
Abstract
Background
A human isolate of Mycobacterium avium subsp. paratuberculosis (M. paratuberculosis 43525) was sequenced and compared genomically to other mycobacterial pathogens. M. paratuberculosis 43525 was recently isolated from a patient with ulcerative colitis and belongs to the M. avium complex, a group known to infect both humans and animals. While M. paratuberculosis is a known pathogen of livestock, there are only 20 human isolates from the last 20 years, therefore we took the opportunity to perform a whole genome comparison between human and animal mycobacterial pathogens. We also compared virulence determinants such as the mycobactin cluster, PE/PPE genes and mammalian cell entry (mce) operons between MAC subspecies that infect animals and those that infect humans. M. tuberculosis was also included in these analyses given its predominant role as a human pathogen.
Results
This genome comparison showed the PE/PPE profile of M. paratuberculosis 43525 to be largely the same as other M. paratuberculosis isolates, except that it had one PPE and one PE_PGRS protein that are only present in human MAC strains and M. tuberculosis. PE/PPE proteins that were unique to M. paratuberculosis 43525, M. avium subsp. hominissuis and a caprine M. paratuberculosis isolate, were also identified. In addition, the mycobactin cluster differed between human and animal isolates and a unique mce operon flanked by two mycobactin genes, mbtA and mbtJ, was identified in all available M. paratuberculosis genomes.
Conclusions
Despite the whole genome comparison placing M. paratuberculosis 43525 as closely related to bovine M. paratuberculosis, key virulence factors were similar to human mycobacterial pathogens. This study highlights key factors of mycobacterial pathogenesis in humans and forms the basis for future functional studies.
Similar content being viewed by others

Background
M. avium subsp. paratuberculosis (M. paratuberculosis), of the M. avium complex (MAC), is one of the slowest growing mycobacteria and like other pathogenic mycobacteria, is difficult to detect and treat. It is widely recognised as the cause of Johne’s disease, a gastrointestinal disease of livestock, and is also implicated in human Crohn’s disease [1–3]. The MAC contain subspecies that infect animals and subspecies pathogenic to humans [4]. The closely related MAC display slight genomic differences depending on their host and comparison of these differences has the potential to identify host specific pathogenicity factors, leading to improved diagnosis and treatment.
In the current study we compared the genome of a newly isolated strain of M. paratuberculosis (M. paratuberculosis 43525) from a female patient with ulcerative colitis [5], to other pathogenic mycobacteria using Single Nucleotide Polymorphism (SNP) analysis, BLASTp (homology based) and phmmer (non-homology based) algorithms. While M. paratuberculosis normally infects animals, its isolation from humans is rare, with less than 20 isolates reported in the last 20 years [6–9]. Like other M. paratuberculosis from humans, M. paratuberculosis 43525 is cattle type (C-Type) [5]. Therefore, further analysis of this strain provides a unique opportunity to explore other possible variations in host pathogenicity factors.
While genomic studies have compared a number of these isolates to other M. paratuberculosis strains and M. hominissuis [10–15], all sequences of M. paratuberculosis to date have been obtained from laboratory strains of unknown subculture number, and often have undergone many years of laboratory passage. Current evidence would suggest that multiple subculture of M. tuberculosis may affect virulence properties with, for example, marked changes in cell wall lipids observed after extensive laboratory passage [16]. Important to note is that M. bovis BCG, widely used in vaccines due to its attenuation in immunocompetent hosts, was produced by multiple subculture in vitro [17]. In contrast, the genome of M. paratuberculosis 43525 was sequenced after only four subcultures and therefore provides a more accurate representation of the wild-type in vivo mycobacterial genome.
The virulence factors explored in this study, the PE/PPE (proline-glutamate/proline-proline-glutamate motif) genes, mammalian cell entry (mce) operons and the mycobactin cluster, were chosen based on studies into M. tuberculosis and M. avium pathogenicity. The analysis of these genomic loci afford the representation of pathogenicity elements present in M. tuberculosis isolated from human infections and M. paratuberculosis isolated from livestock infections [4, 18–22].
The PE/PPE families are unique to mycobacteria and were first identified for their ability to stimulate IFN-γ [19]. They are GC rich and thought to be the main source of strain variability within the MAC [4]. PE and PPE refer to the residues, Proline-Glutamate and Proline-Proline-Glutamate, respectively, located at the N termini of their encoded proteins. The M. tuberculosis genome devotes 10 % of its protein coding potential to this protein family with various functions attributed to them [23]. Similar to M. tuberculosis, some PPE of M. paratuberculosis are expressed on the cell surface, while others are cell wall associated and interact with the immune system via TLR-2 [19], however, this gene family only represents 2.5 % of the M. paratuberculosis genome [24].
The mammalian cell entry (mce) operons of M. tuberculosis were first discovered in studies to elucidate how M. tuberculosis enters non-phagocytic cells [25]. The genes exist in many bacterial species, however, only in the mycobacteria do they exist as operons [26]. The function of these operons is now thought to be diverse and not confined to cell entry, given that they have been found in non-pathogenic, environmental mycobacteria [26]. There are four mce operons in M. tuberculosis, each has two yrbE genes and six mce genes, often coupled to a mce regulator gene. The genes in each operon are contiguous but differentially expressed, depending on growth conditions and/or nutrient supply [21]. In the MAC, additional mce operons have been reported that do not appear to have orthologues in M. tuberculosis [26, 18].
