
Abstract
Background
Genotypic information produced from single nucleotide polymorphism (SNP) arrays has routinely been used to identify genomic regions associated with complex traits in beef and dairy cattle. Herein, we assembled a dataset consisting of 15,815 Red Angus beef cattle distributed across the continental U.S. and a union set of 836,118 imputed SNPs to conduct genome-wide association analyses (GWAA) for growth traits using univariate linear mixed models (LMM); including birth weight, weaning weight, and yearling weight. Genomic relationship matrix heritability estimates were produced for all growth traits, and genotype-by-environment (GxE) interactions were investigated.
Results
Moderate to high heritabilities with small standard errors were estimated for birth weight (0.51 ± 0.01), weaning weight (0.25 ± 0.01), and yearling weight (0.42 ± 0.01). GWAA revealed 12 pleiotropic QTL (BTA6, BTA14, BTA20) influencing Red Angus birth weight, weaning weight, and yearling weight which met a nominal significance threshold (P ≤ 1e-05) for polygenic traits using 836K imputed SNPs. Moreover, positional candidate genes associated with Red Angus growth traits in this study (i.e., LCORL, LOC782905, NCAPG, HERC6, FAM184B, SLIT2, MMRN1, KCNIP4, CCSER1, GRID2, ARRDC3, PLAG1, IMPAD1, NSMAF, PENK, LOC112449660, MOS, SH3PXD2B, STC2, CPEB4) were also previously associated with feed efficiency, growth, and carcass traits in beef cattle. Collectively, 14 significant GxE interactions were also detected, but were less consistent among the investigated traits at a nominal significance threshold (P ≤ 1e-05); with one pleiotropic GxE interaction detected on BTA28 (24 Mb) for Red Angus weaning weight and yearling weight.
Conclusions
Sixteen well-supported QTL regions detected from the GWAA and GxE GWAA for growth traits (birth weight, weaning weight, yearling weight) in U.S. Red Angus cattle were found to be pleiotropic. Twelve of these pleiotropic QTL were also identified in previous studies focusing on feed efficiency and growth traits in multiple beef breeds and/or their composites. In agreement with other beef cattle GxE studies our results implicate the role of vasodilation, metabolism, and the nervous system in the genetic sensitivity to environmental stress.
Similar content being viewed by others
Background
Implementation of genomic selection methods into breeding programs has catalyzed production profitability within the beef cattle industry [1]. In addition to carcass and reproductive traits, the most commonly recorded traits for use in modern breeding programs are growth traits, such as birth weight, weaning weight, and yearling weight. However, genomic selection on these traits should consider that low and high estimated breeding values (EBVs) for birth weight have been found to be associated with reduced calf viability, and increased rates of dystocia events and perinatal mortality, respectively [2, 3]. Therefore, while birth weight has been considered a production indicator and treated as a selection criterion to increase calf viability as well as other economically important traits, modern beef breeding programs and production systems generally strive to increase calving ease while also maximizing both weaning weight and yearling weight [1, 3,4,5].
For at least two decades, studies have sought to identify quantitative trait loci (QTL) influencing bovine growth, body weight, and aspects of stature, including both linkage and modern genome-wide association analyses (GWAA); thereby underscoring the longstanding economic importance of efficient beef cattle production worldwide [6,7,8,9,10,11,12]. Moreover, QTL studies and modern genomic selection programs for economically important traits have been directly enabled by the generation of the bovine genome assembly, development of the Illumina Bovine SNP50 and 778K SNP arrays, and more recently, the demonstrated ability to accurately impute high-density genotypes, thereby enabling high-resolution analyses without the increased costs associated with direct genotyping [13,14,15,16,17,18,19,20]. Notably, several recent studies have established moderate heritability estimates for birth weight, weaning weight, and yearling weight in U.S. Gelbvieh, Angus, Limousin, Simmental, Hereford, and Red Angus beef cattle [20,21,22,23,24,25]. These studies also produce evidence for several relevant QTL and positional candidate genes; including orthologous genes LCORL and PLAG1 that affect both human and bovine height as well as pleiotropic QTL influencing feed efficiency, growth traits, and carcass traits across multiple U.S. beef breeds [6, 10, 12, 20, 26,27,28,29,30,31]. However, the movement of germplasm (animals, semen, and embryos) across the U.S. in conjunction with the lack of tools to select for resilience to abiotic and biotic stressors has likely led to the loss of local adaptation in beef cattle [32]. Understanding genotype-by-environment interactions will allow us to identify the genes and biological processes involved in local adaptation. Genotype-by-environment (GxE) GWAA have been used alongside GWAA with the intent of identifying GxE interactions with complex traits [20, 33,34,35]. GxE GWAA are important to the beef industry as they identify individual ecoregions that could benefit from genomic selection [20, 33, 34].
