
Abstract
Background
In livestock, residual variance has been studied because of the interest to improve uniformity of production. Several studies have provided evidence that residual variance is partially under genetic control; however, few investigations have elucidated genes that control it. The aim of this study was to identify genomic regions associated with within-family residual variance of yearling weight (YW; N = 423) in Nellore bulls with high density SNP data, using different response variables. For this, solutions from double hierarchical generalized linear models (DHGLM) were used to provide the response variables, as follows: a DGHLM assuming non-null genetic correlation between mean and residual variance (rmv ≠ 0) to obtain deregressed EBV for mean (dEBVm) and residual variance (dEBVv); and a DHGLM assuming rmv = 0 to obtain two alternative response variables for residual variance, dEBVv_r0 and log-transformed variance of estimated residuals (ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \)).
Results
The dEBVm and dEBVv were highly correlated, resulting in common regions associated with mean and residual variance of YW. However, higher effects on variance than the mean showed that these regions had effects on the variance beyond scale effects. More independent association results between mean and residual variance were obtained when null rmv was assumed. While 13 and 4 single nucleotide polymorphisms (SNPs) showed a strong association (Bayes Factor > 20) with dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), respectively, only suggestive signals were found for dEBVv_r0. All overlapping 1-Mb windows among top 20 between dEBVm and dEBVv were previously associated with growth traits. The potential candidate genes for uniformity are involved in metabolism, stress, inflammatory and immune responses, mineralization, neuronal activity and bone formation.
Conclusions
It is necessary to use a strategy like assuming null rmv to obtain genomic regions associated with uniformity that are not associated with the mean. Genes involved not only in metabolism, but also stress, inflammatory and immune responses, mineralization, neuronal activity and bone formation were the most promising biological candidates for uniformity of YW. Although no clear evidence of using a specific response variable was found, we recommend consider different response variables to study uniformity to increase evidence on candidate regions and biological mechanisms behind it.
Similar content being viewed by others
Background
Uniformity is becoming increasingly important among livestock species. In meat production systems, this is more evident by the increasing adoption of economic incentives by slaughterhouses to stimulate farmers to deliver animals that meet specific carcass standards. If uniformity is partly under genetic control, genetic selection could be used to improve uniformity of animals. Genetic control of uniformity can be the result of how genotypes respond differently towards unknown micro-environmental factors [1, 2]. Such a phenomenon is called genetic heterogeneity of residual variance or genetic variance in micro-environmental sensitivity. Up to now, several studies support the existence of a genetic component on residual variance and draw attention for its potential to improve uniformity through selection (e.g. [3,4,5,6]). Unraveling the genetic basis of heterogeneity of residual variance through genome-wide association studies (GWAS) will help to understand the biology behind it and increase selection response by identifying candidate genes affecting uniformity and including them in genomic prediction.
Few GWAS for uniformity traits have been performed in livestock and there is no consensus about the most appropriate phenotype to be used in such studies. Phenotypic standard deviation and coefficient of variation was used to address uniformity of egg weight in chickens by Wolc et al. [7] and birth weight in pigs by Wang et al. [8, 9]. Residual variance per individual from double hierarchical generalized linear model (DHGLM; obtained according to Rönnegård et al. [10]) were used as response variable by Mulder et al. [11] to identify genomic regions related to residual variance of somatic cell score in dairy cattle. Estimated breeding values (EBV) from a DHGLM, using an extension developed by Felleki et al. [12], were deregressed (dEBV) and used to identify genomic regions associated with variability of litter size in pigs by Sell-Kubiak et al. [13]. Such variety of phenotypes used as response variables can be explained because uniformity can be measured in different ways depending on trait and data structure. Statistical analysis requires either the within-individual variance based on repeated observation per animal or the within-family variance based on large offspring groups per family. Furthermore, the availability of genotypes influences the choice for the response variable as well. The choice for dEBV in the study by Sell-Kubiak et al. [13] was made because of availability of genotypes on boars with many phenotyped offspring and sows with genotypes and phenotypes for litter size. In the case of Mulder et al. [11], genotypes were only available on cows with many phenotypic observations on experimental farms.
In beef cattle, growth traits are often used as selection criteria in breeding programs due to its economic impact in meat production. Several genes and genomic regions influencing the mean of these traits have been identified (e.g. [14,15,16]). In addition, the existence of a genetic component on residual variance of growth traits was previously observed [17,18,19]. However, no further investigations have been carried out to study the genetic mechanisms underlying uniformity of growth traits. Thus, the aim of this study was to identify genomic regions associated with within-family residual variance of yearling weight (YW) in Nellore cattle through GWAS, using different response variables, and candidate genes to better understand the biology behind genetic control of uniformity.
Results
Our aim was to identify genomic regions (1-Mb windows among the top 20 that explained the largest proportion of genetic variance shared between response variables and 1-Mb windows with SNPs that showed a strong association by BF) associated with within-family residual variance of YW in Nellore cattle, using different response variables in GWAS. For this, we used solutions from DHGLM assuming: i) non-null genetic correlation between mean and residual variance of YW (rmv ≠ 0) to obtain deregressed EBV for mean (dEBVm) and residual variance (dEBVv); and ii) rmv = 0 to obtain dEBVv_r0 and log-transformed variance of estimated residuals (ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \)).
The Pearson’s correlations between the different response variables and the number of common 1-Mb windows (among the top 20 that explained the largest proportion of genetic variance in the association studies) among them are shown in Table 1. As expected, dEBVm was highly correlated with dEBVv, 0.90, given the high and positive rmv (0.76; [19]). As a result, eight out of the top 20 windows were shared between dEBVm and dEBVv: chromosome (Chr) 1 (92 Mb; Chr1_92), 3 (Chr3_42 and Chr3_45), 13 (Chr13_59), 14 (Chr14_24 to 26) and 16 (Chr16_21) (Table 2). The dEBVm was also positively correlated with dEBVv_r0, 0.19, but no overlap was found among the top 20 windows.
The response variables for residual variance of YW had low to moderate correlations with each other (Table 1). The largest correlation was observed between dEBVv and dEBVv_r0 (0.53), which shared one window among the top 20, Chr12_5. On the other hand, the smallest correlation was between dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) (0.08), which also shared only one window among the top 20, Chr22_52. The dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) showed a correlation of 0.24 and had two common regions among the top 20: Chr6_105 and Chr9_8.