Mycobactin dependency in vitro is a major phenotypic difference between M. paratuberculosis and other subspecies of the MAC complex. Mycobactins are siderophores that transport or scavenge iron, particularly in environments where free iron is limited, such as inside a host cell [27]. Like other siderophores, mycobactin is a secondary metabolite, a product of non-ribosomal peptide synthases (NRPS) and polyketide synthases (PKS) (an integrated NRPS-PKS) [28]. The mycobactin gene cluster contains 10 genes (A-J) and the mycobactin operon promoter is active in M. paratuberculosis, with all mycobactin genes able to be transcribed inside bovine macrophages [29]. M. paratuberculosis 43525 has a peculiar mycobactin phenotype as it grows on some types of media, such as Middlebrook 7H10, without the addition of mycobactin [5]. Given this, comparison of M. paratuberculosis 43525 with other MAC strains has the potential to provide unique genomic information and the basis for their pathogenicity.
Results
The general features of the assembled draft genome of M. paratuberculosis 43525 are presented in Table 1. Out of a total of 4433 protein coding sequences (CDS), 1517 (35 %) belonged to recognised subsystems. Of the 2781 non-hypothetical CDS, 1450 belonged to recognised subsystems while 1715 CDS were hypothetical and of these, 67 belonged to subsystems according to RAST.
Comparative blastp searches and clustering analyses executed through Proteinortho [30], suggested that 165 putative protein sequences annotated in the M. paratuberculosis 43525 genome were unique to this strain (Fig. 1). These putatively unique sequences included a large number of hypothetical proteins, as well as PE-PGRS and mce genes that will be described below. In addition, differences were observed between the genes encoding the mycobactin cluster and this cluster was analysed in more detail.
Single nucleotide polymorphism (SNP) analysis
To better characterise M. paratuberculosis 43525, variation between this bacterium and 27 other mycobacterial strains (including two M. avium subsp. avium strains) were compared at the nucleotide level. Of these strains, nine were M. paratuberculosis isolates from humans, one was an ovine isolate and one was a caprine isolate. The SNPs of all strains were concatenated and used for phylogenetic analysis on a genome-wide level with M. paratuberculosis K10 as the reference strain. The rooted tree (Fig. 2) shows M. paratuberculosis 43525 to be closely related to M. paratuberculosis CLIJ644, a bovine isolate from Victoria, Australia [12].

Graphical representation of the 6310 orthologous clusters of annotated protein sequences encoded in 27 mycobacterial strains. Comparative blastp 2.2.22+ searches [61] were conducted using Proteinortho [30] and orthologous clusters were visualised using FriPan (http://www.vicbioinformatics.com/software.fripan.shtml)

Phylogenetic analysis of M. paratuberculosis 43525 relative to other M. paratuberculosis isolates and two M. avium isolates. Neighbour-joining phylogenetic tree based on SNPs of M. paratuberculosis isolates from bovine, ovine and human hosts and M. avium isolates derived from human and avian hosts with M. paratuberculosis K10 as the reference. The colour box indicates the clade in which M. paratuberculosis 43525 belongs and also contains human isolates M. paratuberculosis 4 and 4B. In addition, this clade contains recent (compared to the type strains) bovine isolates, including the bovine isolate CLIJ644. The reference strain M. paratuberculosis K10 is in a different clade to M. paratuberculosis 43525 and the type strain ATCC19698
PE/PPE genes
The nomenclature of the MAC complex PE/PPE genes was used as previously described [4], and a summary of the M. paratuberculosis 43525 genes shared between the MAC and M. tuberculosis is presented in Fig. 3 and Additional file 1. Thirty seven PPE genes were found in M. paratuberculosis 43525, none of which were unique to this strain, while 17 were conserved in all strains examined. In the bovine strain M. paratuberculosis K10, MACPPE15 is a fragmented pseudogene, whereas the full gene is present in the human isolate M. paratuberculosis 43525, and this gene is homologous to Mav2514 from M. avium 104. Although MACPPE41 and MACPPE42 are said to be unique to the M. paratuberculosis subspecies, here only MACPPE42 was found in M. paratuberculosis 43525 [4].

A summary of the presence and absence of the MACPE/PPE. The genes were sorted according to their distribution profiles (Additional file 1). Orthologues in M. tuberculosis were also added for comparison. Blue indicates the gene (listed on the right hand side) is present while yellow indicates the gene is missing. The strain order across the top is determined by the relative presence/absence of PE/PPE genes
Ten PE genes and one PE fragment were present in M. paratuberculosis 43525 (Fig. 3, Additional file 1). PE13 was the only PE gene that was not conserved in all strains studied, being found only in M. paratuberculosis 43525 and M. avium 104. The genome of M. paratuberculosis 43525 also had gene PE_PGRS11 which was also found in M. paratuberculosis K10, M. tuberculosis and M. avium 104, but absent in M. avium ATCC25291.
Mycobactin
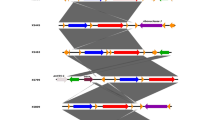
A total of 17 NRPS/ PKS clusters were identified by antiSMASH in the M. paratuberculosis 43525 genome. The cluster identified as the mycobactin cluster was analysed and found to have a different primary structure as compared with that of other MAC strains, with respect to the spacing of genes and gene size (Fig. 4). The mycobactin cluster of M. paratuberculosis has previously been shown to have both NRPS and PKS modules [24]. While the mbtA, mbtC, mbtD, mbtG and mbtI genes of the M. paratuberculosis 43525 mycobactin cluster were found to be identical to the equivalent genes in M. paratuberculosis K10, the remaining 5 genes (3 of which are NRPS modules) were found to encode larger proteins.

