
Abstract
Background
The availability of high-density SNP assays including the BovineSNP50 (50 K) enables the identification of novel quantitative trait loci (QTL) and improvement of the resolution of the locations of previously mapped QTL. We performed a series of genome-wide association studies (GWAS) using 50 K genotypes scored in 18,274 animals from 10 US beef cattle breeds with observations for twelve body weights, calving ease and carcass traits.
Results
A total of 159 large-effects QTL (defined as 1-Mb genome windows explaining more than 1% of additive genetic variance) were identified. In general, more QTL were identified in analyses with bigger sample sizes. Four large-effect pleiotropic or closely linked QTLs located on BTA6 at 37–42 Mb (primarily at 38 Mb), on BTA7 at 93 Mb, on BTA14 at 23–26 Mb (primarily at 25 Mb) and on BTA20 at 4 Mb were identified in more than one breed. Several breed-specific large-effect pleiotropic or closely linked QTL were also identified. Some identified QTL regions harbor genes known to have large effects on a variety of traits in cattle such as PLAG1 and MSTN and others harbor promising candidate genes including NCAPG, ARRDC3, ERGIC1, SH3PXD2B, HMGA2, MSRB3, LEMD3, TIGAR, SEPT7, and KIRREL3. Gene ontology analysis revealed that genes involved in ossification and in adipose tissue development were over-represented in the identified pleiotropic QTL. Also, the MAPK signaling pathway was identified as a common pathway affected by the genes located near the pleiotropic QTL.
Conclusions
This largest GWAS ever performed in beef cattle, led us to discover several novel across-breed and breed-specific large-effect pleiotropic QTL that cumulatively account for a significant percentage of additive genetic variance (e.g. more than a third of additive genetic variance of birth and mature weights; and calving ease direct in Hereford). These results will improve our understanding of the biology of growth and body composition in cattle.
Similar content being viewed by others

Background
Developments in molecular biology and statistical methodologies have made it possible for the dissection of genetic variation and the localization of economically important quantitative trait loci (QTL) in farm animals [1]. Many QTL associated with economically important traits in cattle have been reported and deposited in the AnimalQTLdb (http://www.animalgenome.org). However, not all of the genetic variation in these traits has been captured because of inadequate sample size and the low density of markers historically used in QTL mapping studies. Consequently, confidence intervals obtained for most reported QTL are too large (regions extending over several megabases (Mb)) to allow the identification of appropriate positional candidate gene(s) for the majority of QTL. Recent availability of high-density SNP assays that span the genome at high resolution such as the BovineSNP50 BeadChip [2] now enables the identification of novel QTL and improvement of the resolution of the location of previously mapped QTL.
Genome-wide association studies (GWAS) sometimes identify loci that are not responsible for variation (i.e., false positives) because [3]: 1) stochastic noise can generate false associations in a small sample, and 2) patterns of correlation among loci and factors responsible for trait variation can create indirect associations between markers and traits where no causal relationship exists (known as population structure). The former can be managed by increasing sample size and the latter problem can be solved by validating identified QTL in an independent, but demographically similar population. If the same marker is highly associated across different traits and different populations, then it is very likely to be in strong linkage disequilibrium (LD) with the causal variant.
We performed GWAS using 50 K genotypes scored in 18,274 animals from 10 US beef cattle breeds with observations for twelve economically relevant traits (Tables 1). This large data set enabled us to perform an extensive comparative genome-wide association study in cattle (the largest GWAS in beef cattle ever performed), which had as its goal the identification of regions of the genome that account for at least 1% of genetic variation in these traits, and evaluation of genetic architecture of these traits among breeds. This analysis enabled discovery of several important across-breed and breed-specific large-effect pleiotropic or closely linked QTL.
Gene enrichment analysis provided valuable information as to the biological functions of gene subsets and dominant pathways imposed in the trait of interest [4]. In addition to broad-spectrum GWAS, we used gene ontology (GO) tools to allocate biological roles and common pathways involved in the function of candidate genes within the identified pleiotropic QTL.
Results and discussion
Detected QTL and sample size
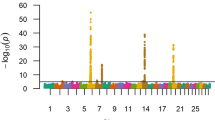
A total of 294 QTL (all considered to be of large-effect and defined as 1-Mb windows of the genome explaining ≥ 1% of additive genetic variance) were identified across twelve traits recorded in ten breeds (Table 2). Some of the identified QTL were associated with more than one trait (pleiotropic QTL) or segregating in more than one breed (across-breed QTL), which leave 159 QTL to be unique (Figure 1). In general, more QTL were identified in the populations (or traits) with bigger sample sizes (Table 2), which simply reflects the increasing statistical power in GWAS by increasing sample size [3, 5]. However, less QTL were identified in Angus and Limousin populations than expected by their sample sizes (Table 2), which could be due to different selection programs practiced in these breeds or due to the biased samples. Long-term directional selection changes the frequencies of causative variants toward fixation, which decreases the genetic variations and consequently the power of GWAS. This could be the case for these populations especially Angus that experienced long-term selection. Also, if the samples are not well distributed across the whole population and only a selected group of animals (usually those with high performances) are sampled then the power of GWAS would decrease due to lower genetic variation available in the selected samples (biased sample). This could be also a possibility with the samples collected in Angus and Limousin populations. Spencer et al. (2009) showed that the patterns of LD and the frequencies of causative variants (common versus rare alleles) also play important roles in the statistical power of GWAS [5].

The QTL network. The genomic locations (BTA_Mb) and the trait(s) associated with each identified QTL. The traits are birth weight (BWT), calving ease direct (CED), calving ease maternal (CEM), carcass weight (CWT), marbling (MRB), mature weight (MWT), ribeye muscle area (REA), weaning weight direct (WWD), weaning weight maternal (WWM), yield grade (YGR) and yearling weight (YWT).
Details concerning support and magnitude of effect for each QTL associated with birth weight, calving ease direct, calving ease maternal, carcass weight, marbling, mature weight, ribeye muscle area, weaning weight direct, weaning weight maternal, yield grade and yearling weight in all ten cattle breeds are in Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12, respectively. The identity of the most strongly associated SNP (denoted throughout as ‘lead-SNP’ and defined as the SNP with the highest posterior probability of inclusion (PPI) within the Bayesian GWAS model within the 1-Mb window most strongly associated with the QTL), its genomic position, its B allele (from the Illumina A/B calling system) frequency and the impact of that allele on the trait (increasing or decreasing) are also reported for each QTL in Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12. In this study, a similar 1-Mb QTL associated with more than one trait considered as a pleiotropic QTL. However, it could be actually two different closely linked QTL. Further analyses using multi-variate models are needed to dissect pleiotropic QTL from closely linked QTL. However, when the B allele at lead-SNP is consistent for direction of affect across different breeds, which was the case for most identified lead-SNPs within identified QTL, then it is more likely to be pleiotropy because with linkage the B allele could detect the QTL1 + \QTL2+ haplotype (favorable alleles at two linked QTL) in one breed and the A allele could detect it in another. Additionally, most of the identified lead-SNPs are located in master regulator genes, which is further evidence for pleiotropy.