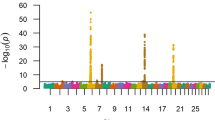
Based on the GWAS results, 40, 13 and 4 single nucleotide polymorphisms (SNPs) showed a strong association (BF > 20) with dEBVm, dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), respectively (Fig. 1 and Additional file 1). Box plots of dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) by genotype of the markers with higher BF were presented to give an overview about the signals that these SNPs are capturing (Fig. 2). In both cases, the first homozygous genotype (0) was more uniform, with lower mean and lower dispersion of the corresponding response variable, compared to the other genotypes, although AA was less frequent in relation to AB and BB. Only a few suggestive association signals (2; BF > 3) were found for dEBVv_r0. All these SNPs are located within the top 20 1-Mb windows that explained the largest proportion of genetic variance in each response variable. The top 20 windows explained together 34.6, 32.6, 4.4 and 20.0% of the genetic variance of dEBVm, dEBVv, dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) (data not shown). The remaining part of genetic variance was explained by the remaining windows, those not included in the top 20. The range of the proportion of variance explained by individual windows and their sum suggest that uniformity of YW behaves as a polygenic trait determined by several genes, as well as its mean.

Manhattan plot for the mean (dEBVm) and residual variance (dEBVv, dEBVv_r0, ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \)) of yearling weight. dEBVm and dEBVv: deregressed EBV for mean and residual variance, respectively; dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \): deregressed EBV for residual variance and log-transformed variance of estimated residuals, respectively, both obtained from solutions of a DHGLM assuming null genetic correlation between mean and residual variance. The horizontal blue and red lines represent Bayes Factor of 3 (suggestive) and 20 (strong), respectively

Box plots for the dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) according to rs133244984 and rs134778206 genotypes, respectively
Within the common windows shared by dEBVm and dEBVv, genes and QTL regions were mapped (Table 2). Several candidate genes for growth traits in beef cattle were identified on chromosome 14 (RB1CC1, NPBWR1, PLAG1 and PENK). Such findings highlight that most of the effects captured by dEBVv can be due to the strong rmv and could be potentially scale effects [19].
In order to prove that these common regions can really affect the variance beyond a simple scale effect, we applied a similar procedure as described in Wolc et al. [7]. For this, only the common SNP with the highest BF in each window was considered. The absolute effect of these SNPs on dEBVm and dEBVv were divided by the mean of YW and mean of \( {\upsigma}_{\mathrm{e}}^2 \), respectively. If SNP would have an effect on mean and variance due to scaling, then the standardized effects should be the same or higher on mean. However, all SNPs showed a greater standardized effect on dEBVv than dEBVm indicating that these regions affect the residual variance beyond a simple scale effect. These results suggest that even with a high and strong rmv it is possible to find regions determining mean growth and its variability simultaneously, while the effects on the variance are larger than can be explained by a simple scale effect.
In the shared windows by dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), Chr6_105 and Chr9_8, genes involved in metabolism (NUDT9, SPARCL1 and MAN2B2), mineralization (DMP1 and DSPP) and neuronal activity (PPP2R2C, JAKMIP1 and ADGRB3) were considered potential candidates for uniformity (Table 3). No gene was identified within Chr12_5, shared by dEBVv and dEBVv_r0. However, several promising genes were found on Chr22_52 shared by dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \); among them, genes related to metabolism (e.g. DPYD, AK7, GMPPB, AMT and GLUD1), stress (IP6K2 and UCN2) and inflammatory and immune responses (UBA7, RNF123, MST1 and RHOA) (Table 3).
For uniformity, promising genes related to metabolism: ATP6V1B2 (Chr8), LPL (Chr8), OSBPL8 (Chr5) and GLI2 (Chr2); immune response: GLIPR1 (Chr5), neuronal plasticity: LZTS1 (Chr8) and stress response: SLC18A1 (Chr8), HIF1A (Chr10), BBS10 (Chr5) and CLASP1 (Chr2) were observed in the windows of the SNPs that showed strong association with ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) (Table 4). For dEBVv, most of the genes in the regions harboring SNPs that showed strong association were related to metabolism: DPYD (Chr3), BTG1 (Chr5), AK7 (Chr21), ADIRF (Chr28), GLUD1 (Chr28), IP6K1, GMPPB, APEH, AMT, USP4, USP19, IMPDH2, NDUFAF3, SLC25A20, PRKAR2A, UQCRC1, PFKFB4 and SHISA5 all located on Chr22 (Table 5). In addition, genes involved in stress response: BDKRB2 and BDKRB1 (Chr21), and IP6K2 and UCN2 (Chr22); inflammatory and immune responses: EEA1 (Chr5), TCL1A and TCL1B (Chr21), UBA7, RNF123, MST1 and RHOA all on Chr11; neuronal plasticity: PLPPR4 and PLPPR5 (Chr3); and bone formation: BMPR1A (Chr28) were also found (Table 5). In summary, most of the regions associated with residual variance of YW harbor interesting candidate genes related to metabolism, stress, inflammatory and immune responses, mineralization, neuronal activity and bone formation.
Discussion
In this study, we identified genomic regions associated with within-family residual variance of YW in Nellore cattle by using different response variables (dEBVv, dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \)). Thirteen and four strong associations (BF > 20) were observed using dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), respectively. Overlapping windows among the top 20 that explained most of the genetic variance were observed between dEBVv and the other response variables including dEBVm. The dEBVv shared 8 out of the top 20 windows with dEBVm, one with dEBVv_r0 and another one with ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \). Potential candidate genes related to metabolism (carbohydrate, energy, lipid, nucleotide and amino acid), stress, inflammatory and immune responses, mineralization, neuronal activity and bone formation were found for ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) and dEBVv.
Common regions between dEBVm and dEBVv
All common regions shared by dEBVm and dEBVv contained previously found QTL, associated with birth, yearling and carcass weight in other breeds [15, 20,21,22]. The region found on chromosome 14 (24 to 26 Mb) was associated with growth, feed efficiency and carcass quality traits in beef cattle and several candidate genes were reported [14,15,16, 23,24,25,26,27]. According to these studies, pleiomorphic adenoma gene 1 (PLAG1) seems to be the major gene due to its role in regulating insulin-like growth factors (IGF; [28]). However, some of these genes, like RB1-inducible coiled-coil 1 (RB1CC1), neuropeptides B/W receptor 1 (NPBWR1) and proenkephalin (PENK), are also involved in processes that can contribute to determine uniformity of growth traits, like YW. RB1CC1 is involved in cell growth and differentiation, senescence, apoptosis and autophagy, and was one of the genes differentially expressed in heat stressed chickens [29, 30]. NPBWR1 modulates feeding behavior and energy homeostasis and PENK is responsible for producing enkephalins in response to stress [31, 32]. Based on these findings and the fact that these shared regions between dEBVm and dEBVv showed effects beyond scale effects, we can conclude that the same gene can affect both, mean and its variability.