Comparison of the mycobactin cluster between members of the MAC complex and M. tuberculosis. Amino acid length is indicated above each gene, gene names are indicated and colour matched to equivalent genes in other strains (M. paratuberculosis 43525, M. paratuberculosis K10, M. paratuberculosis S397 M. avium 104, M. avium ATCC25291 and M. tuberculosis). M. paratuberculosis K10, M. paratuberculosis S397 and M. avium ATCC25291 are of animal origin, the remaining three are from human hosts. The NRPS modules MbtB and MbtE are one gene in M. paratuberculosis 43525 and M. tuberculosis but two genes in other MAC. In addition, all M. paratuberculosis strains had an mce operon (mce6) present in the gap between MbtA and MbtJ (indicated for M. paratuberculosis 43525 and K10)
Furthermore, the mbtB gene, shown by others to be the first gene involved in mycobactin synthesis [31], encodes a polypeptide of 1420 amino acids in M. paratuberculosis 43525, which was larger than the mbtB gene product of strains such as M. paratuberculosis K10 and M. avium 104 but similar in size to the equivalent MbtB in M. tuberculosis. The polypeptide size difference between these strains was due to the thioesterase domain of MbtB being encoded on a separate gene in M. paratuberculosis K10 and other MAC, but not in M. paratuberculosis 43525 or M. tuberculosis (Fig. 4).
The size and organisation of genes encoding MbtE vary greatly across strains (Fig. 4). The AMP binding and peptidyl carrier protein (PCP) domains were encoded on the one mbtE gene in M. paratuberculosis 43525, however, in M. paratuberculosis K10 the AMP binding was split across MAP2172c and MAP2173c, while the PCP domain is encoded on MAP2172c. Confirmation by amplicon sequencing demonstrated a 187 nucleotide indel in M. paratuberculosis 43525 compared to M. paratuberculosis K10 (bases 2411868 to 2412055). The AMP binding domain is a 215 aa protein in M. paratuberculosis K10 as compared with a 394 residue protein in M. paratuberculosis 43525 and a 408 aa protein in M. avium 104. Anti-smash uses a number of databases to predict the substrate for each NRPS domain. The predicted substrate for MbtE of M. paratuberculosis 43525 is tyrosine. In contrast, there was no consensus on the predicted substrate for MbtE of M. paratuberculosis K10 and M. avium 104.
The mbtF gene in M. paratuberculosis 43525 encoded a longer protein than in the M. paratuberculosis K10 equivalent. The main difference was in the epimerisation domain that encodes a shorter polypeptide in M. paratuberculosis K10 (179 aa) as compared with M. paratuberculosis 43525 (299 aa), M. avium 104 (299 aa), M. avium ATCC25291 (299 aa) and M. tuberculosis (288 aa).
Five copies of mbtH were found in M. paratuberculosis 43525, four of which (mbtH_1, mbtH_2, mbtH_3 and phoP) had 100 % sequence similarity with the equivalent genes in M. paratuberculosis K10. The mbtH_3 gene was situated adjacent to the mycobactin cluster. However, the fifth mbtH gene of M. paratuberculosis 43525, adjacent to pstA was found to have an 85 % match to mbtH_2 in K10 but 100 % sequence similarity with 18 other mbtH like genes including the D522_08303 gene in another M. paratuberculosis strain (S5) originally isolated from a goat, MAP4_2610 from M. paratuberculosis MAP4 (a human isolate), MAH_2060 of M. avium TH135 and gene OCQ_31530 in M. intracellulare (strain MOTT-64).
In addition to differences in the size of genes within the mycobactin gene cluster, there were also differences in the spacing between the genes and gene clusters. The gap between mbtA and mbtJ (lipK) was comparable between M. paratuberculosis K10 and M. paratuberculosis 43525 and contained a mce operon 8.7 kbp downstream from mbtA. However, the gap between mbtJ (lipK) and mbtI (trpE2) in M. paratuberculosis 43525 was 2 kb shorter compared with the 6.6 kb spacer region in M. paratuberculosis K10 (Fig. 4).
mce genes
The M. paratuberculosis 43525 genome contained eight mce operons that encode 74 mce proteins, although not all are complete. The four best known mce operons identified in M. tuberculosis are labelled 1–4 in Table 2. Other identified mce operons include mce 5, 6, and 7. Based on these findings it is suggested to include the gene designation mce 8, an operon that was originally described as a duplicate of mce 7. However, mce 8 has low nucleotide and amino acid sequence similarity (72 and 63 %, respectively), when compared to the existing mce7 in M. paratuberculosis [26]. Table 2 shows the amino acid sequence similarity of the mce genes in M. paratuberculosis 43525 compared to equivalent genes in related bacteria of the MAC and M. tuberculosis. Of particular note is that in all M. paratuberculosis isolates included in this study, mce6 was found 8.7 kb downstream from mbtA of the mycobactin cluster.
Four genes of the mce1 operon were longer than the corresponding genes in M. paratuberculosis K10 and the same size as the corresponding genes of M. paratuberculosis MAP4 and other MAC. While the mce2 operon was conserved among M. paratuberculosis, M. avium strains 104 and TH135, the mce3R gene appeared to be missing from M. paratuberculosis 43525 and other M. paratuberculosis strains. As reported by others, the mce4a gene was highly conserved across all mycobacteria [21].