The objective of this study was to identify loci with direct and genotype-by-environment effects on growth traits. Herein, we used 15,815 geographically diverse U.S. Red Angus beef cattle in conjunction with a union set of 836,118 (836K) imputed SNP variants to conduct GWAA and produce marker-based heritability estimates for birth weight, weaning weight, and yearling weight. Additionally, using thirty-year climate data and K-means clustering to assign all Red Angus beef cattle to discrete U.S. climate ecoregions, we estimated the significance of GxE interactions for birth weight, weaning weight, and yearling weight [32]. The present study represents the largest, high-density, single breed report to date that includes both standard GWAA and GxE GWAA for birth weight, weaning weight, and yearling weight; which was facilitated by an industry-supported research framework that includes accurate imputation to high-density genotypes for large-sample analyses [14, 19, 20]. The results of this study are expected to aid existing beef breeding programs and production systems by identifying QTL that may be included in future genotyping assays and genomic selection programs.
Results and discussion
Heritability estimates for growth traits in U.S. Red Angus beef cattle
Marker-based heritability estimates (i.e., chip heritability) were produced for birth weight, weaning weight, and yearling weight using standardized relatedness matrices (GS) with variance component analyses. Collectively, moderate to high heritability estimates with small standard errors (SE) were estimated for birth weight (0.51 ± 0.01), weaning weight (0.25 ± 0.01), and yearling weight (0.42 ± 0.01), respectively (Table 1). Moreover, these moderate to high heritability estimates for birth weight and weaning weight are similar to those produced by another study conducted on Red Angus cattle (0.58 ± 0.01 and 0.29 ± 0.01, respectively) [36]. Likewise, genetic correlations between traits were also high (birth weight and weaning weight = 0.54 ± 0.01; birth weight and yearling weight = 0.50 ± 0.01; weaning weight and yearling weight = 0.84 ± 0.01) (See Additional File 1).
GWAA for birth weight, weaning weight, and yearling weight in U.S. Red Angus beef cattle
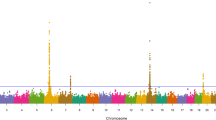
The results of our 836K single-marker GWAA for birth weight are presented in Fig. 1; with detailed summary data for 19 QTL which met a nominal significance threshold for polygenic traits (P ≤ 1e-05) described in Table 2 (Additional File 1) [71]. A comparison of birth weight QTL detected for U.S. Red Angus, Simmental, and Gelbvieh beef cattle as well as Holstein Jersey crossbred dairy cattle, revealed overlapping signals on BTA6, BTA14, and BTA20, suggesting that these birth weight QTL are not breed-specific, but rather, are likely to be more generally involved in bovine species growth processes (Table S1; Additional File 2) [9, 20, 72]. Positional candidate genes detected via birth weight GWAA revealed previous associations with aspects of cattle growth, development, feed efficiency, and carcass traits (i.e., LCORL, LOC782905, NCAPG, PLAG1, LOC104975192, STC2, HERC6, LOC112449660, IMPAD1, SLIT2, LOC101905238, ARRDC3, LOC783392, CPEB4, MMRN1, SH3PXD2B, KCNIP4, GRID2, CCSER1, NSMAF, LOC107133116, ENC1, LOC112443028), as well as milk production (LOC101906669, TRNAG-UCC), and immune response (SLURP1), as detailed in Table 2 [37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70]. Notably, all but two lead SNPs (i.e., 6_37 Mb, 6_35 Mb) were located in noncoding regions, which is concordant with recent studies of feed efficiency and growth traits in beef cattle (Table S1; Additional File 1, Additional File 2) [20, 31]. Additionally, a QTL was detected on BTA6 (42 Mb), but with less statistical support, and included the positional candidate genes LOC782172 and ADGRA3; which have previously been associated with U.S. Gelbvieh growth traits (Table S2; Additional File 2) [20]. The genomic inflation factor for P-value estimates obtained from the birth weight GWAA are presented in Table S3 (Additional File 2).

