevsky M, Levy-Frebault VV: Numerical taxonomy of mycobactin-dependent mycobacteria, emended description of Mycobacterium avium, and description of Mycobacterium avium subsp. avium subsp. nov., Mycobacterium avium. Int J Syst Bacteriol. 1990, 40: 254-260.
Hurley SS, Splitter GA, Welch RA: Development of a diagnostic test for Johne's disease using a DNA hybridization probe. J Clin Microbiol. 1989, 27: 1582-1587.
Saxegaard F, Baess I, Jantzen E: Characterization of clinical isolates of Mycobacterium paratuberculosis by DNA-DNA hybridization and cellular fatty acid analysis. Apmis. 1988, 96: 497-502.
Yoshimura HH, Graham DY: Nucleic acid hybridization studies of mycobactin-dependent mycobacteria. J Clin Microbiol. 1988, 26: 1309-1312.
Harris NB, Barletta RG: Mycobacterium avium subsp. paratuberculosis in Veterinary Medicine. Clin Microbiol Rev. 2001, 14: 489-512. 10.1128/CMR.14.3.489-512.2001.
Horsburgh CR: Mycobacterium avium complex infection in the acquired immunodeficiency syndrome. N Engl J Med. 1991, 324: 1332-1338.
Qin MH, Madiraju MV, Zachariah S, Rajagopalan M: Characterization of the oriC region of Mycobacterium smegmatis. J Bacteriol. 1997, 179: 6311-6317.
Salazar L, Fsihi H, de Rossi E, Riccardi G, Rios C, Cole ST, Takiff HE: Organization of the origins of replication of the chromosomes of Mycobacterium smegmatis, Mycobacterium leprae and Mycobacterium tuberculosis and isolation of a functional origin from M. smegmatis. Mol Microbiol. 1996, 20: 283-293.
Skarstad K, Boye E: The initiator protein DnaA: evolution, properties and function. Biochim Biophys Acta. 1994, 1217: 111-130. 10.1016/0167-4781(94)90025-6.
Smith DW, Yee TW, Baird C, Krishnapillai V: Pseudomonad replication origins: a paradigm for bacterial origins?. Mol Microbiol. 1991, 5: 2581-2587.
Ogasawara N, Yoshikawa H: Genes and their organization in the replication origin region of the bacterial chromosome. Mol Microbiol. 1992, 6: 629-634.
Yoshikawa H, Ogasawara N: Structure and function of DnaA and the DnaA-box in eubacteria: evolutionary relationships of bacterial replication origins. Mol Microbiol. 1991, 5: 2589-2597.
Koonin EV: A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 1993, 21: 2541-2547.
Lobry JR: Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996, 13: 660-665.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B: Artemis: sequence visualization and annotation. Bioinformatics. 2000, 16: 944-945. 10.1093/bioinformatics/16.10.944.
Brown JW, Pace NR: Ribonuclease P RNA and protein subunits from bacteria. Nucleic Acids Res. 1992, 20: 1451-1456.
Courcelle J, Carswell-Crumpton C, Hanawalt PC: recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci U S A. 1997, 94: 3714-3719. 10.1073/pnas.94.8.3714.
Reece RJ, Maxwell A: DNA gyrase: structure and function. Crit Rev Biochem Mol Biol. 1991, 26: 335-375.
Fujita MQ, Yoshikawa H, Ogasawara N: Structure of the dnaA region of Pseudomonas putida: conservation among three bacteria, Bacillus subtilis, Escherichia coli and P. putida. Mol Gen Genet. 1989, 215: 381-387.
Calcutt MJ, Schmidt FJ: Conserved gene arrangement in the origin region of the Streptomyces coelicolor chromosome. J Bacteriol. 1992, 174: 3220-3226.
Madiraju MV, Qin MH, Yamamoto K, Atkinson MA, Rajagopalan M: The dnaA gene region of Mycobacterium avium and the autonomous replication activities of its 5' and 3' flanking regions. Microbiology. 1999, 145 (Pt 10): 2913-2921.