Of particular note was the finding that the conserved hypothetical integral membrane protein yrbe3B was present in M. paratuberculosis 43525 but missing in M. paratuberculosis K10. Interestingly, yrbe3B has been found in a M. paratuberculosis strain (S397) isolated from sheep, M. avium 104, M. avium TH135, M. avium ATCC25291, M. intracellulare and M. tuberculosis.
Discussion
Using comparative genomics a rare human isolate of M. paratuberculosis was compared to both animal and human pathogens of the MAC and M. tuberculosis. After broad analysis by Blast and SNP typing, this study focused on comparisons of PE/PPE genes, the mycobactin cluster and the mce operons, all of which are key virulence factors across the species examined.
When compared at the nucleotide level, M. paratuberculosis 43525 displayed a close relationship to a bovine isolate M. paratuberculosis CLIJ644 (Fig. 2). This requires further investigation, particularly as M. paratuberculosis is shed in the milk of infected cows even at the early subclinical stage and that M. paratuberculosis can survive pasteurisation [32].
As in prior work, it was found that the complement of PPE genes was variable across strains while the PE genes showed a high degree of conservation (Fig. 3 and Additional file 1) [4]. A possible human associated PPE, MACPPE43 was present in M. paratuberculosis 43525 and M. intracellulare, which was orthologous to Rv3621c (PPE65 of M. tuberculosis). In contrast, MACPPE43 was not present in any strains of animal origin, including other M. paratuberculosis isolates. In M. tuberculosis, this gene was not essential for in vitro growth but could be detected in M. tuberculosis H37Rv infected guinea pig lungs at 30 and 90 days post infection suggesting a critical function for this gene product in vivo [33, 34]. PE_PGRS11 was also found to be present in strains isolated from humans, as well as M. paratuberculosis K10 although its function too is unknown. Two PE/PPE genes, MACPPE51 and a Mav2927 orthologue, were only found in the new human isolate M. paratuberculosis 43525, M. hominissuis and a caprine isolate of M. paratuberculosis, S5. Given the significance of the PE/PPE family to virulence, through generation of antigenic variations, the functions of the PE/PPE identified here should be investigated further.
MACPPE42 is unique to M. paratuberculosis and is located on a Large Sequence Polymorphism (LSP)-14 [4, 35]. There is some evidence that LSPs are associated with the cellular immune response [36] and it is co-transcribed with the iron-regulated transporters (irt) A and B equivalents MAP3734-3735 in macrophages [35]. IrtA and B are thought to be involved in the trafficking of carboxymycobactin, which is secreted in contrast to cell wall associated mycobactin [37]. As proposed in a recent study, MACPPE42 may act as a signal transduction protein for the IrtA and B equivalents which in turn form a single ABC transporter for Fe-carboxymycobactin and iron assimilation via ferric iron reduction [35]. Structural studies demonstrating the similarity of M. tuberculosis PPE proteins to signal transduction molecules and the observation that some PE/PPE proteins are up-regulated during iron limitation and repressed by the regulator ideR, form the basis of the above proposal [38, 39]. In addition, the finding that M. tuberculosis mbtB mutants that are unable to synthesise mycobactin or carboxymycobactin, but have irtAB intact, can grow in the presence of exogenous Fe-carboxymycobactin [40], may explain how mycobactin dependent strains of M. paratuberculosis survive the hostile environment of the macrophage as well as the mycobactin independence of other strains in vitro (as long as 1 % ferric ammonium citrate is added) [41, 42]. An attenuated strain of M. paratuberculosis (strain 316FNOR1960) has lost two of the irtA and B orthologues (MAP3734c and MAP3735c) as part of the Large Variable Genomic Island-19 deletion [43]. This strain was used in early vaccine preparations and was extensively subcultured before attenuation on Dubos media with pyruvate [44]. M. paratuberculosis is usually maintained on media containing ferric ammonium citrate rather than pyruvate, as the two are antagonistic once mycobactin is added [45]. MACPPE42 was not part of the vGI-19 deletion in the attenuated strain.
M. paratuberculosis 43525 did not require additional mycobactin on Middlebrook agar, a phenotype that has been described before in M. paratuberculosis isolates from sheep [46]. Therefore the mycobactin cluster required closer scrutiny and was found to differ in its primary structure when compared to M. paratuberculosis K10.
The three NRPS domains of the mycobactin cluster were larger in M. paratuberculosis 43525 as compared to M. paratuberculosis K10 and encode larger proteins. The size of the mbtE gene varies greatly across strains mainly because the AMP binding domain is smaller in M. paratuberculosis K10 as compared to equivalent domains in M. paratuberculosis 43525 and M. avium 104. The increase in product size results in a predicted substrate of tyrosine for this domain, while there is no prediction consensus for the equivalent substrate in M. paratuberculosis K10. Like mbtB, the mbtE gene has been shown to be crucial in the biosynthesis of both mycobactin and carboxymycobactin, with disruption of this gene in M. tuberculosis resulting in the loss of mycobactin and carboxymycobactin production and a drastically reduced ability to grow on agar [47]. However, unlike the mbtB mutant, the mbtE mutant of M. tuberculosis is unable to grow on iron replete media [47, 27]. Given that iron availability in an infected macrophage is thought to fluctuate [48, 49], mutations in mbtE resulting in the loss of mycobactin and carboxymycobactin production would likely hamper the ability of the pathogen to adapt and persist in this environment.