Four pleiotropic QTL segregate in several phylogenetically distinct US cattle breeds
Among all identified QTL, four pleiotropic QTL were detected in two or more breeds (across-breed pleiotropic QTL). These pleiotropic QTL were identified on BTA6 at 37–42 Mb (varies across breeds but primarily detected at 38 Mb), on BTA7 at 93 Mb, on BTA14 at 23 – 26 Mb (primarily at 24 or 25 Mb) and on BTA20 at 4 Mb (Table 3). In general, the QTL on BTA6 had a larger impact than any of the other QTL and was associated with a greater number of traits in the greatest number of breeds (Table 3). This QTL is associated with body weights (birth, carcass, direct weaning, mature, and yearling weights), calving ease direct and weaning weight maternal, in all breeds except Angus. It explained more than 10% of additive genetic variance of birth weight in Hereford, Limousin, Red Angus and Simmental; of calving ease direct in Hereford; of carcass weight in Red Angus and Simmental; of mature weight in Hereford; of weaning weight direct in Limousin and Red Angus; and of yearling weight in Hereford, Limousin and Red Angus. The largest effect found for this QTL was for calving ease in Hereford where it explained 32% of additive genetic variance. The 1-Mb QTL region on BTA6 most strongly associated with the traits varied from 38 to 39 Mb and was not unique across breeds or traits. Further, there were six different lead-SNPs at 38 Mb and four different lead-SNPs at 39 Mb detected for different traits and breeds. However, the more frequently model-selected SNPs in this region were rs81131471 at 38.91 Mb, rs110834363 at 38.94 Mb and rs81151923 at 39.26 Mb (near the boundary separating the 38 and 39 Mb windows). Many studies have pointed to the presence of QTL on BTA6 for body weights, growth and carcass traits [6–11], calving ease direct [12], milk traits [13–15], reproductive traits [16–18] and feed efficiency traits [8, 9, 19] in cattle. Three interesting genes located in this region have been suggested as positional candidate genes: 1) LAP3 (leucine aminopeptidase 3), located at 38.57 - 38.60 Mb, which is an aminopeptidase which catalyzes the removal of N-terminal amino acids and is involved in protein maturation and degradation [15]; 2) NCAPG (non-SMC condensing I complex, subunit G), located at 38.74 - 38.81 Mb, which has a catalytic function in the mammalian condensin I complex and is important during mitotic cell division [20]; 3) LCORL (ligand dependent nuclear receptor corepressor-like), located at 38.84 - 38.99 Mb, which is associated with stature in human and cattle [21]. A nonsynonymous substitution in NCAPG, NCAPG c.1326 T > G that leads to the amino acid change p.Ile442Met in the NCAPG protein has been suggested as the candidate causative variant associated with prenatal [22] and postnatal growth traits [10, 20] in cattle. However, Bongiorni et al. (2012) has suggested that LAP3 is the more probable gene to affect calving ease direct in Piedmontese cattle [12]. Whether the p.Ile442Met variant is responsible for the pleiotropic QTL on BTA6 detected in this study or if different mutation(s) in NCAPG or other genes are involved is currently unknown.
The QTL on BTA7 at 93 Mb is pleiotropic and associated with birth weight, calving ease direct and maternal in Angus and Hereford; carcass weight in Angus and Simmental; mature weight in Hereford; ribeye muscle area in Angus, Gelbvieh, Hereford, Red Angus and Simmental; weaning weight direct and yearling weight in Angus, Hereford and Simmental. This is the most important pleiotropic QTL in Angus and is the largest-effect QTL associated with ribeye muscle area in several breeds. This QTL explained more than 16% of additive genetic variance in ribeye muscle area in Gelbvieh (Additional file 8). Associations of this QTL with composition of fatty acids in beef has also been reported [23, 24]. Only one lead-SNP (rs110059753) was detected for this QTL in all associated traits in Angus, Brangus, Hereford and Simmental animals. That SNP, located at 93.22 Mb, was also lead-SNP for a weaning weight direct QTL in Red Angus (Additional file 9). The effect of the B allele at this lead-SNP was in the same direction (increasing or decreasing) for the associated traits across different breeds, indicating that linkage phase between this lead-SNP and the causal mutation is conserved across breeds (Additional files 1, 2, 3, 4, 7, 8, 10 and 12) suggesting that the SNP is very close to the causal mutation. There is a very good candidate gene located close to this lead-SNP. ARRDC3 (arresting domain containing 3), located at 93.24 - 93.25 Mb, is a member of the arrestin superfamily that regulates obesity in mice and human males [25, 26]. Arrestins are signaling proteins that control metabolism usually through desensitization of the beta-adrenergic receptors which are present on the surface of almost every type of mammalian cell. These receptors are stimulated physiologically by the neurotransmitter, norepinephrine and the adrenal medullary hormone, epinephrine [27]. It has been shown that oral administration of some beta-adrenergic agonists increases muscle and decreases fat accretion in cattle, pigs, poultry, and sheep [27, 28]. Consequently, if the casual mutation(s) associated with body weights and carcass traits can be established as influencing ARRDC3 expression or activity, it could be considered a natural beta-adrenergic agonist in cattle. The physiological roles of ARRDC3 in cattle are unknown.
The QTL on BTA14 at 24 – 26 Mb is segregating in Brangus, Gelbvieh and Simmental. This QTL was associated with five body weights (birth, carcass, and yearling weights; weaning weights direct and maternal) and calving ease in Gelbvieh and Simmental and explained 1 – 2% of additive genetic variance in carcass, weaning and yearling weights in Brangus. No unique lead-SNP was detected for this pleiotropic QTL in either the 24 or 25 Mb windows. Three different lead-SNPs at 24 Mb and four different lead-SNPs at 25 Mb (extending from 24.44 to 25.70 Mb) were detected in Gelbvieh and Simmental. This QTL region harbors PLAG1 (pleiomorphic adenoma gene 1) which has been shown to be associated with stature in a Holstein × Jersey F2 cross [29]; carcass weight in Japanese Black cattle [30]; and early life body weight, peripubertal weight and growth in New Zealand Holstein–Friesian cattle [31]. Utsunomiya et al. (2013) reported a QTL on BTA14 in the vicinity of PLAG1 associated with variation in birth weight in Nellore cattle suggesting that PLAG1 may also be responsible for variation in body weights in Bos indicus cattle [32]. Our results suggest that mutations in PLAG1 may also be important in Brangus, Gelbvieh and Simmental cattle. Whether the same mutation is associated with body weights in Brangus, Gelbvieh and Simmental or if different mutation(s) in PLAG1 or nearby genes cause this variation is unclear.
The pleiotropic QTL on BTA20 at 4 Mb was segregating in several breeds including Angus, Shorthorn, Hereford, Red Angus and Simmental. This QTL was associated with most body weights and with calving ease direct in these breeds. The telomeric adjacent 1-Mb window (at 5 Mb) also explained 1.9 and 1.1% of additive genetic variance of carcass weight in Shorthorn and yield grade in Simmental, respectively (Additional files 4 and 11). This QTL explained more than 5% of additive genetic variance for birth, mature and yearling weights; weaning weight direct; calving ease and more than 18% of additive genetic variance of mature weight in Hereford (Additional file 7). Several lead-SNPs spanning the region from 4.50 to 4.75 Mb were identified. The lead-SNP rs43350564, located at 4.62 Mb, was the most frequent model-selected SNP within this region across the different traits and breeds. The effect of the B allele at this SNP was in the same direction for the associated traits across the different breeds, indicating that this SNP is in the same linkage phase with the casual mutation across breeds. This lead-SNP is a downstream gene variant (a sequence variant located 3’ of a gene) of ERGIC1 (endoplasmic reticulum-Golgi intermediate compartment protein 1). ERGIC1 is a cycling membrane protein contributing to the membrane traffic and selective transport of cargo between the endoplasmic reticulum, the intermediate compartment, and the Golgi apparatus, which plays important roles in the organization and function of the early secretory pathway [33]. It is unlikely that protein sorting is the sole function of the ERGIC family, but other functions are less clear [33]. The window defining the pleiotropic QTL on BTA20 contains another interesting candidate gene, SH3PXD2B (SH3 and PX domain 2B) located at 4.01 - 4.14 Mb. The SH3PXD2B protein is essential for normal postnatal growth and development [34]. In two independent mouse models, the absence of SH3PXD2B was found to profoundly impair normal development causing runted growth, craniofacial and skeletal abnormalities, hearing impairment, glaucoma and the virtual absence of white adipose tissue [34, 35]. In human, SH3PXD2B deficiency is responsible for the development of Frank-Ter Haar syndrome, an autosomal-recessive disorder characterized by skeletal, cardiovascular, and eye abnormalities [35]. It has been also shown that SH3PXD2B plays an important role in cellular attachment and cell spreading [36].