Birth weight QTL. Manhattan plot with -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 15,815 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 2
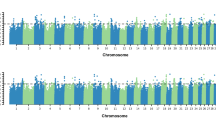
Single-marker GWAA (836K) for weaning weight in U.S. Red Angus beef cattle produced evidence for 14 QTL (P ≤ 1e-05), as defined by their lead SNPs (Table 3, Fig. 2; Additional File 1). Similar to a recent analysis of U.S. Gelbvieh beef cattle [20], the weaning weight QTL regions detected for U.S. Red Angus cattle suggest extensive pleiotropy with birth weight, as would be expected due to high genetic correlations between the two traits [4]. This includes the shared positional candidate genes on BTA6 (LCORL, LOC782905, HERC6, CCSER1, SLIT2, GRID2), BTA14 (LOC112449660), and BTA20 (LOC104975192, STC2, SH3PXD2B) (Table 2, Table 3, Table S1; Additional File 2). Additional positional candidate genes identified for weaning weight QTL include those associated with growth and development (FAM184B, LOC112447052, NSG2, LOC112449630, MOS, PENK, MIR3660, CETN3) (Table 3) [73,74,75,76,77]. All weaning weight QTL detected by GWAA were located in noncoding regions. An additional pleiotropic QTL was noted on BTA6 (42 Mb), but with less statistical support, which is the same QTL detected in the birth weight GWAA (Table S2, Table S4; Additional File 2). The genomic inflation factor for P-value estimates obtained from the weaning weight GWAA are displayed in Table S3 (Additional File 2).

Weaning weight QTL. Manhattan plot with -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 15,620 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 3
Similar to a recent study of U.S. Gelbvieh growth traits [20], our GWAA for yearling weight in U.S. Red Angus beef cattle also identified positional candidate genes shared across all three investigated traits on BTA6 (LCORL, LOC782905, HERC6, SLIT2, and CCSER1), BTA14 (LOC112449660), and BTA20 (STC2 and SH3PXD2B) (Table 4, Fig. 3, Table S1; Additional File 1, Additional File 2). Evidence for pleiotropic QTL influencing birth weight (Table 2) and yearling weight (Table 4) was also noted via overlapping positional candidate genes for these traits on BTA6 (KCNIP4, MMRN1), BTA7 (LOC101905238, ARRDC3, LOC112447488, LOC112447489), BTA14 (PLAG1), and BTA20 (LOC783392, CPEB4) (Table S1; Additional File 2). Likewise, a comparison of U.S. Red Angus QTL detected for weaning weight (Table 3) and yearling weight (Table 4) also revealed evidence for pleiotropic QTL influencing these traits via shared positional candidate genes on BTA6 and BTA14, including FAM184B and PENK, respectively. Positional candidate genes for QTL on BTA7, BTA20, and BTA21 which were only detected for yearling weight have been associated with general growth and development in Xenopus laevis (KCNIP1) [78], as well as bovine milk production (LOC112447488) (Table 4, Fig. 3, Table S1; Additional File 2) [46]. Collectively, two of the 16 lead SNPs (20_05 Mb, STC2; 6_35 Mb, MMRN1) noted for yearling weight QTL were located within coding