While other NRPS domains were larger in the M. paratuberculosis 43525 mycobactin cluster, none of these resulted in different substrate predictions. Currently there is no complete consensus on the substrate for the equivalent mbtE gene in M. tuberculosis, although a recent study has obtained the soluble megasynthase components (including MbtE) by co-producing them with MbtH [28]. Although the growth requirements of M. paratuberculosis 43525 suggest that it does produce a functional mycobactin, a similar functional study is needed to confirm this hypothesis as well as determine the structure of the isolate M. paratuberculosis 43525 mycobactin and whether this differs to mycobactins produced by other M. paratuberculosis strains.
M. paratuberculosis 43525 did, however, have an additional mbtH gene compared to M. paratuberculosis K10, with orthologues of this gene present in 18 other mycobacterial strains, and all with 100 % amino acid sequence homology. The MbtH proteins are thought to play a vital role in mycobactin precursor biosynthesis [50, 28]. In vitro, the activity of NRPS adenylating enzymes is stimulated by the addition of MbtH and further they have been shown to act as activators and/or chaperones in the NRPS assembly line [51, 50]. This may explain the different mycobactin phenotype apparent in M. paratuberculosis 43525 given that several mbtH-like genes can functionally replace each other [52].
A surprising link between the mycobactin cluster and the mce operons was observed in this study. A mce operon (mce6) exists 8.7 kbp downstream from mbtA and upstream of mbtJ and mbtI. The mce are thought to be involved in transport particularly under nutrient deplete conditions and each operon can be expressed at different stages of the growth cycle in M. tuberculosis [53, 21]. The mce also have high amino acid homology with ABC transport permeases which exist 15 kbp downstream of the M. tuberculosis mycobactin cluster [35]. The significance of the close proximity of this operon to the mycobactin cluster is currently unknown, however, further work to determine if this operon is co-transcribed with the mycobactin cluster is currently underway.
Also of note, mce3R was missing from M. paratuberculosis 43525 and other M. paratuberculosis genomes, a finding that may explain why the mce3 operon appears to be non-functional in this subspecies. mce3R belongs to the TetR family and controls the expression of genes involved in β-oxidation and lipid metabolism in M. tuberculosis in vitro [54]. In M. tuberculosis, mce3 mutants have been shown to grow slower than in the wild-type, thus providing a possible explanation for the longer doubling time of M. paratuberculosis [55].
M. paratuberculosis 43525, along with sheep strains of M. paratuberculosis, M. avium 104, M. avium TH135, M. avium ATCC25291 and M. intracellulare, was found to have the yrbE3B orthologue, unlike M. paratuberculosis K10. The function of yrbE3B is largely unknown but it is thought to be the permease component of an ABC-type transport system involved in resistance to organic solvents [21]. As yet the individual functions of the mce3 genes have not been determined due to the fact that generating mutants for the genes in question has been extremely difficult [56]. The variable gene composition of the mce3 operon between MAC strains may allow further studies to be performed to elucidate these functions.
Conclusions
This study investigated human specific virulence genes of the mycobacteria and explored differences in the PE/PPE, mce and mycobactin cluster present in animal and human isolates of the MAC complex. Although M. paratuberculosis has long been thought of as the poor cousin when it comes to scavenging iron, the current study has shown for the first time the presence of unique PPE and mce genes that are possibly involved in both mycobactin and carboxymycobactin synthesis. Strains exist that appear to have only one mechanism of sequestering iron and M. paratuberculosis strains that display differing phenotypes form the basis of future functional studies designed to elucidate how pathogenic mycobacteria survive for long periods inside the host cells.
Given that the M. paratuberculosis 43525 genome is now publicly available, investigation of a range of other virulence factors present in the Mycobacteria, including mmp, the esx secretion pathway and the fatty acid synthesis genes can be undertaken which would shed further light on the ability of specific mycobacterial strains to colonise and cause disease in different tissues of different hosts.
Methods
Bacterial growth and genome sequencing
M. paratuberculosis 43525, isolated from a female with ulcerative colitis in 2009 [5], was grown on a slope of Middlebrook 7H10 agar supplemented with 10 % oleic acid-albumin-dextrose-catalase (OADC) (Difco) and 2 μg/mL mycobactin J (Allied Monitor) for 3 months. DNA was extracted as previously reported and the concentration and quality of DNA was measured using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies) [57]. The genome of M. paratuberculosis 43525 was sequenced, using an Illumina HiSeq sequencer with the TruSeq SBS v4 GA kit. Paired-end indexed libraries were prepared from purified DNA fragments of approximately 320 bp in length generating raw reads of 100 bp in length. Sequencing was performed at the Ramaciotti Centre for Gene Function Analysis, University of New South Wales (Sydney, Australia). The sequence reads were submitted to the Sequence Read Database (http://www.ncbi.nlm.nih.gov/sra) and the SRA study accession for the M. paratuberculosis 43525 genome sequence is SRP033522.
Genome assembly
Read quality was controlled by FASTQC (Babraham Bioinformatics) (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) using default values. Raw reads were filtered for quality (mean phred > 20) and trimmed 10 bp on each end using custom Perl scripts, reducing each read to 80 bp. Paired-reads were then used to estimate the genome size using the program khmerfreq (kmer = 17). The trimmed reads were then assembled using Velvet 1.0.09 [58] and SoapDenovo [59] with a range of kmer lengths (57–64) the final assembly being based on assembly size, number of contigs and contig size compared to M. paratuberculosis K10 (Accession number AE016958).