None of these four pleiotropic QTL was associated with marbling in any breed or with any trait in Charolais. Besides these four pleiotropic QTL, several QTL were identified that were associated with more than one trait, but segregating in different breeds (Table 3).
Breed-specific pleiotropic QTL
Besides the four pleiotropic QTL that were found to segregate in more than one breed, several breed-specific pleiotropic QTL (associated with more than one trait but segregating in only one breed) were identified (Table 3). A pleiotropic QTL on BTA2 at 6 Mb associated with birth weight, calving ease direct, marbling, rib eye area, weaning weight direct, weaning weight maternal, and yield grade was identified in Limousin. This QTL explained 4.7, 1.2, 5.4, 11.8, 1.4, 5.3 and 13.8% of additive genetic variance in these traits, respectively (Additional files 1, 2, 6, 8, 9, 10 and 11). The telomeric adjacent 1-Mb window (at 7 Mb) also explained 3.8% of additive genetic variance in calving ease direct in Limousin (Additional file 2). Two lead-SNPs (rs110233897 and rs41638273) in close proximity, separated by less than 0.02 Mb (at 6.68 and 6.70 Mb, respectively), were detected. This QTL window harbors myostatin (MSTN) a major gene responsible for double-muscled phenotype in cattle [37]. This phenotype occurs at high frequency in some breeds of cattle such as Belgian Blue and Piedmontese [38]. There are numerous mutations in this gene including non-synonymous, missense, premature stop codons and frame-shift variants that are associated with different levels of change in muscle morphology [38]. A specific MSTN mutation identified as F94L, has been shown to be associated with increased muscle growth and beef traits in Limousin [39].
A pleiotropic QTL on BTA5 at 48 – 50 Mb associated with birth weight, calving ease direct, marbling and ribeye muscle area was identified in Brangus. Cumulatively (over three contiguous 1-Mb windows), this QTL explained 6.9, 5.2, 8.4 and 2.8% of additive genetic variance in these traits, respectively (Additional files 1, 2, 6 and 8). This QTL also associated with ribeye muscle area in Angus (Table 3). However, there was no overlap between identities of lead-SNPs in Angus and Brangus. This QTL window harbors some very good candidate genes. The lead-SNP rs29016809, detected for birth weight in Brangus (Additional file 1), is located in an intron of HMGA2 (high mobility group protein A2). The HMGA proteins are architectural transcription factors that regulate transcription of a variety of genes and direct cellular growth, proliferation and differentiation [40]. HMGA2 variants are associated with human height [41], body size in dogs [42] and horses [43], and meat quality and carcass traits in pigs [44]. HMGA2 knockout mice had 40% of the mean body weight of controls [45]. The other lead-SNP, rs109566520, which was detected for calving ease direct in Brangus (Additional file 2), is located in an intron of MSRB3 (methionine-S-sulphoxide reductase 3). Lee et al. (2012) have shown that methylation of MSRB3 is associated in humans with changes in gestational age at birth [46]. Mutations in MSRB3 are also associated with hereditary deafness and primary tooth development during infancy in human [47, 48]. The lead-SNP for ribeye muscle area in Brangus (rs41657459, Additional file 8) is located in an intron of LEMD3 (LEM domain containing 3), which is responsible for bone density disorders [49]. Loss of function mutations in LEMD3 result in increased bone density, namely osteopoikilosis in human [50]. Although many QTL associated with body weights, meat and milk traits have been reported in this region [6, 18, 51], no associations between traits and variants in HMGA2, MSRB3 and LEMD3 have been reported in cattle.
A pleiotropic QTL on BTA5 at 106 Mb in Hereford explained 2.6, 3.9, 2.0 and 4.9% of additive genetic variance in birth weight, mature weight, weaning weight direct and yearling weight, respectively (Additional files 1, 7, 9 and 12). Lead-SNP rs41654528 located on BTA5 at 106.23 Mb was associated with birth weight, weaning weight direct and yearling weight while rs110421124 located 0.04 Mb telomeric of rs41654528 at 106.27 Mb was associated with mature weight in Hereford. Lead-SNP rs41654528 is located in an intron of the Fructose-2,6-bisphosphotase TIGAR (TP53-induced glycolysis and apoptosis regulator). TIGAR, which also known as the chromosome 5 open reading frame or C12orf5 in human, is a recently identified fructose-2,6-bisphosphatase that contributes to regulation of glucose metabolism [52]. Knockdown of TIGAR increased glycolysis with elevated fructose-2,6-biphosphate levels and reduced myocyte apoptosis whereas overexpression of TIGAR reduced glucose utilization and increased myocyte apoptosis in mice [53]. There is a missense variant (rs207837488) within this gene, which causes a V94A substitution. That variant could be one of the potential candidate quantitative trait nucleotides for the body weight associations in Hereford.
Another breed-specific pleiotropic QTL was identified in Hereford on BTA15 at 38–39 Mb and was associated with mature weight and ribeye muscle area explaining 1.2 and 2.5% of additive genetic variance in each of these traits, respectively. There was no overlap in the identity of lead-SNPs within this QTL window. A pleiotropic QTL on BTA15 was reported in the range 37.96 to 54.29 Mb associated with carcass weight, mature height and ribeye muscle area in Angus [51].
Two breed-specific pleiotropic QTL associated with carcass, weaning and yearling weights were identified in Maine-Anjou, one on BTA4 at 61 Mb and the other on BTA29 at 30 Mb. The BTA4 QTL explained less than 2% of additive genetic variance in these three traits, while the BTA29 QTL explained 3.3, 1.7 and 3.8% of additive genetic variance (Additional files 4, 10 and 12). Lead-SNP rs43400956 located on BTA4 at 61.73 Mb was detected for weaning weight direct and yearling weight in Maine-Anjou. This lead-SNP is located just 0.02 Mb telomeric of SEPT7 (61.61 - 61.71 Mb), a member of the septin gene family. The septins are conserved GTP-binding proteins that form filaments during cell divisions or cytokinesis [54, 55]. The septins are also important in embryonic and nervous system development in animals [54]. There are several reported QTL in this region that are associated with body weights in Angus cattle [51, 56], carcass traits in Japanese Black cattle [57] and social behaviors in Charolais and Holstein [58]. Therefore, SEPT7 may be a good candidate gene for the QTL associated with body weights in Maine-Anjou.
Lead-SNP rs41651735 located on BTA29 at 30.70 Mb was also detected for carcass weight, weaning weight direct and yearling weight in Maine-Anjou. This lead-SNP is located in KIRREL3 (kin of IRRE like 3), which is essential for the formation of synapses for cell-cell communication [59]. Variants in KIRREL3 are associated with intellectual disability in human [60]. Many QTL associated with carcass, production, reproduction and behavior traits have been reported in different cattle breeds in this region [6, 18, 51].
Trait-specific QTL
Only one trait-specific QTL (associated with only one trait but segregating in more than one breed) was identified and it was on BTA1 at 2 Mb (Table 3). It was associated with weaning weight maternal in Charolais, Hereford and Simmental and explained 1.3, 3.0 and 1.0% of additive genetic variance in these breeds, respectively (Additional file 10). The lead-SNP rs109378326 at 2.52 Mb was detected for weaning weight maternal in Charolais and Simmental while rs109205247 at 2.46 Mb was found in Hereford. The adjacent telomeric 1-Mb window to this QTL window (at 3 Mb) explained 0.7% of additive genetic variance in weaning weight maternal in Angus and Gelbvieh (data not shown). The milk production ability of a cow in terms of total energy available to the calf (comprising milk volume and composition) is a key factor affecting weaning weight in beef cattle. Many QTL associated with milk production traits (fat, protein and yield) have been reported in this region [61, 62]. McClure et al. (2010) also reported a QTL on BTA1 at 1–1.8 Mb associated with weaning weight maternal in Angus cattle [51]. MRAP2 (melanocortin 2 receptor accessory protein), which is located in this QTL window encodes a melanocortin receptor-interacting protein that regulates trafficking and function of all members of the melanocortin receptor family in the adrenal gland [63]. Associations among polymorphisms in different melanocortin receptor family members and milk production traits in cattle and sheep have now been established [64, 65]. Loss of function of MRAP2 is associated with mammalian obesity [66].