regions (Table 4). Interestingly, STC2 has previously been associated with body size, feed efficiency, and growth in cattle [20, 24, 31, 40, 42, 47]; whereas MMRN1 has been associated with growth, feed efficiency, and metabolic stability during weather stress in cattle (Table 4, Table S1; Additional File 1, Additional File 2) [24, 43, 56,57,58]. Despite less statistical support overall, the QTL on BTA6 at 42 Mb (i.e., LOC782172 and ADGRA3) was detected for birth weight, weaning weight, and yearling weight in the Red Angus GWAA (Table S2, Table S4, Table S5; Additional File 2); as was the QTL on BTA6 at 32 Mb (Table 2, Table 3, Table S5; Additional File 2), and the QTL at BTA7 at 91 Mb (Table 2, Table 4, Table S5; Additional File 2). Finally, it should be noted that a pleiotropic QTL was also detected on BTA7 at 90 Mb for both weaning weight and yearling weight; albeit with less overall statistical support (Table 3, Table S5; Additional File 2). Genetic correlations estimated for all growth traits are summarized in Additional File 1. The genomic inflation factor for yearling weight GWAA is reported in Table S3 (Additional File 2). Genomic inflation factors (λ) larger than 1 are expected for well-powered studies of polygenic traits [79, 80], reflecting the large number of genomic loci influencing variation in these traits.

Yearling weight QTL. Manhattan plot with -log10 P-values. Lead and supporting SNPs for QTL represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 12,388 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 4
GxE GWAA for birth weight, weaning weight, and yearling weight in U.S. Red Angus beef cattle
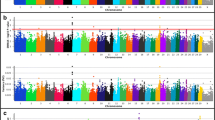
To investigate GxE interactions in relation to birth weight, weaning weight, and yearling weight in U.S Red Angus beef cattle, we conducted additional single-marker (836K) analyses. All analyses included a variable for U.S. geographic ecoregion of origin, which was generated via K-means clustering using thirty-year climate data and treated as an interaction term, as previously described [20, 32, 72]. GxE GWAA for birth weight produced evidence for three interactions on BTA26 and BTA22 interacting with two ecoregions (Table 5, Fig. 4; Additional File 1). Positional candidate genes identified by GxE interactions for birth weight have been previously associated with cattle feed efficiency (PRKG1, LOC531679, SEC61G, and NEK10) (Table S1; Additional File 2) [81,82,83,84,85,86,87]. Additionally, PRKG1 is involved in vasodilation (Table 5) [82]. Notably, only one interaction detected by GxE GWAA for birth weight was identified as a coding variant (Additional File 1). More specifically, the lead SNP within the positional candidate gene NEK10 encodes a nonsynonymous mutation in exon 2 (Ser → Thr). Four additional interactions were also noted with less statistical support, as described in Table S6 (Additional File 2). Genomic inflation factors for P-value estimates obtained from GxE GWAA for birth weight are presented in Table S7 (Additional File 2).