Promising genes associated with ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), dEBVv and dEBVv_r0
Genes involved in metabolism (ATP6V1B2, LPL, OSBPL8 and GLI2), immune response (GLIPR1), neuronal plasticity (LZTS1) and stress response (SLC18A1, HIF1A and BBS10 and CLASP1) were considered the most promising biological candidates for uniformity using ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \). For dEBVv, genes related to metabolism (DPYD, BTG1, AK7, ADIRF, GLUD1, IP6K1, GMPPB, APEH, AMT, USP4, USP19, IMPDH2, NDUFAF3, SLC25A20, PRKAR2A, UQCRC1, PFKFB4 and SHISA5), stress response (BDKRB2, BDKRB1, IP6K2 and UCN2), inflammatory and immune responses (TCL1A, TCL1B, UBA7, RNF123, MST1 and RHOA), neuronal plasticity (PLPPR4 and PLPPR5) and bone formation (BMPR1A) were identified as potential candidates. Such findings support partially the hypothesis that most likely the mean of a trait and its uniformity share processes, like those related to metabolism. However, uniformity may be a trait which is even more complex and may be controlled by several processes and mechanisms beyond metabolism, in order to reach homeostasis and maintain the body in balance.
Among the metabolism-related genes for ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), ATPase, H+ transporting, V1 subunit B2 (ATP6V1B2) encodes a component of the vacuolar ATPase (V-ATPase), which is activated according to the energy level, like during glucose starvation [33]. Previously, the ATP6V1B2 gene was found to be associated with feed efficiency traits in beef cattle [34]. Lipoprotein lipase (LPL) and oxysterol binding protein-like 8 (OSBPL8) genes are involved in lipid metabolism. LPL is responsible for hydrolysis of circulating triglycerides and very low-density lipoprotein [35]. Associations between LPL and growth and carcass quality traits were also reported previously [36,37,38,39]. OSBPL8 modulates lipid homeostasis through sterol regulatory element binding proteins (SREBPs; [40]). In addition, glioma-associated oncogene family zinc finger 2 (GLI2) gene is a transcription factor of the Hedgehog (Hh) signaling pathway, which is involved in muscle development mainly during embryonic development, but also in adult tissue homeostasis and repair [41, 42].
For dEBVv, most of the genes are involved in metabolic pathways or related processes to it, such as metabolism of carbohydrate, energy and lipid, nucleotide and amino acid (GMPPB, UQCRC1, PFKFB4, AMT, GLUD1, AK7, IMPDH2, NDUFAF3, DPYD, SLC25A20, PRKAR2A and IP6K1). Genes regulating muscle cell proliferation, differentiation and myogenesis (BTG1 [43], USP4 and USP19 [44, 45], and SHISA5), adipogenic differentiation (ADIRF [46]) and proteolysis (APEH) also showed metabolic functions that potentially can contribute to uniformity of YW.
Genes involved in the energy and protein metabolism may play an important role to keep body homeostasis against environmental perturbations. Usually, metabolic responses to these stressors involve catabolic processes such as energy mobilization and protein degradation [47, 48]. In such situations, genes related to metabolism are required to recover homeostasis and consequently part of the energy is diverted from growth which can potentially affect performance and therefore explain the potential trade-off between energy for growth and energy for homeostasis.
Genes related to stress response were also found using ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) (SLC18A1, HIF1A, BBS10 and CLASP1) and dEBVv (BDKRB2, BDKRB1, IP6K2 and UCN2). Stress is a response to new environmental conditions, like nutrition, housing or any stimuli, that threatens homeostasis. Therefore, stress genes may have a direct effect on uniformity, such that animals that are worse in dealing with environmental perturbations tend to be less uniform (e.g. [49]).
Vesicular monoamine transporter 1 (SLC18A1) and urocortin 2 (UCN2) genes are involved in the hypothalamic-pituitary-adrenal (HPA) axis. SLC18A1 acts in the final stage of the HPA-axis, transporting catecholamines like dopamine, noradrenaline and adrenaline. Catecholamines are released as a physiological stress response [50]. UCN2 is a member of the corticotropin-releasing hormone (CRH) family, which is a key mediator of the stress response by activating the HPA axis [51, 52].
The hypoxia-inducible factor 1-alpha (HIF1A) gene encodes a transcription factor that is induced by hypoxia, promoting angiogenesis, cell proliferation/survival and glucose/iron metabolism [53]. Angiogenesis is the process of new vessel formation, which is related to heat stress. Recently, HIF1A was highly expressed in the ruminal epithelium of highly-efficient animals [54], which can be explained by its functions, mainly angiogenesis and glucose metabolism. It indicates that animals can differ in relation to absorption and energy generation, changing individual growth performance.
The Bardet-Biedl Syndrome 10 (BBS10) gene, a member of heat shock protein (HSP) family, encodes a chaperonin-like protein [55]. The HSP family plays a crucial role in response to several stressful conditions in addition to heat stress [56], and it is not the first time that a member of this family is related to uniformity. Previously, Hsp90 was considered a candidate gene for developmental stability of morphological traits in Drosophila melanogaster and Arabidopsis thaliana (e.g. [57,58,59,60]) and variability of litter size in pigs [13]. It supports the evidence that the function of chaperones is extended to several organisms, making these HSP protein genes promising candidates for uniformity.
Another gene related to HSP family is the inositol hexakisphosphate kinase 2 (IP6K2), a metabolism related-gene that also plays a role in apoptosis [61]. However, its catalytic activity is regulated by HSP90 gene [62], which suggests that cellular response can also be expected due to environmental stimuli.
Cytoplasmic linker associated protein 1 (CLASP1) and bradykinin receptor B1 and B2 (BDKRB1 and BDKRB2) genes are involved in regulation of wound healing and blood pressure regulation, mechanisms that can be used to deal with environmental stressors. Previously, Coble et al. [63] found BDKRB1 among up-regulated genes during heat stress in chickens.
Some genes involved in inflammatory and immune responses, GLIPR1, EEA1, TCL1A, TCL1B, UBA7, RNF123, MST1 and RHOA, were also found. GLIPR1, UBA7, RNF123 and EEA1 participate in several immune system-related pathways. T-cell leukemia/lymphoma 1A and 1B (TCL1A and TCL1B) genes are involved in T- and B-cell development [64]. The macrophage stimulating 1 (MST1) gene regulates macrophage activity during inflammation [65, 66]. Ras homolog family member A (RHOA) is a small GTPases of the Rho family, which regulates several cellular processes including actin cytoskeleton reorganization and inflammation [67, 68].