Trait-breed specific QTL
Several trait-breed specific QTL (associated with only one trait and segregating in only one breed) were identified (Figure 1). Most of these QTL explained less than 2% of additive genetic variance of their corresponding trait in their respective breed. However, some of them explained a larger percentage of genetic variance. For example, a trait-breed specific QTL on BTA6 associated with birth weight explained 8.1% of additive genetic variance in Brangus (Additional file 1). The lead-SNP rs110561712 on BTA6 at 35.56 Mb is an intronic variant within CCSER1. CCSER1 deficiency creates a cell division defect in human [67]. Experimental knockdown of CCSER1 expression caused cytokinesis defects, multipolar mitosis and multinuclearity [67]. A trait-breed specific QTL associated with calving ease maternal (explaining more than 8% of additive genetic variance) was identified in Angus on BTA10 at 73 Mb (Additional file 3). The lead-SNP, rs43638895 located on BTA10 at 73.83 Mb, is an intronic variant within PRKCH protein kinase C. Laramée et al. (2002) [68] showed that parathyroid-related peptide and parathyroid hormone, which have important roles in maternal-fetal calcium homeostasis, operate through activation of protein kinase C.
Five trait-breed specific QTL associated with marbling were identified in Gelbvieh on BTA5 at 11 Mb and 43 Mb; on BTA13 at 72 Mb; on BTA16 at 46 Mb; and on BTA24 at 39 Mb (Additional file 6). These cumulatively explained about 30% of additive genetic variance in marbling in Gelbvieh and the largest-effect QTL on BTA24 explained 10.5% of additive genetic variance (Additional file 6). There are few reported QTL in these regions associated with marbling in cattle. McClure et al. (2010) reported a similar QTL on BTA5 at 11 Mb associated with marbling score in Angus cattle [51]. Casas et al. (2000) reported overlapping QTL associated with fat thickness, ribeye muscle area and tenderness score on BTA5 [69].
Two adjacent trait-breed specific QTL on BTA3 at 112 and 113 Mb cumulatively explained 7.1% of additive genetic variance in ribeye muscle area in Shorthorn (Additional file 8). Another QTL on BTA5 at 34 Mb explained 3.9% of additive genetic variance of ribeye muscle area in Shorthorn (Additional file 8). A trait-breed specific QTL located on BTA26 at 42 Mb explaining 3.9% of additive genetic variance of yield grade was identified in Red Angus (Additional file 11). No QTL associated with yield grade have previously been reported in this region. We should point out that the reported trait-breed specific QTL are not validated QTL as they were identified only in one population and associated only with one trait. Independent populations are needed to validate these QTL.
Genetic architecture of traits
Figure 2 shows the proportions of additive genetic variance explained by each of the across-breed and breed-specific pleiotropic QTL; and all other identified QTL. The four across-breeds pleiotropic QTL (on BTAs 6, 7, 14 and 20) and the breed-specific pleiotropic QTL explained most of the detected additive genetic variances of body weights (birth, carcass, yearling weights; and weaning weight direct) and calving ease direct in ten cattle breeds. The pleiotropic QTL explained a significant percentage of additive genetic variances of these traits cumulatively (more than a third of additive genetic variance of birth weight in Hereford and Simmental, calving ease direct in Herford, carcass weight in Red Angus and Simmental, mature weight in Hereford, weaning weight direct and yearling weight in Simmental, Figure 2). If the causal mutations of these pleiotropic QTL were found by further genotyping and statistical analyses, then knowledge of such mutations would create new opportunities for cattle breeders to select their animals with appropriate body weights for harvest or maternal purposes based on the few number of genes.

Proportions of additive genetic variance explained by detected QTL. The proportions of additive genetic variance explained by detected QTL (1-Mb windows of the genome explaining ≥ 1% of additive genetic variance) for each trait in 10 US cattle breeds. The traits are birth weight (BWT), calving ease direct (CED), calving ease maternal (CEM), carcass weight (CWT), marbling (MRB), mature weight (MWT), ribeye muscle area (REA), weaning weight direct (WWD), weaning weight maternal (WWM), yield grade (YGR) and yearling weight (YWT). The breeds are shown as AAN (Angus), BRG (Brangus), BSH (Shorthorn), CHA (Charolais), GVH (Gelbvieh), HER (Hereford), LIM (Limousin), RAN (Red Angus), RDP (Maine-Anjou) and SIM (Simmental).
Different genetic architectures were observed for meat quality (fat thickness, marbling, ribeye muscle area and yield grade) and maternal (calving ease and weaning weight) traits. The detected QTL for these traits explained a lower percentage of genetic variances than those detected for body weights. This may reflect the fact that more selection pressure is on meat quality traits than growth traits in beef cattle, which may change the frequency of favorable allele(s) toward fixation, which decreases the genetic variance explained by a given QTL. Moderate body weights are more desirable for many producers because of increased calving problems and higher maintenance feed costs for animals with high body weights. The more likely reason could be the lack of power to detect QTL for meat quality traits due to less accurate data for carcass than growth traits. The accuracy of deregressed estimated breeding values (DEBV), used as response variables to estimate SNP effects, are generally lower for carcass traits versus growth traits due to lower number of observations and indirect measurement. For example, the average of reliability of DEBV for carcass weight, marbling and ribeye area in Angus was 0.41, 0.44 and 0.47, while it was 0.79, 0.69 and 0.70 for birth, weaning and yearling weights, respectively.
Most of the detected additive genetic variances for meat quality traits were explained by trait or trait-breed specific QTL and the pleiotropic QTL had less impact on these traits except for the pleotropic QTL on BTA 7 for ribeye muscle area and the Limousin-specific pleiotropic QTL on BTA 2. The QTL associated with ribeye muscle area had little overlap across the ten breeds. The pleiotropic QTL on BTA7 at 93 Mb was the only QTL associated with ribeye muscle area segregating in more than one breed (Angus, Gelbvieh, Hereford and Simmental). The pleiotropic QTL on BTA2 at 6 Mb explained 11.8% of additive genetic variance in ribeye muscle area in Limousin (Additional file 8) while a second QTL on BTA2 at 38 Mb explained 1.8% of additive genetic variance. Only ten large-effect QTL were identified for yield grade in the five breeds recorded for the trait. The QTL on BTA2 at 6 Mb in Limousin (pleiotropic QTL associated with many traits in Limousin) was the largest-effect QTL explaining 13.8% of additive genetic variance (Additional file 11). Five of these QTL were identified in Red Angus, three in Simmental and one in Gelbvieh and none overlapped (Additional file 11). There was also no overlap in marbling QTL across different breeds, which could be partly due to lack of power to detect all QTL associated with marbling in different breeds. Obviously not all QTL are discovered and those that are detected are different partly due to chance in different breeds. The QTL associated with weaning weight and calving ease maternal also had little overlap across the ten different breeds. The QTL on BTA1 at 2 Mb was the only QTL associated with weaning weight maternal segregating in more than one breed (Charolais, Hereford and Simmental, Additional file 10). The detected QTL explained less than 50% of additive genetic variances of the traits across all ten breeds (except for carcass weight in Simmental). This shows that a large number of loci of small effect are necessary to capture the remaining genetic variation as has been shown previously [70]. Here, we should acknowledge that the reported percentage of additive genetic variance explained for the identified QTL could be biased upward (or downward) due to influence of the prior used in the Bayesian regression model [71].