Birth weight genotype-by-environment interactions. Manhattan plots with -log10 P-values for U.S. Desert Ecoregion (a), U.S. Southeast Ecoregion (b), U.S. High Plains Ecoregion (c), U.S. Arid Prairie Ecoregion (d), U.S. Foothills Ecoregion (e), U.S. Forested Mountains Ecoregion (f), U.S. Fescue Belt Ecoregion (g), and U.S. Upper Midwest and Northeast Ecoregion (h). Lead and supporting SNPs for interactions represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 15,815 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 5
GxE GWAA for weaning weight in U.S. Red Angus beef cattle produced evidence for six significant interactions; thereby implicating positional candidate genes related to growth and development (DNAJC12), milk production (LOC112447568, TRNAE-UUC, LOC112447164, COX18), carcass traits (LOC112447496, LOC112447497, LOC782092), cellular proliferation and metabolism (SIRT1), and feed efficiency (LCLAT1), as defined by relevant lead SNPs (Table 6, Table S1, Fig. 5; Additional File 2) [88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109]. Additionally, positional candidate genes identified on BTA28 (DNAJC12 and SIRT1) have been associated with bovine maturity rate, milk production, and meat quality traits [46, 48, 91]; as well as promoting cellular proliferation and regulation in humans and mice [90, 92]. Interestingly, interactions on BTA7 (101 Mb) and BTA6 (88 Mb) revealed previous associations with aspects of bovine heat stress and thermotolerance, and both showed significant GxE interactions in the U.S. Desert Ecoregion (Table 6; Additional File 1) [89, 104, 105]. All interactions identified in the GxE GWAA for weaning weight in Red Angus cattle were located in noncoding regions (Table 6; Additional File 1). Sixteen additional interactions with less overall statistical support are noted in Table S8, with only one lead SNP encoding a nonsynonymous change within the positional candidate gene ANKK1 (Table S8; Additional File 1, Additional File 2). Genomic inflation factors for P-value estimates obtained from a GxE GWAA for weaning weight are summarized in Table S7 (Additional File 2).

Weaning weight genotype-by-environment interactions. Manhattan plots with -log10 P-values for U.S. Desert Ecoregion (a), U.S. Southeast Ecoregion (b), U.S. High Plains Ecoregion (c), U.S. Arid Prairie Ecoregion (d), U.S. Foothills Ecoregion (e), U.S. Forested Mountains Ecoregion (f), U.S. Fescue Belt Ecoregion (g), and U.S. Upper Midwest and Northeast Ecoregion (h). Lead and supporting SNPs for interactions represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 15,620 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 6
We conducted a final GxE GWAA for yearling weight in U.S. Red Angus beef cattle and found evidence for five interactions which met a nominal significance threshold for polygenic traits (P ≤ 1e-05), as summarized in Table 7 and Fig. 6 (Additional File 1) [71]. The positional candidate genes corresponding to each interaction have been associated with cellular proliferation and differentiation (EDNRB, POU4F1, and DNAJC12), bovine development (PARD3B, NRP2, and SIRT1), neural development (PARD3B and NRP2), and carcass traits (ZHX3) (Table 7, Table S1; Additional File 2) [110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125]. All lead SNPs defining GxE associations for yearling weight via GWAA were located in noncoding regions. Positional candidate genes underlying an interaction detected on BTA28 at 24 Mb (i.e., DNAJC1 and SIRT1) strongly suggest that pleiotropic GxE interactions exist with respect to weaning weight (Table 6) and yearling weight (Table 7) for U.S. Red Angus beef cattle. Moreover, pleiotropic GxE interactions for weaning weight and yearling weight were also detected on BTA8 at 15 Mb, and on BTA27 at 39 Mb, including positional candidate genes LINGO2, LOC112447774, and PSD3 (Tables S8-S9; Additional File 2), but with less overall statistical support. All pleiotropic associations detected for U.S. Red Angus beef cattle are summarized in Table S1 (Additional File 2). Genomic inflation factors for P-value estimates obtained from a GxE GWAA for yearling weight are displayed in Table S7 (Additional File 2).