Four other potential candidate genes for uniformity were identified. Leucine zipper putative tumor suppressor 1 (LZTS1), phospholipid phosphatase related 4 and 5 (PLPPR4 and PLPPR5) are involved in neuronal plasticity [69, 70], and bone morphogenetic protein receptor type 1A (BMPR1A) playing an important role in bone formation, which was recently found associated with body size in sheep [71]. Neuronal plasticity is the ability of the nervous system to adapt to environmental changes, which denotes another potential class of genes determining uniformity.
In the common regions between ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) and dEBVv_r0, genes related to metabolism (NUDT9, SPARCL1 and MAN2B2) and mineralization of tissues like bones and teeth (DMP1 and DSPP [72]), are likely to be linked to uniformity on Chr6_105. It is not unusual to find common genes related to stature, height and body weight in beef cattle (e.g. [14, 73,74,75]). Such findings highlight the strong relationship between height and weight, partially explained by common regulatory mechanisms.
Additionally, both common regions (Chr6_105 and 9_8) contain genes with neuronal functions: PPP2R2C, JAKMIP1 and ADGRB3. Protein phosphatase 2, regulatory subunit B, gamma (PPP2R2C) and adhesion G protein-coupled receptor B3 (ADGRB3) were previously related to learning and memory (synaptic plasticity) and synaptogenesis in mice [76, 77]. Janus kinase and microtubule-interacting protein 1 (JAKMIP1) was associated with social behavior in mice through its function on neuronal translation and was related to T cell-mediated cytotoxicity [78, 79]. These results together with dEBVv and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) provide evidence that the nervous system is also modulated according to environmental perturbations. Previously, studies reporting such neuronal plasticity in response to changes in nutrition and temperature were observed in Drosophila [80,81,82]. Therefore, together with other tissues, the nervous system seems to be a potential modulator of the mechanisms underlying uniformity.
Response variables
The high correlation between dEBVm and dEBVv resulted in 8 common regions affecting simultaneously mean and residual variance. Such findings are in line with Wolc et al. [7] who found correlations ranging from 0.54 to 0.74 and a region explaining a large proportion of the genetic variance of the mean and standard deviation (SD) of egg weight at different ages. The authors noted that it was not a simple scale effect, because the effect of the QTL on mean egg weight (measured between 26 and 28 weeks of age) was about 4% of the mean egg weight and on SD egg weight at the same age was about 5% of the mean SD. More recently, Sell-Kubiak et al. [13] also reported a high genetic correlation, 0.49, between litter size and its variation, but no overlapped regions were found.
The dEBVv not only captured common effects with the mean of YW but also its residual variance by sharing regions (among top 20 by variance explained) with dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), Chr12_5 and Chr22_52, respectively. These results suggest that the mean and uniformity of YW are partially under different genetic control. Though some of the genes found on chromosome 22 and most of those in common between dEBVm and dEBVv are involved in metabolism (e.g. PLAG1, DPYD, AK7, GMPPB, AMT and GLUD1), a process most probably related to both, mean and uniformity.
The weak correlations among dEBVm and the response variables that assumed null rmv emphasize the role of the latter to find genomic regions that are specifically affecting residual variance and not the mean of YW. Generally, data transformation is used as the main tool to reduce mean-variance relationship in studies on uniformity (e.g. [83,84,85]). This is the first study on uniformity reporting null rmv as a strategy to deal with scale effects and showing the differences in terms of associated regions when comparing to GWAS using dEBVv when non-null rmv was assumed.
Knowing the importance of considering potential scale effects and confounding between mean and residual variance, the next step is to discuss the suitability of the different response variables to be used in a GWAS for uniformity. Based on our results, we recommend considering the following aspects regarding the response variable and data structure. Firstly, we need to take into account the nature of the trait because small genetic variance and low heritabilities are often found for uniformity. Previously we estimated a heritability for residual variance of YW even smaller (0.007; [19]) than has been reported for similar traits in other livestock populations [86]. Probably this also may have contributed to the small (or none) number of SNPs that reached the significance threshold (BF > 20) when we analyzed the response variables for residual variance. In this context, irrespective of the response variable, a larger sample size is required to increase the power of GWAS [87].
Secondly, we used two different procedures to obtain response variables to perform GWAS for uniformity. The first one was the deregressed EBV; deregression, which removes both the contribution of parents and the shrinkage present in the EBV. On the other hand, the second one resulted in a measure, ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), with less adjustments compared to deregression. However, some non-genetic effects, such as the contemporary group (CG) effect on the variance may have affected the within-family variance estimate ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), while they are accounted for when we used dEBV since non-genetic effects, were fitted in the DHGLM in the residual variance part of the model. In addition, the assumptions of null and non-null rmv were assumed to measure the influence of the mean and/or potential scale effects on the residual variance. Therefore, analyzing each response variable, we can conclude that: i) dEBVv was greatly affected by the mean; ii) dEBVv_r0 had low accuracy by assuming null rmv, which requires a large amount of phenotypic information and/or genotyped sires; and iii) ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) was less related to the mean and less regressed compared to dEBV, but it may include some non-genetic effects, like CG. It is clear that all three approaches have advantages and disadvantages. Given the fact that ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) is likely more independent to the mean than dEBVv, there may be in this case a small preference for this response variable as it behaved also better in the Markov chain Monte Carlo (MCMC). Furthermore, considering different response variables can be beneficial, windows that appear in multiple approaches may have a higher credibility and may help in understanding the role of genes affecting the mean, the residual variance or both and whether genes controlling the variance beyond scale effects.
Conclusions
Using solutions from a DHGLM that assumed null genetic correlation between mean and residual variance was a suitable strategy to identify genomic regions affecting uniformity less dependent on the mean. Thirteen and four SNPs were associated with dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), respectively, and additional common regions were observed among the responses variables. In all these regions, promising biological candidate genes for uniformity of YW were related not only with metabolic genes, but also stress, inflammatory and immune responses, mineralization, neuronal activity and bone formation. No strong evidence was found in favor of using a specific response variable for GWAS, while using different response variables was beneficial from the biological point of view, since more evidence on candidate regions can be found.
Methods
The phenotypic data used in this study are described in Iung et al. [19]. Here, genotypic information from 423 influential Nellore sires from the Alliance Nellore database with a large number of evaluated progeny were available. The number of progeny per sire considered for the study ranged from 50 to 10,180, with an average of 411 (see distribution of progeny per sire in Additional file 2). In total, 194,628 yearling weight measurements from the progeny were used to estimate the response variables used in the GWAS.