One might expect to find largely the same variants segregating in phylogenetically similar breeds due to their limited number of generations since divergence (see e.g., [72]). However, we did not find overwhelming evidence that this was the case since, for example, the first of the across-breed pleiotropic QTL was found to segregate in all breeds except Angus, yet Angus is closely related to other British breeds, namely Red Angus, Hereford and Shorthorn breeds. This result cannot be explained by small sample size since the Angus sample was the second largest studied. However, inadequate sample size may explain why QTL were not detected in Charolais, Maine-Anjou and Shorthorn. We suspect that resolution as to the concordance of causal mutations across breeds will not be resolved until multiple individuals from each of these breeds are sequenced to identify the extent of sharing of variation among breeds. Additional genotyping for variants discovered by sequencing and additional statistical analyses will be required to resolve identities of casual mutations. Elucidation of even a few of these mutations will create new opportunities for selection of animals with appropriate harvest body weights and meat quality specifications as well as for creation of maternal lines which could decrease dystocia and increase growth rate of calves in the US cattle population.
Genes involved in ossification and in adipose tissue development are over-represented in the pleiotropic QTL
Four across-breed pleiotropic QTL (on BTAs 6, 7, 14 and 20), Limousin-specific pleiotropic QTL on BTA 2 and Hereford-specific pleiotropic QTL on BTA 5 were associated with the greater number of traits (these represent the core of the QTL network, Figure 1). Genes within these pleiotropic QTL along with the genes within Brangus-specific pleiotropic QTL (in total 83 genes, Additional file 13) were interrogated for GO (gene ontology) category and KEGG (Kyoto encyclopedia of genes and genomes) pathway enrichment using the web-based tool g:Profiler (http://biit.cs.ut.ee/gprofiler/) [73] GO analysis of the genes within these pleiotropic QTL showed significant enrichment for functional categories related to tissue development especially ossification and adipose tissue development (Table 4). Interestingly, all of the 8 pleiotropic QTL had genes involved in tissue development, where four of them include genes involved in the ossification process and 3 others involved our candidate genes (HMGA2, ARRDC3 and SH3PXD2B) in adipose tissue development (Table 4). The GO categories related to the cellular responses to the chemical stimulus, and the regulations of cation (especially calcium) transporter activities were also significantly over-represented in the pleiotropic QTL (Table 4). The pathway enrichment revealed that 5 genes (FGF6, FGF23, C-MOS, RRAS2, DUSP1) from 4 pleiotropic QTL (on BTAs 5, 14, 15 and 20) are significantly associated with the Mitogen-Activated Protein Kinase (MAPK) signaling pathway (KEGG:04010). The MAPK cascade is a highly conserved module that controls many cellular events from complex programs, such as embryogenesis, cell differentiation, cell proliferation, and cell death, to short-term changes required for homeostasis and acute hormonal responses [74].
Some of the significant GO categories are not convincing to have pleiotropic roles (e. g. cellular responses to the chemical stimulus or the regulations of cation, Table 4) or some of the genes listed in the significant GO categories have a wider role than just that significant category (e. g. HMGA2 and ARRDC3 genes listed in adipose tissue GO term). Also, some of the significant GO categories do not include any candidate genes identified from association study in the pleiotropic QTL (e. g. ossification or MAPK signaling pathway, Table 4). This could be partly due to the GO analysis approach that we used in this study. Here, all genes within a 1-Mb were simply considered as significant genes. This assumption limits the analysis to find biological meaning.
Conclusions
Although many quantitative trait loci (QTL) associated with economically important traits in beef cattle have been identified, not all of the genetic variation in these traits has been captured because of inadequate sample size and insufficient density of markers historically used in QTL mapping studies. Here, we took advantage of a high-density SNP assay that spans the genome at moderate resolution in conjunction with large sample size (18,274 animals from 10 US beef cattle breeds) for the identification of novel QTL and improvement of the resolution of the location of previously mapped QTL. We found a total of 159 unique large-effect QTL (defined as 1-Mb genome windows explaining more than 1% of additive genetic variance), where four QTL have pleiotropic effects (associated with more than one trait) and segregate in more than one breed. We also found pleiotropic QTL that segregate in single breeds. GO analysis revealed that genes involved in ossification and in adipose tissue development were over-represented in the identified pleiotropic QTL. Also, GO analysis showed that our identified pleiotropic QTL harbor genes that involved in the MAPK signaling pathway. Our results will improve understanding of the biology of growth and body composition in cattle.
Methods
Animal Care and Use Committee approval was not required or obtained for data that were extracted from existing breed association databases. Blood, semen or hair samples collected on Angus, Hereford, Gelbvieh, Limousin, Simmental and Shorthorn animals for genotyping at the University of Missouri were collected under protocol 7505 approved by the University of Missouri Animal Care and Use Committee.
Genotype and phenotype data
A total of 18,274 animals from 10 US cattle breeds (3,570 Black Angus, 1,328 Brangus, 200 Charolais, 1,374 Gelbvieh, 2,779 Hereford, 2,239 Limousin, 1,761 Red Angus, 328 Shorthorn, 574 Maine-Anjou and 4,124 Simmental) were genotyped with either BovineSNP50 BeadChip versions B or C (Illumina, San Diego, CA) or (less than 3%) with the BovineHD BeadChip (Illumina, San Diego, CA). For animals that were genotyped with the BovineHD BeadChip, genotypes for markers that were in common with the BovineSNP50 BeadChip were extracted. The marker quality control tests were performed using data available in the University of Missouri database at the time of analysis. Genotypes at a particular locus were filtered from further analysis according to the following criteria: average heterozygosity more than 0.52 in more than 10 breeds; average call rate less than 0.80 in more than 10 breeds; monomorphic in more than 7 breeds; minor allele frequency less than 0.001 and Hardy-Weinberg equilibrium test p-value less than 3 × 10- 9. Only markers that passed quality control and that were uniquely assigned to bovine autosomes or the X chromosome on the UMD3.1 assembly were used for analysis (54,555 markers). Missing genotypes (less than 3%) were replaced with the average value (on a 0–2 scale) for each SNP within each breed.
In total, twelve traits were analyzed (birth, carcass, mature and yearling weights; weaning weight and calving ease direct and maternal; fat thickness; marbling; ribeye muscle area and yield grade), however, some traits were recorded in only some breeds (Table 1). Expected progeny differences (EPD, which is one half of the estimated breeding value, EBV) and their Beef Improvement Federation (BIF) accuracies were obtained from each breed association for all genotyped animals and their sires and dams. The EPD were transformed to EBV by multiplying by 2 and corresponding reliabilities (R2) were obtained as R2 = 1 - (1 - BIF _ Accuracy)2.
Next, deregressed estimated breeding values (DEBV) were derived [75] and used as response variables to estimate SNP effects in a weighted analysis which used weights corresponding to the amount of information that was available for the estimation of each DEBV. This method results in DEBV that are free of parent average effects and the weights can be used to appropriately account for heterogeneous variance due to differences in reliabilities of individual and parent average EBV and therefore of corresponding DEBV. The proportion of additive genetic variance not explained by markers (parameter c of [75]) was assumed to be 0.40 [76–78] and heritabilities that were used to derive the weighting factors are in Table 5. The number of genotyped animals with DEBV varied among traits because some animals had no individual or offspring information contributing to their EPD or because some traits were recently introduced after some of the older animals had already had their progeny recorded. The numbers of genotyped animals with DEBV for each trait in each breed are in Table 1.
Statistical model
In this study, all 54,555 SNP markers were simultaneously considered as predictors of the response variables in order to estimate partial SNP effects. The “Bayes-B” method [79] which fits a mixture model in which non-zero SNP effects are drawn from distributions with marker specific variances and some known fraction of markers (π) have no effect on the trait was used to estimate marker effects. For each trait, the following model was fit to estimate marker effects:
where y is the vector of observations (i.e., DEBV); b is the vector of fixed effects which comprised only the population mean because DEBV are free of systematic environmental effects such as herds, years and seasons of data origin; u is a vector of random marker substitution effects, where element j of u has effect greater than zero (with probability 1 - π) or effect equal to zero (with probability π) as described by [80]; X and Z are design matrices which relate observations to the fixed and marker effects, respectively, with each element of Z representing an allelic state (i.e., centered number of B alleles from the Illumina A/B calling system); and e is the vector of random residuals ~ N(0, ) where D is a diagonal matrix whose inverse elements are the weights described by [75]. The DEBV for maternal traits (calving ease and weaning weight maternal) were derived from EBV reported by respective breed association similar to other traits. In this study, parameter π was set to 0.99 for all analyses. MCMC methods with 41,040 iterations were used to obtain samples of marker effects and variances after discarding the first 1,000 samples to allow for burn-in. The estimates of genetic and residual variances for constructing priors of genetic and residual scale parameters for Bayes-B analysis [80], were obtained from preliminary Bayes-C analyses with π = 0.95 [81], which is less sensitive to prior assumptions than Bayes-B.