Ogasawara N, Moriya S, Yoshikawa H: Initiation of chromosome replication: structure and function of oriC and DnaA protein in eubacteria. Res Microbiol. 1991, 142: 851-859. 10.1016/0923-2508(91)90065-I.
Rajagopalan M, Qin MH, Nash DR, Madiraju MV: Mycobacterium smegmatis dnaA region and autonomous replication activity. J Bacteriol. 1995, 177: 6527-6535.
Sutton MD, Kaguni JM: The Escherichia coli dnaA gene: four functional domains. J Mol Biol. 1997, 274: 546-561. 10.1006/jmbi.1997.1425.
Pustell J, Kafatos FC: A convenient and adaptable package of computer programs for DNA and protein sequence management, analysis and homology determination. Nucleic Acids Res. 1984, 12: 643-655.
Kapur V, Kanjilal S, Hamrick MR, Li LL, Whittam TS, Sawyer SA, Musser JM: Molecular population genetic analysis of the streptokinase gene of Streptococcus pyogenes: mosaic alleles generated by recombination. Mol Microbiol. 1995, 16: 509-519.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ: Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999, 397: 176-180. 10.1038/16495.
Bannantine JP, Baechler E, Zhang Q, Li L, Kapur V: Genome Scale Comparison of Mycobacterium avium subsp. paratuberculosis with Mycobacterium avium subsp. avium Reveals Potential Diagnostic Sequences. J Clin Microbiol. 2002, 40: 1303-1310. 10.1128/JCM.40.4.1303-1310.2002.
Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR: Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001, 409: 529-533. 10.1038/35054089.
Foley-Thomas EM, Whipple DL, Bermudez LE, Barletta RG: Phage infection, transfection and transformation of Mycobacterium avium complex and Mycobacterium paratuberculosis. Microbiology. 1995, 141: 1173-1181.
Gonzalez-y-Merchand JA, Garcia MJ, Gonzalez-Rico S, Colston MJ, Cox RA: Strategies used by pathogenic and nonpathogenic mycobacteria to synthesize rRNA. J Bacteriol. 1997, 179: 6949-6958.
Kirsebom LA, Baer MF, Altman S: Differential effects of mutations in the protein and RNA moieties of RNase P on the efficiency of suppression by various tRNA suppressors. J Mol Biol. 1988, 204: 879-888.
Fujita MQ, Yoshikawa H, Ogasawara N: Structure of the dnaA and DnaA-box region in the Mycoplasma capricolum chromosome: conservation and variations in the course of evolution. Gene. 1992, 110: 17-23. 10.1016/0378-1119(92)90439-V.
Roth A, Messer W: The DNA binding domain of the initiator protein DnaA. Embo J. 1995, 14: 2106-2111.
Messer W, Blaesing F, Jakimowicz D, Krause M, Majka J, Nardmann J, Schaper S, Seitz H, Speck C, Weigel C, Wegrzyn G, Welzeck M, Zakrzewska-Czerwinska J: Bacterial replication initiator DnaA. Rules for DnaA binding and roles of DnaA in origin unwinding and helicase loading. Biochimie. 2001, 83: 5-12. 10.1016/S0300-9084(00)01216-5.
Bannantine JP, Stabel JR: HspX is present within Mycobacterium paratuberculosis-infected macrophages and is recognized by sera from some infected cattle. Vet Microbiol. 2000, 76: 343-358. 10.1016/S0378-1135(00)00247-9.
Acknowledgements
This work was funded by Agricultural Research Service intramural funds and USDA-NRI grants to V.K. and J.P.B. We thank Robert Fleischmann and coworkers at TIGR for their efforts in sequencing the M. avium genome.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Bannantine, J.P., Zhang, Q., Li, LL. et al. Genomic homogeneity between Mycobacterium avium subsp. avium and Mycobacterium avium subsp. paratuberculosis belies their divergent growth rates. BMC Microbiol 3, 10 (2003). https://doi.org/10.1186/1471-2180-3-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-3-10