Response variables in the association studies
All responses variables used solutions or residuals from DHGLM fitted in our previous study [19]. The DHGLM is an iterative approach that estimates simultaneously genetic parameters for the mean and residual variance. In our previous study a sire DHGLM was used as follows: \( \left[\begin{array}{c}\mathbf{y}\\ {}\boldsymbol{\uppsi} \end{array}\right]=\left[\begin{array}{cc}\mathbf{X}& \mathbf{0}\\ {}\mathbf{0}& {\mathbf{X}}_{\mathbf{v}}\end{array}\right]\left[\begin{array}{c}\mathbf{b}\\ {}{\mathbf{b}}_{\mathbf{v}}\end{array}\right]+\left[\begin{array}{cc}\mathbf{Z}& \mathbf{0}\\ {}\mathbf{0}& {\mathbf{Z}}_{\mathbf{v}}\end{array}\right]\left[\begin{array}{c}\mathbf{s}\\ {}{\mathbf{s}}_{\mathbf{v}}\end{array}\right]+\left[\begin{array}{c}\mathbf{e}\\ {}{\mathbf{e}}_{\mathbf{v}}\end{array}\right] \), where y and ψ are vectors of response variables for the mean and residual variance models (denoted with the subscript v), respectively, b and bv are vectors of fixed effects and covariates (contemporary group, linear and quadratic effects of animal age, nested within sex), s and sv are vectors of random sire genetic effects, e and ev are vectors of random residual effects, X, Xv, Z and Zv are corresponding incidence matrices. This approach was chosen because of its better predictability compared to a two-step approach, as previously stated by Iung et al. [19]. Mean of YW was evaluated through dEBVm obtained following the deregression procedure proposed by Garrick et al. [88] and using EBV of the mean from DHGLM [12]. For the residual variance of YW, different response variables were analyzed: i) dEBVv: deregressed EBV of the residual variance from a DHGLM assuming non-null genetic correlation between mean and residual variance (rmv ≠ 0); ii) dEBVv_r0: deregressed EBV of the residual variance from a DHGLM assuming rmv = 0; and iii) ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \), log-transformed variance of estimated residuals for each sire family, using residuals from the mean model of a DHGLM also assuming rmv = 0. The log-transformation was used to reduce mean-variance relationship and correct for non-normality of the residual variances. Fitting the DHGLM assuming null rmv was intended to reduce the impact of the mean of YW may have on the EBV in the residual variance part of the model, given the strong estimate of rmv on YW previously reported for this population [19]. For instance, the EBVv and EBVm would be even higher correlated than the rmv because of using the information on mean YW for both sets of EBV. When using null rmv, it is expected to have a higher possibility to find regions only affecting the residual variance. Descriptive statistics of each response variable are presented in Table 6 and the distribution of them in Additional file 2.
Genotypes
Genotypic information from 423 Nellore bulls was used, being 415 genotyped with the Illumina® BovineHD chip (HD) and 8 with Illumina BovineSNP50 BeadChip (Illumina Inc., San Diego, CA, USA) and then imputed to HD (777 k) using FImpute software [89]. In the quality control of genotypes, SNPs located on the sex chromosomes, with minor allele frequency (MAF) lower than 0.02, p-value for Hardy-Weinberg equilibrium test (HWE) less than 10− 5 and highly correlated SNPs (r2 > 0.99) within a window of 100 consecutive SNPs were removed, so that 333,877 SNPs remained for analysis. All genotyped bulls had a call rate higher than 0.90, passing the quality control.
GWAS
The SNP effects were estimated using Bayes C method [90], a mixture model which assumes that there is a smaller fraction of SNPs (1-π) with large effects and a large proportion (π) with zero or near zero effects on the trait, as follows:
where y is the vector of response variables (dEBVm, dEBVv, dEBVv_r0 and ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \)), μ is the overall mean, 1 is a vector of ones, zi is the vector of genotypes of the animals for the ith SNP, ai is the allele substitution effect of the ith SNP, δi is an indicator variable set to 1 if the ith SNP has an non-zero effect on the trait and to 0 otherwise, e is the vector of random residual effects and N is the number of SNPs. It was assumed ai ~ N(0,\( {\upsigma}_{\mathrm{a}}^2 \)) and e ~ N(0, R\( {\upsigma}_{\mathrm{e}}^2 \)), where \( {\upsigma}_{\mathrm{a}}^2 \) is the variance of SNP effects, \( {\upsigma}_{\mathrm{e}}^2 \) is the residual variance and R is a diagonal matrix whose elements account for heterogeneous residual variance across observations. When dEBV was the response variable, the diagonal elements of R were derived following Garrick et al. [88], whereas when ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) was the response, such elements were equal to the reciprocal of the number of progeny of each sire. The R-accounted for differences in sampling variance due to different numbers of progeny per sire, even observing low correlations between the number of progeny per sire and each response variable (0.021 to 0.145). In addition, scaled inverse chi-squared prior distributions with v degrees of freedom and scale parameter S were assumed for \( {\upsigma}_{\mathrm{a}}^2 \) and \( {\upsigma}_{\mathrm{e}}^2 \). In our study, π was fixed at 0.999, which means that 0.1% of the SNPs fitted in the model, i.e. about 334, have an effect on the trait. Such a criterion was assumed aiming to obtain stronger signals of candidate QTL and also due to the limiting number of genotypes.
The analyses were performed using the GS3 software [91]. Chains of 550,000 and 250,000 iterations, after discarding the first 150,000 and 50,000 as burn-in, were generated for analyses pertaining to the three response variables for residual variance and mean of YW, respectively, saving samples every 50 iterations in both cases. Convergence was assessed through Geweke test [92] using CODA R package [93].
The significance of each association was evaluated through the Bayes Factor (BF):
where p and π are the posterior and prior probability of a SNP being included in the model, i.e. having a non-zero effect, respectively. Suggestive and strong evidences were assigned to SNPs with BF greater than 3 and 20, respectively [94, 95]. In addition, the proportion of genetic variance explained by SNPs within non-overlapping 1-Mb windows were calculated. A window size of 1-Mb was chosen since SNPs within this distance present moderate to high (> 0.2) pairwise linkage disequilibrium (LD; measured by r2) (see Additional file 3). In total, SNPs were allocated to 2522 windows, with an average (SD) of 132 (42) SNPs. The top 20 windows that explained the largest proportion of genetic variance were compared between responses variables.