Genome analysis
Annotation of the M. paratuberculosis 43525 genome was performed using the Rapid Annotation and Subsystem Technology (RAST) web application server [60].
Proteinortho was used to conduct comparative blastp searches and clustering analyses [30, 61], which were visualised using Fripan (http://www.vicbioinformatics.com/software.fripan.shtml). Single Nucleotide Polymorphisms (SNPs) were called and used to infer phylogeny using the program CSI Phylogeny 1.0a (https://cge.cbs.dtu.dk/services/CSIPhylogeny/). The parameters for SNP calling were: Minimum depth 10x, minimum relative depth > 10 %, minimum distance between SNPs > 10 bp, minimum SNP quality = 30, read mapping quality score > 25 and minimum Z score 1.96. The phylogenetic tree was imported to FigTree (http://tree.bio.ed.ac.uk/software/figtree/) for visualisation.
Probable orthologues in M. paratuberculosis 43525 for PE, PPE and mce genes were defined using both the BLASTp algorthrim and Hmmer3 (http://hmmer.janelia.org/) [62]. Orthologues with > 70 % amino acid identity and over 50 % of the sequence length compared to public sequences of the MAC complex and M. tuberculosis were considered. Protein databases such as the PFAM database were also used for comparative purposes [63].
In order to investigate the mycobactin cluster of M. paratuberculosis 43525 the annotated genome was uploaded to Version 2.0 of the antiSMASH (Antibiotics and Secondary Metabolite Analysis SHell) program [64]. The antiSMASH algorithm identifies backbone enzymes, usually polyketide synthase (PKS), nonribosomal synthetase (NRPS), hybrid PKS-NRPS, or NRPS-like enzymes. Adjacent genes are scanned for the presence of common secondary metabolite gene domains and boundaries are predicted for each cluster. The clusters were then manually analysed and synteny of the mycobactin cluster was visually evaluated by examining whether a gene had orthologues in other mycobacterial species.
Given that the mbtE gene of M. paratuberculosis 43525 was found to be different to other mycobacterial species. PCR primers mbtE fwd (5′ gttacttccccgtcgatccc) and mbtE rev (5′ gtagtagagctcccccacca) were designed to amplify the region of mbtE that differed from the equivalent gene in M. paratuberculosis K10. Automated sequencing to identify PCR products was carried out using the PRISM BigDye™ cycle sequencing system v3.1 and ABI 3730 capillary sequencer (Applied Biosystems).
The mce and mycobactin cluster genes were compared across MAC and M. tuberculosis with emphasis on members of the MAC complex that infect animals; M. paratuberculosis K10 (bovine), M. avium ATCC 25291 (avian), M. paratuberculosis S397 (sheep) and those that infect humans; M. paratuberculosis 43525, M. avium 104, M. avium TH135. The PE/PPE genes were compared to defined PE/PPE genes from completed genomes only.
Ethics statement
Ethics approval was not required for this study. All experiments were conducted according to the regulations of the University of New South Wales.
Availability of supporting data
All supporting data for this article are included as additional files.
References
Chiodini RJ, Rossiter CA. Paratuberculosis: a potential zoonosis? Vet Clin North Am Food Anim Pract. 1996;12(2):457–67.
Autschbach F, Eisold S, Hinz U, Zinser S, Linnebacher M, Giese T, et al. High prevalence of Mycobacterium avium subspecies paratuberculosis IS900 DNA in gut tissues from individuals with Crohn’s disease. Gut. 2005;54(7):944–9.
Behr MA, Kapur V. The evidence for Mycobacterium paratuberculosis in Crohn’s disease. Curr Opin Gastroenterol. 2008;24(1):17–21.
Mackenzie N, Alexander DC, Turenne CY, Behr MA, De Buck JM. Genomic comparison of PE and PPE genes in the Mycobacterium avium complex. J Clin Microbiol. 2009;47(4):1002–11. doi:10.1128/JCM.01313-08.
Timms VJ, Gehringer MM, Mitchell HM, Daskalopoulos G, Neilan BA, editors. Isolation of Mycobacterium sp. from patients with Inflammatory Bowel Disease. 11th International Colloquium on Paratuberculosis. Sydney, Australia: Proceedings of the 11th Colloquim on Paratuberculosis; 2012.
Kirkwood CD, Wagner J, Boniface K, Vaughan J, Michalski WP, Catto-Smith AG, et al. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn’s disease. Inflamm Bowel Dis. 2009;15(11):1643–55. doi:10.1002/ibd.20967.
Naser SA, Schwartz D, Shafran I. Isolation of Mycobacterium avium subsp paratuberculosis from breast milk of Crohn’s disease patients. Am J Gastroenterol. 2000;95(4):1094–5.
Naser SA, Ghobrial G, Romero C, Valentine JF. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet. 2004;364(9439):1039–44. doi:10.1016/S0140-6736(04)17058-XS014067360417058X.
Chiodini RJ, Van Kruiningen HJ, Thayer WR, Merkal RS, Coutu JA. Possible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn’s disease. Dig Dis Sci. 1984;29(12):1073–9.