Yearling weight genotype-by-environment interactions. Manhattan plots with -log10 P- values for U.S. Desert Ecoregion (a), U.S. Southeast Ecoregion (b), U.S. High Plains Ecoregion (c), U.S. Arid Prairie Ecoregion (d), U.S. Foothills Ecoregion (e), U.S. Forested Mountains Ecoregion (f), U.S. Fescue Belt Ecoregion (g), and U.S. Upper Midwest and Northeast Ecoregion (h). Lead and supporting SNPs for interactions represented at or above the blue line (P ≤ 1e-05; −log10 P-values ≥ 5.00) for n = 12,388 U.S. Red Angus beef cattle. A summary of all markers passing the nominal significance threshold is presented in Table 7
Conclusions
Herein, we present evidence for pleiotropic QTL resulting from traditional GWAA influencing birth weight, weaning weight, and yearling weight in U.S. Red Angus beef cattle, and further confirm the involvement of genomic regions on BTA6 from 34 to 41 Mb and BTA14 from 23 to 25 Mb in various aspects of bovine growth, feed efficiency, carcass traits, and stature across breeds. Additionally, the results from our GWAA and GxE GWAA for birth weight, weaning weight, and yearling weight in U.S. Red Angus beef cattle provide compelling comparative evidence for at least 35 statistically well-supported associations segregating across multiple cattle breeds; including three U.S. Red Angus growth associations that were previously detected for mid-test metabolic weight, two QTL previously detected for average daily gain in U.S. SimAngus and Hereford beef cattle, and 11 U.S. Red Angus growth associations also detected in U.S. Gelbvieh beef cattle. Examination of GxE GWAA associations for U.S. Red Angus growth traits revealed ecoregion-specific positional candidate genes with suggested pleiotropy for genes DNAJC12, and SIRT1 for weaning weight and yearling weight. Similar to previous GWAA and GxE GWAA on U.S. Gelbvieh and Simmental beef cattle, significant GxE associations detected for birth weight, weaning weight, and yearling weight in U.S. Red Angus cattle were not overlapping; thereby suggesting that although the majority of the main effect QTL were conserved between breeds, GxE interactions were not conserved. In agreement with previous GxE and local adaptation results in beef cattle, we find GxE effects associated with vasodilation and neural development. Identification of pleiotropic growth QTL and breed specific GxE interactions may potentially serve to benefit beef breeding programs across diverse U.S. climates via creation of region-specific genomic predictions. Moreover, the results of this study further demonstrate that imputation to a union set of high-density SNPs (i.e., 836K) can directly facilitate future studies at a fraction of the cost associated with direct genotyping; thus providing a research framework that directly enables large-scale analyses for economically important livestock species, and the potential for identifying causal variants via genome sequence-level imputed genotypes.
Methods
Birth weight (n = 17,320; in 5803 contemporary groups), weaning weight (n = 17,306; in 6478 contemporary groups), and yearling weight (n = 13,648; in 4809 contemporary groups) phenotypes were obtained from the Red Angus Association of America for animals with genotypes. These animals had 2102 unique sires and 12,124 unique dams. Analyzed animals were born from 1975 to 2017. Phenotypes were pre-adjusted by Red Angus Association of America for age of animal and age of the dam (i.e., 205-day weight for weaning weight). Phenotypes were further adjusted using the mmer() function in the sommer package v3.9.3 in R v3.5.2 [126, 127] to account for contemporary group effects using contemporary group IDs supplied by Red Angus Association of America. Details regarding Red Angus contemporary groups are summarized in Additional File 1. Birth weight and weaning weight were adjusted for maternal effect using pedigree files provided by the Red Angus Association of America. Discrete climate ecoregions were designated for each individual using K-means clustering with three continuous climate variables (mean temperature in Fahrenheit, precipitation in inches, and elevation in feet) gathered from the PRISM climate dataset from thirty years of normalized records [128]. The pamk function in conjunction with the kmeans algorithm in the fpc (Flexible Procedures for Clustering) [129] package and the RStoolbox package [127, 130] in R assigned every four kilometer (km) square of the continental U.S. to one of 9 clusters, denoted as ecoregions. These designated ecoregions consist of the Upper Midwest & Northeast, Fescue Belt, Rainforest, Forested Mountains, High Plains, Foothills, Desert, Southeast, and Arid Prairie. Animals were assigned to ecoregions by breeder zip-code as recorded in the U.S. Red Angus Association of America herdbook [32]. If the breeder’s zip-code overlapped with two or more ecoregions, the animal was filtered from further analysis.