Functional annotation
The shared 1-Mb windows (among the top 20 that explained the largest proportion of genetic variance) between response variables and windows with SNPs that showed a strong association (BF > 20) were screened to identify potential candidate genes using Ensembl Genome Browser (http://www.ensembl.org), UMD3.1 bovine genome assembly [96]. Previously reported QTL regions overlapping such windows were identified from Cattle QTLdb [97]. Functional enrichment based on Gene Ontology (GO) terms and pathway analysis using Blast2GO PRO and Reactome [98, 99], respectively, was performed on these regions in order to help us to determine potential candidate genes for uniformity. Only the promising candidate genes, according to their function, pathway or related biological process, were discussed here (the complete gene list and annotations are shown in Additional file 1). The LD maps within each 1-Mb window discussed here were obtained using the R package LDheatmap [100] (Additional file 3).
Abbreviations
- BF:
-
Bayes factor
- Chr:
-
Chromosome
- dEBV:
-
Deregressed EBV
- DHGLM:
-
Double hierarchical generalized linear model
- EBV:
-
Estimated breeding value
- GWAS:
-
Genome-wide association study
- HWE:
-
Hardy-Weinberg equilibrium
- ln_\( {\upsigma}_{\widehat{\mathrm{e}}}^2 \) :
-
log-transformed variance of estimated residuals
- MAF:
-
Minor allele frequency
- Mb:
-
Mega bases
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- YW:
-
Yearling weight
References
Falconer DS, Mackay TFC. Introduction to quantitative genetics. Pearson: Essex; 1996.
Mulder HA, Rönnegård L, Fikse WF, Veerkamp RF, Strandberg E. Estimation of genetic variance for macro- and micro-environmental sensitivity using double hierarchical generalized linear models. Genet Sel Evol. 2013;45:23.
Vandenplas J, Bastin C, Gengler N, Mulder HA. Genetic variance in micro-environmental sensitivity for milk and milk quality in Walloon Holstein cattle. J Dairy Sci. 2013;96:5977–90.
Sell-Kubiak E, Bijma P, Knol EF, Mulder HA. Comparison of methods to study uniformity of traits: application to birth weight in pigs. J Anim Sci. 2015;93:900–11.
Marjanovic J, Mulder HA, Khaw HL, Bijma P. Genetic parameters for uniformity of harvest weight and body size traits in the GIFT strain of Nile tilapia. Genet Sel Evol. 2016;48:41.
Mulder HA, Visscher J, Fablet J. Estimating the purebred-crossbred genetic correlation for uniformity of eggshell color in laying hens. Genet Sel Evol. 2016;48:39.
Wolc A, Arango J, Settar P, Fulton JE, O’Sullivan NP, Preisinger R, et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim Genet. 2012;43:87–96.
Wang X, Liu X, Deng D, Yu M, Li X. Genetic determinants of pig birth weight variability. BMC Genet. 2016;17:41.
Wang Y, Ding X, Tan Z, Ning C, Xing K, Yang T, et al. Genome-wide association study of piglet uniformity and farrowing interval. Front Genet. 2017;8:1–9.
Rönnegård L, Felleki M, Fikse F, Mulder HA, Strandberg E. Genetic heterogeneity of residual variance - estimation of variance components using double hierarchical generalized linear models. Genet Sel Evol. 2010;42:8.
Mulder HA, Crump RE, Calus MPL, Veerkamp RF. Unraveling the genetic architecture of environmental variance of somatic cell score using high-density single nucleotide polymorphism and cow data from experimental farms. J Dairy Sci. 2013;96:7306–17.
Felleki M, Lee D, Lee Y, Gilmour AR, Rönnegård L. Estimation of breeding values for mean and dispersion, their variance and correlation using double hierarchical generalized linear models. Genet Res. 2012;94:307–17.
Sell-Kubiak E, Duijvesteijn N, Lopes MS, Janss LLG, Knol EF, Bijma P, et al. Genome-wide association study reveals novel loci for litter size and its variability in a large white pig population. BMC Genomics. 2015;16:1049.
Utsunomiya YT, do Carmo AS, Carvalheiro R, HHR N, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442.
Seabury CM, Oldeschulte DL, Saatchi M, Beever JE, Decker JE, Halley YA, et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genomics. 2017;18:386.
Neves HHR, Carvalheiro R, Roso VM, Queiroz SA. Genetic variability of residual variance of production traits in Nellore beef cattle. Livest Sci. 2011;142:164–9.
Neves HHR, Carvalheiro R, Queiroz SA. Genetic and environmental heterogeneity of residual variance of weight traits in Nellore beef cattle. Genet Sel Evol. 2012;44:19.
Iung LHS, Neves HHR, Mulder HA, Carvalheiro R. Genetic control of residual variance of yearling weight in Nellore beef cattle. J Anim Sci. 2017;95:1425–33.
Casas E, Shackelford SD, Keele JW, Koohmaraie M, Smith TPL, Stone RT. Detection of quantitative trait loci for growth and carcass composition in cattle. J Anim Sci. 2003;81:2976–83.
McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607.
Casas E, Keele JW, Shackelford SD, Koohmaraie M, Stone RT. Identification of quantitative trait loci for growth and carcass composition in cattle. Anim Genet. 2004;35:2–6.
Lindholm-Perry AK, Kuehn LA, Smith TPL, Ferrell CL, Jenkins TG, Freetly HC, et al. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle. Anim Genet. 2012;43:216–9.
Júnior GAF, Costa RB, de Camargo GMF, Carvalheiro R, Rosa GJM, Baldi F, et al. Genome scan for postmortem carcass traits in Nellore cattle. J Anim Sci. 2016;94:4087–95.
Magalhães AFB, de Camargo GMF, Fernandes GA, Gordo DGM, Tonussi RL, Costa RB, et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS One. 2016;11:e0157845.
Silva RMO, Stafuzza NB, Fragomeni BO, de Camargo GMF, Ceacero TM, Cyrillo JNSG, et al. Genome-wide association study for carcass traits in an experimental nelore cattle population. PLoS One. 2017;12:e0169860.
Ramayo-Caldas Y, Fortes MRS, Hudson NJ, Porto-Neto LR, Bolormaa S, Barendse W, et al. A marker-derived gene network reveals the regulatory role of PPARGC1A, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J Anim Sci. 2014;92:2832–45.
Voz ML, Agten NS, Van de Ven WJM, Kas K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000;60:106–13.
Nishimura I, Chano T, Kita H, Matsusue Y, Okabe H. RB1CC1 protein suppresses type II collagen synthesis in chondrocytes and causes dwarfism. J Biol Chem. 2011;286:43925–32.
Luo QB, Song XY, Ji CL, Zhang XQ, Zhang DX. Exploring the molecular mechanism of acute heat stress exposure in broiler chickens using gene expression profiling. Gene. 2014;546:200–5.
Li H, Kentish SJ, Wittert GA, Page AJ. The role of neuropeptide W in energy homeostasis. Acta Physiol. 2018;222:e12884.