For each 40th iteration of the post burn-in chain (1,000 samples in total), sampled values for the effects of the SNPs within each 1-Mb window were used to compute samples of the direct genomic breeding value (DGV) of every animal for that window (by multiplying the number of copies of B alleles by the sample of their corresponding SNP effect, and summing these values over all marker loci located within the 1-Mb window). The variance of window DGV across all animals within the breed was then used to obtain a sample of the additive genetic variance for that window. The percentage of additive genetic variance explained by each 1-Mb window was calculated as the proportion of the 1-Mb window variance for that sample relative to the sample in the same iteration of the whole genome additive genetic variance. Any 1-Mb window for which the posterior mean percentage of additive genetic variance explained was ≥ 1% (~25 fold greater than the expected value of 0.04% for each of 2,677 1-Mb windows genome-wide assuming a polygenic model for which all genomic regions explain the same amount of variance) was selected as a window containing (or defining) a large-effect QTL. Those QTL that were associated with at least two traits in more than one breed were considered to be pleiotropic. The posterior probability of inclusion (PPI) for a given window, which is the proportion of samples in which at least one SNP from a given window was included in the model with a non-zero effect, was used for significance testing [82]. All analyses were performed using GenSel software [82].
Individual 1-Mb windows that explained the largest proportions of additive genetic variation were visualized in GBrowse [83] for detailed inspection of the chromosomal region containing the 1-Mb window. The overlapping QTL for a given 1-Mb window were obtained from cattle QTLdb (http://www.animalgenome.org/cgi-bin/gbrowse/bovine/). The SNP with the highest PPI within a given 1-Mb QTL window was selected as the lead-SNP or most strongly associated SNP for that QTL window. Further information for strongly associated SNP was obtained using NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp/) and Ensemble (http://www.ensembl.org/) databases. Gene searches were performed for these genomic regions using the NCBI gene database (http://www.ncbi.nlm.nih.gov/gene/). The sfdp algorithm from Graphviz software was used to draw the QTL network [84]. GO term enrichment analysis was performed for gene sets that existed within the pleiotropic QTL (those with available GO ID, Additional file 13) over all known genes in the GO database using the web tool g:Profiler (http://biit.cs.ut.ee/gprofiler/) [73] with Bonferroni corrected p-value cut off 0.05. Only GO terms from the categories biological processes and KEGG pathway enrichments were retained.
Availability of supporting data
All association results have been deposited in the AnimalQTLdb (http://www.animalgenome.org/cgi-bin/QTLdb/BT/qabstract?PUBMED_ID=ISU0069).
Abbreviations
- BIF:
-
Beef Improvement Federation
- DEBV:
-
Deregressed estimated breeding values
- DGV:
-
Direct genomic breeding value
- EPD:
-
Expected progeny differences
- GO:
-
Gene ontology
- GWAS:
-
Genome-wide association studies
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LD:
-
Linkage disequilibrium
- Mb:
-
Megabases
- MCMC:
-
Markov chain Monte Carlo
- PPI:
-
Posterior probability of inclusion
- QTL:
-
Quantitative trait loci.
References
Andersson L: Genetic dissection of phenotypic diversity in farm animals. Nat Rev Genet. 2001, 2: 130-138.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O’Connell J, Moore SS, Smith TP, Sonstegard TS, Van Tassell CP: Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009, 4: e5350-
Platt A, Vilhjálmsson BJ, Nordborg M: Conditions under which genome-wide association studies will be positively misleading. Genetics. 2010, 186: 1045-1052.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology The Gene Ontology Consortium. Nat Genet. 2000, 25 (1): 25-29.
Spencer CC, Su Z, Donnelly P, Marchini J: Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009, 5 (5): e1000477-
Gutiérrez-Gil B, Williams JL, Homer D, Burton D, Haley CS, Wiener P: Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J Anim Sci. 2009, 87: 24-36.
Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, Hansen C, Moore SS: Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci. 2004, 82: 3405-3414.
Lindholm-Perry AK, Kuehn LA, Oliver WT, Sexten AK, Miles JR, Rempel LA, Cushman RA, Freetly HC: Adipose and Muscle Tissue Gene Expression of Two Genes (NCAPG and LCORL) Located in a Chromosomal Region Associated with Cattle Feed Intake and Gain. PLoS One. 2013, 8 (11): e80882-
Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, Wheeler TL, Ferrell CL, Jenkins TG, Snelling WM, Freetly HC: Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011, 12: 103-
Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, Takasuga A: Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009, 10: 43-
Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T, Smith TP, Sonstegard TS, Thallman RM, Bennett GL: Genome-wide association study of growth in crossbred beef cattle. J Anim Sci. 2010, 88: 837-848.
Bongiorni S, Mancini G, Chillemi G, Pariset L, Valentini A: Identification of a short region on chromosome 6 affecting direct calving ease in Piedmontese cattle breed. PLoS ONE. 2012, 7: e50137-
Olsen HG, Lien S, Gautier M, Nilsen H, Roseth A, Berg PR, Sundsaasen KK, Svendsen M, Meuwissen TH: Mapping of a milk production quantitative trait locus to a 420-kb region on bovine chromosome 6. Genetics. 2005, 169: 275-283.
Schrooten C, Bink MC, Bovenhuis H: Whole genome scan to detect chromosomal regions affecting multiple traits in dairy cattle. J Dairy Sci. 2004, 87: 3550-3560.
Zheng X, Ju Z, Wang J, Li Q, Huang J, Zhang A, Zhong J, Wang C: Single nucleotide polymorphisms, haplotypes and combined genotypes of LAP3 gene in bovine and their association with milk production traits. Mol Biol Rep. 2011, 38: 4053-4061.
Daetwyler HD, Schenkel FS, Sargolzaei M, Robinson JA: A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J Dairy Sci. 2008, 91 (8): 3225-3236.
Holmberg M, Andersson-Eklund L: Quantitative trait loci affecting fertility and calving traits in Swedish dairy cattle. J Dairy Sci. 2006, 89 (9): 3664-3671.
Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A: Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein x Jersey crossbred population. Anim Genet. 2009, 40 (1): 27-34.
Snelling WM, Allan MF, Keele JW, Kuehn LA, Thallman RM, Bennett GL, Ferrell CL, Jenkins TG, Freetly HC, Nielsen MK, Rolfe KM: Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle. J Anim Sci. 2011, 89: 1731-1741.
Weikard R, Altmaier E, Suhre K, Weinberger KM, Hammon HM, Albrecht E, Setoguchi K, Takasuga A, Kühn C: Metabolomic profiles indicate distinct physiological pathways affected by two loci with major divergent effect on Bos taurus growth and lipid deposition. Physiol Genomics. 2010, 42A: 79-88.
Pryce JE, Hayes BJ, Bolormaa S, Goddard ME: Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011, 187: 981-984.
Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, Klopp N, Fürbass R, Weikard R, Kühn C: Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC Condensin I complex, subunit G (NCAPG) gene. Genetics. 2009, 183: 951-964.
Alexander LJ, MacNeil MD, Geary TW, Snelling WM, Rule DC, Scanga JA: Quantitative trait loci with additive effects on palatability and fatty acid composition of meat in a Wagyu–Limousin F2 population. Anim Genet. 2007, 38: 506-513.