Bannantine JP, Baechler E, Zhang Q, Li L, Kapur V. Genome scale comparison of Mycobacterium avium subsp. paratuberculosis with Mycobacterium avium subsp. avium reveals potential diagnostic sequences. J Clin Microbiol. 2002;40(4):1303–10.
Semret M, Zhai G, Mostowy S, Cleto C, Alexander D, Cangelosi G, et al. Extensive genomic polymorphism within Mycobacterium avium. J Bacteriol. 2004;186(18):6332–4. doi:10.1128/JB.186.18.6332-6334.2004186/18/6332.
Wynne JW, Bull TJ, Seemann T, Bulach DM, Wagner J, Kirkwood CD, et al. Exploring the zoonotic potential of Mycobacterium avium subspecies paratuberculosis through comparative genomics. PLoS One. 2011;6(7), e22171. doi:10.1371/journal.pone.0022171PONE-D-11-06874.
Ghosh P, Hsu C, Alyamani EJ, Shehata MM, Al-Dubaib MA, Al-Naeem A, et al. Genome-wide analysis of the emerging infection with Mycobacterium avium subspecies paratuberculosis in the Arabian camels (Camelus dromedarius). PLoS One. 2012;7(2), e31947. doi:10.1371/journal.pone.0031947PONE-D-11-17590.
Hsu CY, Wu CW, Talaat AM. Genome-Wide Sequence Variation among Mycobacterium avium Subspecies paratuberculosis Isolates: a better understanding of Johne’s Disease Transmission Dynamics. Front Microbiol. 2011;2:236. doi:10.3389/fmicb.2011.00236.
Paustian ML, Amonsin A, Kapur V, Bannantine JP. Characterization of novel coding sequences specific to Mycobacterium avium subsp. paratuberculosis: implications for diagnosis of Johne’s Disease. J Clin Microbiol. 2004;42(6):2675–81.
Domenech P, Reed MB. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology. 2009;155(Pt 11):3532–43. doi:10.1099/mic.0.029199-0.
Calmette A. La Vaccination Preventive Contre la Tuberculose. Masson Paris. 1927.
Uchiya K, Takahashi H, Yagi T, Moriyama M, Inagaki T, Ichikawa K, et al. Comparative genome analysis of Mycobacterium avium revealed genetic diversity in strains that cause pulmonary and disseminated disease. PLoS One. 2013;8(8), e71831. doi:10.1371/journal.pone.0071831PONE-D-13-12459.
Sampson SL. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol. 2011;2011:497203. doi:10.1155/2011/497203.
McGuire AM, Weiner B, Park ST, Wapinski I, Raman S, Dolganov G, et al. Comparative analysis of Mycobacterium and related Actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis. BMC Genomics. 2012;13:120. doi:10.1186/1471-2164-13-120.
Zhang F, Xie JP. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol Cell Biochem. 2011;352(1-2):1–10. doi:10.1007/s11010-011-0733-5.
Whittington RJ, Begg DJ, de Silva K, Plain KM, Purdie AC. Comparative immunological and microbiological aspects of paratuberculosis as a model mycobacterial infection. Vet Immunol Immunopathol. 2012;148(1-2):29–47. doi:10.1016/j.vetimm.2011.03.003.
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393(6685):537–44. doi:10.1038/31159.
Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, et al. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci U S A. 2005;102(35):12344–9.
Arruda S, Bomfim G, Knights R, Huima-Byron T, Riley LW. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science. 1993;261(5127):1454–7.
Casali N, Riley LW. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics. 2007;8:60. doi:10.1186/1471-2164-8-60.
De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry 3rd CE. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci U S A. 2000;97(3):1252–7.
McMahon MD, Rush JS, Thomas MG. Analyses of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in Mycobacterium tuberculosis. J Bacteriol. 2012;194(11):2809–18. doi:10.1128/JB.00088-12.
Janagama HK, Senthilkumar TM, Bannantine JP, Rodriguez GM, Smith I, Paustian ML, et al. Identification and functional characterization of the iron-dependent regulator (IdeR) of Mycobacterium avium subsp. paratuberculosis. Microbiology. 2009;155(Pt 11):3683–90. doi:10.1099/mic.0.031948-0.
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi:10.1186/1471-2105-12-124.
Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5(11):631–45.
Grant IR. Mycobacterium paratuberculosis and milk. Acta Vet Scand. 2003;44(3-4):261–6.
Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7(9), e1002251. doi:10.1371/journal.ppat.1002251PPATHOGENS-D-11-00689.
Kruh NA, Troudt J, Izzo A, Prenni J, Dobos KM. Portrait of a pathogen: the Mycobacterium tuberculosis proteome in vivo. PLoS One. 2010;5(11), e13938. doi:10.1371/journal.pone.0013938.
Lamont EA, Xu WW, Sreevatsan S. Host-Mycobacterium avium subsp. paratuberculosis interactome reveals a novel iron assimilation mechanism linked to nitric oxide stress during early infection. BMC Genomics. 2013;14(1):694. doi:10.1186/1471-2164-14-694.
Deb R, Goswami PP. Expression of a gene encoding 34.9 kDa PPE antigen of Mycobacterium avium subsp. paratuberculosis in E. coli. Mol Biol Int. 2010;2010:628153. doi:10.4061/2010/628153.
Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi:10.1146/annurev.micro.54.1.88154/1/881.
Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103(21):8060–5. doi:10.1073/pnas.0602606103.
Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. ideR, An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002;70(7):3371–81.
Rodriguez GM, Smith I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J Bacteriol. 2006;188(2):424–30. doi:10.1128/JB.188.2.424-430.2006.
Farhana A, Kumar S, Rathore SS, Ghosh PC, Ehtesham NZ, Tyagi AK, et al. Mechanistic insights into a novel exporter-importer system of Mycobacterium tuberculosis unravel its role in trafficking of iron. PLoS One. 2008;3(5), e2087. doi:10.1371/journal.pone.0002087.
Ryndak MB, Wang S, Smith I, Rodriguez GM. The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J Bacteriol. 2010;192(3):861–9. doi:10.1128/JB.00223-09.
Bull TJ, Schock A, Sharp JM, Greene M, McKendrick IJ, Sales J, et al. Genomic variations associated with attenuation in Mycobacterium avium subsp. paratuberculosis vaccine strains. BMC Microbiol. 2013;13:11. doi:10.1186/1471-2180-13-11.
Saxegaard F, Fodstad FH. Control of paratuberculosis (Johne’s disease) in goats by vaccination. Vet Rec. 1985;116(16):439–41.
Merkal RS, Curran BJ. Growth and metabolic characteristics of Mycobacterium paratuberculosis. Appl Microbiol. 1974;28(2):276–9.
Aduriz JJ, Juste RA, Cortabarria N. Lack of mycobactin dependence of mycobacteria isolated on Middlebrook 7H11 from clinical cases of ovine paratuberculosis. Vet Microbiol. 1995;45(2-3):211–7.
Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, et al. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J Infect Dis. 2013;208(8):1255–65. doi:10.1093/infdis/jit250.
Pandey R, Rodriguez GM. IdeR is required for iron homeostasis and virulence in Mycobacterium tuberculosis. Mol Microbiol. 2014;91(1):98–109. doi:10.1111/mmi.12441.
Wagner D, Maser J, Lai B, Cai Z, Barry 3rd CE, Honer Zu Bentrup K, et al. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J Immunol. 2005;174(3):1491–500.
Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, et al. MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry. 2010;49(41):8815–7. doi:10.1021/bi1012854.
Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. N-acylation during glidobactin biosynthesis by the tridomain nonribosomal peptide synthetase module GlbF. Chem Biol. 2010;17(10):1077–83. doi:10.1016/j.chembiol.2010.08.007.
Lautru S, Oves-Costales D, Pernodet JL, Challis GL. MbtH-like protein-mediated cross-talk between non-ribosomal peptide antibiotic and siderophore biosynthetic pathways in Streptomyces coelicolor M145. Microbiology. 2007;153(Pt 5):1405–12. doi:10.1099/mic.0.2006/003145-0.
Kumar A, Chandolia A, Chaudhry U, Brahmachari V, Bose M. Comparison of mammalian cell entry operons of mycobacteria: in silico analysis and expression profiling. FEMS Immunol Med Microbiol. 2005;43(2):185–95. doi:10.1016/j.femsim.2004.08.013.
Santangelo MP, Blanco FC, Bianco MV, Klepp LI, Zabal O, Cataldi AA, et al. Study of the role of Mce3R on the transcription of mce genes of Mycobacterium tuberculosis. BMC Microbiol. 2008;8:38. doi:10.1186/1471-2180-8-38.
Gioffre A, Infante E, Aguilar D, Santangelo MP, Klepp L, Amadio A, et al. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 2005;7(3):325–34. doi:10.1016/j.micinf.2004.11.007.
Klepp LI, Forrellad MA, Osella AV, Blanco FC, Stella EJ, Bianco MV, et al. Impact of the deletion of the six mce operons in Mycobacterium smegmatis. Microbes Infect. 2012;14(7-8):590–9. doi:10.1016/j.micinf.2012.01.007.
Bull TJ, McMinn EJ, Sidi-Boumedine K, Skull A, Durkin D, Neild P, et al. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s disease. J Clin Microbiol. 2003;41(7):2915–23.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–9. doi:10.1101/gr.074492.107.
Tang Z, Wu H, Cort JR, Buchko GW, Zhang Y, Shao Y, et al. Constraint of DNA on functionalized graphene improves its biostability and specificity. Small. 2010;6(11):1205–9. doi:10.1002/smll.201000024.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi:10.1186/1471-2164-9-75.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi:10.1016/S0022-2836(05)80360-2.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(Web Server issue):W29–37. doi:10.1093/nar/gkr367.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–301. doi:10.1093/nar/gkr1065.
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39(Web Server issue):W339–46. doi:10.1093/nar/gkr466.
Acknowledgments
The authors thank Rati Sinha for assistance with genome assembly. This work was funded by the Australian Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VJT: conceived and planning of experiments, data collection, analysis and manuscript preparation, KH: data analysis and manuscript preparation, HMM: supervision and planning of experiments, manuscript preparation, BAN: conceived, supervision and planning of experiments, manuscript preparation. All authors read and approved the final manuscript.
Additional file
Additional file 1:
The amino acid percent homology between the PE and PPE genes of M. paratuberculosis 43525 compared to other MAC species. (XLSX 15 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Timms, V.J., Hassan, K.A., Mitchell, H.M. et al. Comparative genomics between human and animal associated subspecies of the Mycobacterium avium complex: a basis for pathogenicity. BMC Genomics 16, 695 (2015). https://doi.org/10.1186/s12864-015-1889-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1889-2