Bali A, Randhawa PK, Jaggi AS. Stress and opioids: role of opioids in modulating stress-related behavior and effect of stress on morphine conditioned place preference. Neurosci Biobehav Rev. 2015;51:138–50.
Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, et al. The lysosomal v-ATPase-ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526–40.
Abo-Ismail MK, Kelly MJ, Squires EJ, Swanson KC, Bauck S, Miller SP. Identification of single nucleotide polymorphisms in genes involved in digestive and metabolic processes associated with feed efficiency and performance traits in beef cattle. J Anim Sci. 2013;91:2512–29.
Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl). 2002;80:753–69.
Wang XP, Luoreng ZM, Li F, Wang JR, Li N, Li SH. Genetic polymorphisms of lipoprotein lipase gene and their associations with growth traits in Xiangxi cattle. Mol Biol Rep. 2012;39:10331–8.
Oh D, La B, Lee Y, Byun Y, Lee J, Yeo G, et al. Identification of novel single nucleotide polymorphisms (SNPs) of the lipoprotein lipase (LPL) gene associated with fatty acid composition in Korean cattle. Mol Biol Rep. 2013;40:3155–63.
Ding XZ, Liang CN, Guo X, Xing CF, Bao PJ, Chu M, et al. A novel single nucleotide polymorphism in exon 7 of LPL gene and its association with carcass traits and visceral fat deposition in yak (Bos grunniens) steers. Mol Biol Rep. 2012;39:669–73.
Gui L, Jia C, Zhang Y, Zhao C, Zan L. Association studies on the bovine lipoprotein lipase gene polymorphism with growth and carcass quality traits in Qinchuan cattle. Mol Cell Probes. 2016;30:61–5.
Zhou T, Li S, Zhong W, Vihervaara T, Béaslas O, Perttilä J, et al. Osbp-related protein 8 (ORP8) regulates plasma and liver tissue lipid levels and interacts with the nucleoporin Nup62. PLoS One. 2011;6:e21078.
Straface G, Aprahamian T, Flex A, Gaetani E, Biscetti F, Smith RC, et al. Sonic hedgehog regulates angiogenesis and myogenesis during post-natal skeletal muscle regeneration. J Cell Mol Med. 2009;13:2424–35.
Petrova R, Joyner AL. Roles for hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141:3445–57.
Rodier A, Rochard P, Berthet C, Rouault JP, Casas F, Daury L, et al. Identification of functional domains involved in BTG1 cell localization. Oncogene. 2001;20:2691–703.
Il YS, Kim KK. Ubiquitin-specific protease 4 (USP4) suppresses myoblast differentiation by down regulating MyoD activity in a catalytic-independent manner. Cell Signal. 2017;35:48–60.
Wing SS. Deubiquitinating enzymes in skeletal muscle atrophy - an essential role for USP19. Int J Biochem Cell Biol. 2016;79:462–8.
Ni Y, Ji C, Wang B, Qiu J, Wang J, Guo X. A novel pro-adipogenesis factor abundant in adipose tissues and over-expressed in obesity acts upstream of PPARγ and C/EBPα. J Bioenerg Biomembr. 2013;45:219–28.
Brockman RP, Laarveld B. Hormonal regulation of metabolism in ruminants; a review. Livest Prod Sci. 1986;14:313–34.
Belhadj Slimen I, Najar T, Ghram A, Abdrrabba M. Heat stress effects on livestock: molecular, cellular and metabolic aspects, a review. J Anim Physiol Anim Nutr. 2016;100:401–12.
Blackshaw JK, Blackshaw AW. Heat stress in cattle and the effect of shade on production and behaviour: a review. Aust J Exp Agric. 1994;34:285–95.
Möstl E, Palme R. Hormones as indicators of stress. Domest Anim Endocrinol. 2002;23:67–74.
Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–7.
Bale TL, Vale WW. CRF AND CRF R ECEPTORS: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–57.
Lee J-W, Bae S-H, Jeong J-W, Kim S-H, Kim K-W. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med. 2004;36:1–12.
Elolimy AA, McCann JC, Shike DW, Loor JJ. 443 Residual feed intake in beef cattle and its association with ruminal epithelium gene expression. J Anim Sci. 2017;95:217–8.
Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–11.
Sørensen JG, Kristensen TN, Loeschcke V. The evolutionary and ecological role of heat shock proteins. Ecol Lett. 2003;6:1025–37.
Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–24.
Rutherford S, Hirate Y, Swalla BJ. The Hsp90 capacitor, developmental remodeling, and evolution: the robustness of gene networks and the curious Evolvability of metamorphosis. Crit Rev Biochem Mol Biol. 2007;42:355–72.
Sangster TA, Bahrami A, Wilczek A, Watanabe E, Schellenberg K, McLellan C, et al. Phenotypic diversity and altered environmental plasticity in Arabidopsis thaliana with reduced Hsp90 levels. PLoS One. 2007;2:e648.
Sangster TA, Salathia N, Undurraga S, Milo R, Schellenberg K, Lindquist S, et al. HSP90 affects the expression of genetic variation and developmental stability in quantitative traits. Proc Natl Acad Sci U S A. 2008;105:2963–8.
Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol Hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-β in ovarian carcinoma cells. J Biol Chem. 2001;276:24965–70.
Chakraborty A, Koldobskiy MA, Sixt KM, Juluri KR, Mustafa AK, Snowman AM, et al. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2008;105:1134–9.
Coble DJ, Fleming D, Persia ME, Ashwell CM, Rothschild MF, Schmidt CJ, et al. RNA-seq analysis of broiler liver transcriptome reveals novel responses to high ambient temperature. BMC Genomics. 2014;15:1–12.
Kang SM, Narducci MG, Lazzeri C, Mongiovì AM, Caprini E, Bresin A, et al. Impaired T- and B-cell development in Tcl1-deficient mice. Blood. 2005;105:1288–94.
Leonard JE, Johnson DE, Felsen RB, Tanney LE, Royston I, Dillman RO. Establishment of a human B-cell tumor in athymic mice. Cancer Res. 1987;47:2899–902.
Skeel A, Leonard EJ. Action and target cell specificity of human macrophage-stimulating protein (MSP). J Immunol. 1994;152:4618–23.
Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14.
Singh R, Wang B, Shirvaikar A, Khan S, Kamat S, Schelling JR, et al. The IL-1 receptor and rho directly associate to drive cell activation in inflammation. J Clin Invest. 1999;103:1561–70.
Konno D. The postsynaptic density and dendritic raft localization of PSD-Zip70, which contains an N-myristoylation sequence and leucine-zipper motifs. J Cell Sci. 2002;115:4695–706.