Saatchi M, Garrick DJ: Tait Jr RG, Mayes MS, Drewnoski M, Schoonmaker J, Diaz C, Beitz DC. Reecy JM: Genome-wide association and prediction of direct genomic breeding values for composition of fatty acids in Angus beef cattle. BMC Genomics. 2013, 14: 730-
Patwari P, Emilsson V, Schadt EE, Chutkow WA, Lee S, Marsili A, Zhang Y, Dobrin R, Cohen DE, Larsen PR, Zavacki AM, Fong LG, Young SG, Lee RT: The arrestin domain containing 3 protein regulates body mass and energy expenditure. Cell Metab. 2011, 14: 671-683.
Patwari P, Lee RT: An expanded family of arrestins regulate metabolism. Trends Endocrinol Metab. 2012, 23: 216-222.
Mersmann HJ: Overview of the effects of beta-adrenergic receptor agonists on animal growth including mechanisms of action. J Anim Sci. 1998, 76: 160-172.
Beermann DH: Beta-Adrenergic receptor agonist modulation of skeletal muscle growth. J Anim Sci. 2002, 80: E18-E23.
Karim L, Takeda H, Lin L, Druet T, Arias JA, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, Harris BL, Keehan MD, Littlejohn MD, Spelman RJ, Georges M, Coppieters W: Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011, 43: 405-413.
Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, Sugimoto Y, Takasuga A: Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13: 40-
Littlejohn M, Grala T, Sanders K, Walker C, Waghorn G, Macdonald K, Coppieters W, Georges M, Spelman R, Hillerton E, Davis S, Snell R: Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos taurus. Anim Genet. 2012, 43: 591-594.
Utsunomiya YT, Carmo AS, Carvalheiro R, Neves HH, Matos MC, Zavarez LB, Pérez O'Brien AM, Sölkner J, McEwan JC, Cole JB, Van Tassell CP, Schenkel FS, da Silva MV, Porto Neto LR, Sonstegard TS, Garcia JF: Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14: 52-
Breuza L, Halbeisen R, Jenö P, Otte S, Barlowe C, Hong W, Hauri HP: Proteomics of endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membranes from brefeldin A-treated HepG2 cells identifies ERGIC-32, a new cycling protein that interacts with human Erv46. J Biol Chem. 2004, 279: 47242-47253.
Mao M, Thedens DR, Chang B, Harris BS, Zheng QY, Johnson KR, Donahue LR, Anderson MG: The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development. Mamm Genome. 2009, 20: 462-475.
Iqbal Z, Cejudo-Martin P, de Brouwer A, van der Zwaag B, Ruiz-Lozano P, Scimia MC, Lindsey JD, Weinreb R, Albrecht B, Megarbane A, Alanay Y, Ben-Neriah Z, Amenduni M, Artuso R, Veltman JA, van Beusekom E, Oudakker A, Millán JL, Hennekam R, Hamel B, Courtneidge SA, van Bokhoven H: Disruption of the podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of Frank-Ter Haar syndrome. Am J Hum Genet. 2010, 86: 254-261.
Lányi Á, Baráth M, Péterfi Z, Bogel G, Orient A, Simon T, Petrovszki E, Kis-Tóth K, Sirokmány G, Rajnavölgyi É, Terhorst C, Buday L, Geiszt M: The homolog of the five SH3-domain protein (HOFI/SH3PXD2B) regulates lamellipodia formation and cell spreading. PLoS ONE. 2011, 6: e23653-
Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Ménissier F, Massabanda J, Fries R, Hanset R, Georges M: A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997, 17: 71-74.
Bellinge RH, Liberles DA, Iaschi SP, O'brien PA, Tay GK: Myostatin and its implications on animal breeding: a review. Anim Genet. 2005, 36 (1): 1-6.
Alexander LJ, Kuehn LA, Smith TP, Matukumalli LK, Mote B, Koltes JE, Reecy J, Geary TW, Rule DC, MacNeil MD: A Limousin specific myostatin allele affects longissimus muscle area and fatty acid profiles in a Wagyu-Limousin F 2 population. J Anim Sci. 2009, 87: 1576-1581.
Cleynen I, van de Ven WJ: The HMGA proteins: a myriad of functions (Review). Int J Oncol. 2008, 32: 289-305.
Weedon MN, Lettre G, Freathy RM, Lindgren CM, Voight BF, Perry JR, Elliott KS, Hackett R, Guiducci C, Shields B, Zeggini E, Lango H, Lyssenko V, Timpson NJ, Burtt NP, Rayner NW, Saxena R, Ardlie K, Tobias JH, Ness AR, Ring SM, Palmer CN, Morris AD, Peltonen L, Salomaa V: Diabetes Genet Initiative, Wellcome Trust Case Control Consortium, Davey Smith G, Groop LC, Hattersley AT, McCarthy MI, et al.: A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet. 2007, 39: 1245-1250.
Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, VonHoldt BM, Cargill M, Auton A, Reynolds A, Elkahloun AG, Castelhano M, Mosher DS, Sutter NB, Johnson GS, Novembre J, Hubisz MJ, Siepel A, Wayne RK, Bustamante CD, Ostrander EA: A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010, 8: e1000451-
Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, Loredo AI, Bellone RR, Mezey JG, Brooks SA, Sutter NB: Four loci explain 83% of size variation in the horse. PLoS ONE. 2012, 7: e39929-
Rehfeldt C, Te Pas MF, Wimmers K, Brameld JM, Nissen PM, Berri C, Valente LM, Power DM, Picard B, Stickland NC, Oksbjerg N: Advances in research on the prenatal development of skeletal muscle in animals in relation to the quality of muscle based food. II – Genetic factors related to animal performance and advances in methodology. Animal. 2011, 5: 718-730.
Zhou X, Benson KF, Ashar HR, Chada K: Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nat. 1995, 376 (6543): 771-774.
Lee H, Jaffe AE, Feinberg JI, Tryggvadottir R, Brown S, Montano C, Aryee MJ, Irizarry RA, Herbstman J, Witter FR, Goldman LR, Feinberg AP, Fallin MD: DNA methylation shows genome-wide association of NFIX, RAPGEF2 and MSRB3 with gestational age at birth. Int J Epidemiol. 2012, 41: 188-199.
Ahmed ZM, Yousaf R, Lee BC, Khan SN, Lee S, Lee K, Husnain T, Rehman AU, Bonneux S, Ansar M, Ahmad W, Leal SM, Gladyshev VN, Belyantseva IA, Van Camp G, Riazuddin S, Friedman TB, Riazuddin S: Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet. 2011, 88 (1): 19-29.
Pillas D, Hoggart CJ, Evans DM, O'Reilly PF, Sipilä K, Lähdesmäki R, Millwood IY, Kaakinen M, Netuveli G, Blane D, Charoen P, Sovio U, Pouta A, Freimer N, Hartikainen AL, Laitinen J, Vaara S, Glaser B, Crawford P, Timpson NJ, Ring SM, Deng G, Zhang W, McCarthy MI, Deloukas P, Peltonen L, Elliott P, Coin LJ, Smith GD, Jarvelin MR: Genome-wide association study reveals multiple loci associated with primary tooth development during infancy. PLoS Genet. 2010, 6 (2): e1000856-
Ben-Asher E, Zelzer E, Lancet D: LEMD3: The gene responsible for bone density disorders (osteopoikilosis). Isr Med Assoc J. 2005, 7: 273-274.
Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T, Janssens K, Menten B, Van Roy N, Vermeulen SJ, Savarirayan R, Van Hul W, Vanhoenacker F, Huylebroeck D, De Paepe A, Naeyaert JM, Vandesompele J, Speleman F, Verschueren K, Coucke PJ, Mortier GR: Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004, 36 (11): 1213-1218.
McClure MC, Morsci MS, Schnabel RD, Kim JW, Yao P, Rolf MM, McKay SD, Gregg SJ, Chapple RH, Northcutt SL, Taylor JF: A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010, 41: 597-607.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH: TIGAR, a p53 inducible regulator of glycolysis and apoptosis. Cell. 2006, 126: 107-120.