Genotypes from 22,932 U.S. Red Angus cattle were provided by Neogen GeneSeek (Lincoln, NE, U.S.A). The ARS-UCD1.2 Bos taurus assembly [131] was used for SNP positions. The genotypes underwent filtering using PLINK 1.9 to remove individuals with call rates < 0.90 on an assay-by-assay basis (i.e., GeneSeek GGP-LDv3, GeneSeek GGP-LDv4, GeneSeek GGP-90KT, GeneSeek GGP-HDv3, GeneSeek Bovine-GGP-F250, Illumina Bovine SNP50, and Illumina HD 778K), removal of variants with call rates < 0.90 and Hardy-Weinberg Equilibrium (HWE) P-values < 1e-20 to exclude poorly genotyped loci [132]. Only autosomal chromosomes were utilized in these analyses. The remaining 22,457 individuals and associated genotypes were then merged and phased using PLINK and EagleV2.4, respectively [133]. Phased haplotypes for 8622 diverse individuals genotyped using the Illumina HD (778K SNPs; Illumina, San Diego, CA) and 28,114 individuals genotyped using the Bovine-GGP-F250 (250K SNPs; GeneSeek, Lincoln, NE) were used as a multi-breed reference panel for imputation in minimac4 as previously described [19, 134]. The 22,457 Red Angus genotypes from various assays were imputed for all markers contained on the two high-density research chips in this multi-breed reference panel. A total of 6642 cattle had only genotype information, thus providing 15,815 individuals with 836,118 markers each (ARS-UCD1.2) to be utilized as the final dataset for GWAA and GxE GWAA. Minimac4 reported imputed dosage genotypes to account for any potential uncertainty during imputation processes, as previously described [19, 134].
Imputed genotypes (836K markers) and the adjusted phenotypes for Red Angus cattle were used to conduct univariate linear mixed model GWAA for birth weight (15,815 individuals), weaning weight (15,620 individuals), and yearling weight (12,388 individuals) using the program GEMMA. Prior to the execution of all GWAA, GEMMA filtered all SNP loci as follows: MAF (< 0.01 excluded), polymorphism (monomorphic SNPs excluded), and Hardy-Weinberg Equilibrium (HWE; P-values < 0.001 excluded), thereby producing genotypic sets of 675,115 SNPs for birth weight, 675,060 SNPs for weaning weight, and 674,493 SNPs for yearling weight. Genomic relationship matrices (Gs) were computed with the imputed genotypes in GEMMA to control for dependence between samples due to relatedness. The linear mixed models implemented in GEMMA also estimate the proportion of variance explained (PVE) by the genomic relationship matrix. The PVE is also referred to as “chip heritability” [135]. The univariate linear mixed model implemented for GWAA can be generally specified as: y = Wα + xβ + u + ϵ; where y is a n-vector of quantitative traits (i.e., birth weight, weaning weight, and yearling weight) for n-Red Angus individuals, W is an n x c matrix of specified covariates (i.e., fixed effects) including a column of 1s, α is a c-vector of corresponding coefficients including the intercept, x is an n-vector of SNP genotypes, β is the effect size of the SNP, u is an n-vector of random effects, and ϵ represents an n-vector of errors [20, 135]. Additionally, u ∼ MVNn(0, λτ−1Κ) and ϵ ∼ MVNn(0, τ−1I), where MVN denotes multivariate normal distribution, τ−1 is the variance of the residual errors, λ is the ratio between the two variance components, Κ is the n x n genomic relatedness matrix, and I represents an n x n identity matrix [20, 135]. Specifically, GEMMA performed a Wald test using -lmm 1 as follows: \({F}_{Wald}=\frac{{\hat{\beta}}^2}{V\left(\hat{\beta}\right)}\), tests the alternative hypothesis (\({H}_1:\hat{\beta}\ne 0\Big)\) for each SNP against the null hypothesis for each SNP (\({H}_0:\hat{\beta}=0\)). Moreover, \(\hat{\beta}={\left({x}^T{P}_c\left({\hat{\lambda}}_r\right)x\right)}^{-1}\left({x}^T{P}_c\left({\hat{\lambda}}_r\right)y\right)\) is the estimate for β obtained using the restricted