Savaskan NE, Bräuer AU, Nitsch R. Molecular cloning and expression regulation of PRG-3, a new member of the plasticity-related gene family. Eur J Neurosci. 2004;19:212–20.
Kominakis A, Hager-Theodorides AL, Zoidis E, Saridaki A, Antonakos G, Tsiamis G. Combined GWAS and “guilt by association”-based prioritization analysis identifies functional candidate genes for body size in sheep. Genet Sel Evol BioMed Central. 2017;49:1–16.
Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res. 2003;44(Suppl1):33–40.
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, et al. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese black cattle. BMC Genet. 2012;13:40.
Cao XK, Zhan ZY, Huang YZ, Lan XY, Lei CZ, Qi XL, et al. Variants and haplotypes within MEF2C gene influence stature of chinese native cattle including body dimensions and weight. Livest Sci. 2016;185:106–9.
Han YJ, Chen Y, Liu Y, Liu XL. Sequence variants of the LCORL gene and its association with growth and carcass traits in Qinchuan cattle in China. J Genet. 2017;96:9–17.
Backx L, Vermeesch J, Pijkels E, de Ravel T, Seuntjens E, Van Esch H. PPP2R2C, a gene disrupted in autosomal dominant intellectual disability. Eur J Med Genet. 2010;53:239–43.
Sigoillot SM, Iyer K, Binda F, González-Calvo I, Talleur M, Vodjdani G, et al. The secreted protein C1QL1 and its receptor BAI3 control the synaptic connectivity of excitatory inputs converging on cerebellar purkinje cells. Cell Rep. 2015;10:820–32.
Penney J, Tsai LH. JAKMIP1: translating the message for social behavior. Neuron. 2015;88:1070–2.
Libri V, Schulte D, van Stijn A, Ragimbeau J, Rogge L, Pellegrini S. Jakmip1 is expressed upon T cell differentiation and has an inhibitory function in cytotoxic T lymphocytes. J Immunol. 2008;181:5847–56.
Lin S, Marin EC, Yang CP, Kao CF, Apenteng BA, Huang Y, et al. Extremes of lineage plasticity in the drosophila brain. Curr Biol. 2013;23:1908–13.
Lanet E, Gould AP, Maurange C. Protection of neuronal diversity at the expense of neuronal numbers during nutrient restriction in the Drosophila visual system. Cell Rep. 2013;3:587–94.
Mellert DJ, Williamson WR, Shirangi TR, Card GM, Truman JW. Genetic and environmental control of neurodevelopmental robustness in Drosophila. PLoS One. 2016;11:e0155957.
Yang Y, Christensen OF, Sorensen D. Analysis of a genetically structured variance heterogeneity model using the box–cox transformation. Genet Res. 2011;93:33–46.
Sonesson AK, Ødegård J, Rönnegård L. Genetic heterogeneity of within-family variance of body weight in Atlantic salmon (Salmo salar). Genet Sel Evol. 2013;45:41.
Sae-Lim P, Kause A, Janhunen M, Vehviläinen H, Koskinen H, Gjerde B, et al. Genetic (co) variance of rainbow trout (Oncorhynchus mykiss) body weight and its uniformity across production environments. Genet Sel Evol. 2015;47:46.
Hill WG, Mulder HA. Genetic analysis of environmental variation. Genet Res. 2010;92:381–95.
Spencer CCA, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477.
Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:55.
Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478.
Habier D, Fernando RL, Kizilkaya K, Garrick DJ. Extension of the bayesian alphabet for genomic selection. BMC Bioinformatics. 2011;12:186.
Legarra A, Ricard A, Filangi O. GS3: Genomic Selection, Gibbs Sampling, Gauss Seidel (and BayesCπ). 2014; http://snp.toulouse.inra.fr/~alegarra/manualgs3_last.pdf.
Geweke J. Evaluating the accuracy of sampling-based approaches to the calculation of posterior moments. Bayesian Stat 4. 1992;8:169–93.
Plummer M, Best N, Cowles K, Vines K. CODA: convergence diagnostics and output analysis for MCMC. R News. 2006;6:7–11.
Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773.
Vidal O, Noguera JL, Amills M, Varona L, Gil M, Jiménez N, et al. Identification of carcass and meat quality quantitative trait loci in a landrace pig population selected for growth and leanness. J Anim Sci. 2005;83:293–300.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, et al. A whole-genome assembly of the domestic cow. Bos taurus Genome Biol. 2009;10:R42.
Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:871–9.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Croft D, Mundo A, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42:D472–7.
Shin J-H, Blay S, McNeney B, Graham J. LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J Stat Softw. 2006;16:1–10.
Sikora KM, Magee DA, Berkowicz EW, Berry DP, Howard DJ, Mullen MP, et al. DNA sequence polymorphisms within the bovine guanine nucleotide-binding protein Gs subunit alpha (Gsα)-encoding (GNAS) genomic imprinting domain are associated with performance traits. BMC Genet. 2011;12:4.
Acknowledgements
We thank GenSys for providing phenotypic and genotypic data. Tom Berghof and Camila Urbano Braz are gratefully acknowledged for their help with the functional analysis and determination of potential candidate genes.
Funding
National Council for Scientific and Technological Development (CNPq; grant number: 308636/2014–7) and Coordination for the Improvement of Higher Education Personnel (CAPES, PDSE: 88881.131673/2016–01) for providing fellowships to LHSI.
Availability of data and materials
All the data supporting the results of this article are included within the article. The phenotypic and genotypic data cannot be shared because of being part of commercial breeding programs.
Author information
Authors and Affiliations
Contributions
RC conceived the study. RC and HAM led the coordination of the study. LHSI, HAM, HHRN and RC designed the study. LHSI and HHRN were responsible for phenotypic and genotypic data edition and statistical analysis. LHSI led the manuscript preparation. All authors interpreted the results, contributed to edit the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval
Animal Care and Use Committee approval was not required for this study because the data were obtained from an existing database. No permissions were required for the use of the samples in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Complete list of the genes located within 1-Mb windows harboring SNPs with strong association by trait. Gene Ontology (GO) terms and pathways of each gene were obtained using Blast2GO and Reactome. (XLSX 99 kb)
Additional file 2:
Distribution of number of progeny per sire and responses variables. (DOCX 242 kb)
Additional file 3:
Manhattan plot and linkage disequilibrium (LD) map in each 1-Mb window discussed in our study. (PDF 419 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Iung, L.H.S., Mulder, H.A., Neves, H.H.R. et al. Genomic regions underlying uniformity of yearling weight in Nellore cattle evaluated under different response variables. BMC Genomics 19, 619 (2018). https://doi.org/10.1186/s12864-018-5003-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5003-4