Kimata M, Matoba S, Iwai-Kanai E, Nakamura H, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Mita Y, Okigaki M, Ikeda K, Tatsumi T, Matsubara H: p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. Am J Physiol Heart Circ Physiol. 2010, 299: H1908-H1916.
Kinoshita M: The septins. Genome Biol. 2003, 4: 236-
Sirajuddin M, Farkasovsky M, Hauer F, Kühlmann D, Macara IG, Weyand M, Stark H, Wittinghofer A: Structural insight into filament formation by mammalian septins. Nature. 2007, 449: 311-317.
Imumorin IG, Kim EH, Lee YM, De Koning DJ, van Arendonk JA, De Donato M, Taylor JF, Kim JJ: Genome scan for parent-of-origin QTL effects on bovine growth and carcass traits. Front Genet. 2011, 2: 44-
Yokouchi K, Mizoguchi Y, Watanabe T, Iwamoto E, Sugimoto Y, Takasuga A: Identification of a 3.7-Mb region for a marbling QTL on bovine chromosome 4 by identical by descent and association analysis. Anim Genet. 2009, 40: 945-951.
Gutierrez-Gil B, Ball N, Burton D, Haskell M, Williams JL, Wiener P: Identification of quantitative trait loci affecting cattle temperament. J Hered. 2008, 99: 629-638.
Prince JE, Brignall AC, Cutforth T, Shen K, Cloutier JF: Kirrel3 is required for the coalescence of vomeronasal sensory neuron axons into glomeruli and for male-male aggression. Development. 2013, 140: 2398-2408.
Bhalla K, Luo Y, Buchan T, Beachem MA, Guzauskas GF, Ladd S, Bratcher SJ, Schroer RJ, Balsamo J, DuPont BR, Lilien J, Srivastava AK: Alterations in CDH15 and KIRREL3 in patients with mild to severe intellectual disability. Am J Hum Genet. 2008, 83: 703-713.
Nadesalingam J, Plante Y, Gibson JP: Detection of QTL for milk production on Chromosomes 1 and 6 of Holstein cattle. Mamm Genome. 2001, 12: 27-31.
Zhang Q, Boichard D, Hoeschele I, Ernst C, Eggen A, Murkve B, Pfister-Genskow M, Witte LA, Grignola FE, Uimari P, Thaller G, Bishop MD: Mapping quantitative trait loci for milk production and health of dairy cattle in a large outbred pedigree. Genet. 1998, 149: 1959-1973.
Chan LF, Webb TR, Chung TT, Meimaridou E, Cooray SN, Guasti L, Chapple JP, Egertová M, Elphick MR, Cheetham ME, Metherell LA, Clark AJ: MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc Natl Acad Sci U S A. 2009, 106: 6146-6151.
Fontanesi L, Beretti F, Dall'Olio S, Portolano B, Matassino D, Russo V: A melanocortin 1 receptor (MC1R) gene polymorphism is useful for authentication of Massese sheep dairy products. J Dairy Res. 2011, 78: 122-128.
Maudet C, Taberlet P: Holstein’s milk detection in cheeses inferred from melanocortin receptor 1 (MC1R) gene polymorphism. J Dairy Sci. 2002, 85: 707-715.
Asai M, Ramachandrappa S, Joachim M, Shen Y, Zhang R, Nuthalapati N, Ramanathan V, Strochlic DE, Ferket P, Linhart K, Ho C, Novoselova TV, Garg S, Ridderstråle M, Marcus C, Hirschhorn JN, Keogh JM, O'Rahilly S, Chan LF, Clark AJ, Farooqi IS, Majzoub JA: Loss of function of the melanocortin 2 receptor accessory protein 2 is associated with mammalian obesity. Science. 2013, 341: 275-278.
Patel K, Scrimieri F, Ghosh S, Zhong J, Kim MS, Ren YR, Morgan RA, Iacobuzio-Donahue CA, Pandey A, Kern SE: FAM190A deficiency creates a cell division defect. Am J Pathol. 2013, 183: 296-303.
Laramée M, Simoneau L, Lafond J: Phospholipase C axis is the preferential pathway leading to PKC activation following PTH or PTHrP stimulation in human term placenta. Life Sci. 2002, 72: 215-225.
Casas E, Shackelford SD, Keele JW, Stone RT, Kappes SM, Koohmaraie M: Quantitative trait loci affecting growth and carcass composition of cattle segregating alternate forms of myostatin. J Anim Sci. 2000, 78: 560-569.
Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME: Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010, 6 (9): e1001139-
Gianola D: Priors in whole-genome regression: Bayesian alphabet returns. Genetics. 2013, 194: 573-596.
Decker JE, McKay SD, Rolf MM, Kim JW, Alcalá AM, Sonstegard TS, Hanotte O, Götherström A, Seabury CM, Praharani L, Babar ME, Regitano LC, Yildiz MA, Heaton MP, Lui W, Lei CZ, Reecy JM, Saif-Ur-Rehman M, Schnabel RD, Taylor JF: Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle. PLoS Genetics. 2014, 10 (3): e1004254-
Reimand J, Arak T, Vilo J: g:Profiler–a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res. 2011, 39: W307-315.
Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb M: MAP kinases. Chem Rev. 2001, 101: 2449-2476.
Garrick DJ, Taylor JF, Fernando RL: Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009, 41: 55-
Saatchi M, McClure MC, McKay SD, Rolf MM, Kim J, Decker JE, Taxis TM, Chapple RH, Ramey HR, Northcutt SL, Bauck S, Woodward B, Dekkers JC, Fernando RL, Schnabel RD, Garrick DJ, Taylor JF: Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet Sel Evol. 2011, 43: 40-
Saatchi M, Schnabel RD, Rolf MM, Taylor JF, Garrick DJ: Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet Sel Evol. 2012, 44: 38-
Saatchi M, Ward J, Garrick DJ: Accuracies of direct genomic breeding values in Hereford beef cattle using national or international training populations. J Anim Sci. 2013, 91 (4): 1538-1551.
Meuwissen TH, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Habier D, Fernando RL, Kizilkaya K, Garrick DJ: Extension of the bayesian alphabet for genomic selection. BMC Bioinformatics. 2011, 12: 186-
Kizilkaya K, Fernando RL, Garrick DJ: Genomic prediction of simulated multibreed and purebred performance using observed fifty thousand single nucleotide polymorphism genotypes. J Anim Sci. 2010, 88: 544-551.
Garrick DJ, Fernando RL: Implementing a QTL detection study (GWAS) using genomic prediction methodology. Genome-Wide Association Studies and Genomic Prediction. Edited by: Gondro C, van der Werf J, Hayes B. 2013, Springer: Humana Press, 275-298. ISBN: 978-1-62703-446-3
Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, Lewis S: The generic genome browser: a building block for a model organism system database. Genome Res. 2002, 12: 1599-1610.
Ellson J, Gansner E, Koutsofios L, North S, Woodhull G: Graphviz and Dynagraph – static and dynamic graph drawing tools. Graph Drawing Software. 2004, Berlin Heidelberg: Springer, 127-148.
Acknowledgements
We gratefully acknowledge the provision of semen samples from breeders, breed associations and semen distributors. This project was supported by Iowa State University, the University of Missouri, National Research Initiative grants number 2008-35205-04687 and 2008-35205-18864 from the USDA Cooperative State Research, Education and Extension Service and National Research Initiative grants number 2009-35205-05100, 2009-65205-05635, 2011-68004-30214, 2012-67015-19420 and 2013-68004-20364 from the USDA National Institute of Food and Agriculture.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare they have no competing interests.
Authors’ contributions
MS and DJG conceived of the experiment. DJG, JFT and RDS collected the samples. MS performed the statistical analysis and wrote the first draft of the manuscript. JFT, DJG and RDS critically contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Saatchi, M., Schnabel, R.D., Taylor, J.F. et al. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics 15, 442 (2014). https://doi.org/10.1186/1471-2164-15-442
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-442