maximum likelihood (REML) estimate \({\hat{\lambda}}_r\) in the alternative model; and \(V\ \left(\hat{\beta}\right)={\left(n-c-1\right)}^{-1}{\left({x}^T{P}_c\left({\hat{\lambda}}_r\right)x\right)}^{-1}\left({y}^T{P}_x\left({\hat{\lambda}}_r\right)y\right)\) is the variance for \(\hat{\beta}\) [20, 135]. Under the null hypothesis, the Wald test statistics (FWald) come from an F(1, n − c − 1) distribution [20, 135]; with GEMMA producing marker-based REML estimates and corresponding P-values. For all GxE GWAAs, discrete geographic ecoregion (i.e., the environmental variable) was specified as an interaction term using the -gxe command. GxE GWAA were computed using a mixed model which can be generally specified as: y = Wα + xsnpβsnp + xenvβenv + xsnp × xenvβsnp × env + u + ϵ; where y is an n-vector of quantitative traits (i.e., birth weight, weaning weight, and yearling weight) for n-Red Angus individuals, W represents an n x c matrix of specified covariates, α is a c-vector of corresponding coefficients including the intercept, xsnp represents an n-vector of SNP genotypes, βsnp is the effect size of the SNP, xenv is an n-vector of membership in a single ecoregion, βenv represents the fixed effect of the ecoregion, βsnp × env is the estimated interaction between SNP genotype and ecoregion, u is an n-vector of random effects, and ϵ is an n-vector of errors [20, 135]. As above, u ∼ MVNn(0, λτ−1Κ) and ϵ ∼ MVNn(0, τ−1I). Each discrete ecoregion was compared against the remaining U.S. dataset using binary (0, 1) coding as an environmental variable with one exception; the Rainforest ecoregion had insufficient sample size for GxE GWAA, and thus eight separate GxE GWAA were computed (Additional File 1). GEMMA evaluated the alternative hypothesis for each interaction (H1 : βsnp × env ≠ 0) in comparison to the null hypothesis (H0 : βsnp × env = 0) using linear mixed models while controlling for population stratification, SNP main effect, and environmental effect while examining the interaction effect of each ecoregion [20, 135]. Single-marker P-value results produced by GEMMA using the -lmm 1 and -gxe commands were further adjusted using chi-squared test statistics divided by a constant for additional genomic control [37, 136]. Adjusted P-value results were utilized to produce Manhattan plots using the manhattan command in R [137]. All SNPs meeting the nominal significance threshold (P ≤ 1e-05) were rounded to the nearest Mb and strongly supported QTL were defined by ≥ 5 significant SNP loci with MAF ≥ 0.01 (i.e., a lead SNP plus four or more additional supporting SNPs within the same rounded Mb) [31, 71]. Additional QTL were also noted with less overall statistical support in Tables S2, S4-S6, and S8-S9; thereby representing QTL defined by ≥ 2 but ≤ 4 SNP loci which met the nominal significance threshold (P ≤ 1e-05) within the same rounded Mb (Additional File 2). Positional candidate genes were implicated by location of the lead SNP. Genomic inflation factors (λ) were estimated using observed and expected P-values via regression for all GWAA and GxE GWAA in R [80, 138, 139]. The proportion of variance explained (PVE) by bovine SNPs was estimated as previously described [140]. Genetic correlations were estimated using the multivariate approach implemented in GEMMA [135, 141], as previously described [142].
Availability of data and materials
Third party data analyzed in the present study are available for non-commercial use via data use agreement (DUA) with the Red Angus Association of America.
References
Barwick SA, Henzell AL. Development successes and issues for the future in deriving and applying selection indexes for beef breeding. Aust J Exp Agric. 2005;45:923. https://doi.org/10.1071/EA05068.
Eriksson S, Näsholm A, Johansson K, Philipsson J. Genetic parameters for calving difficulty, stillbirth, and birth weight for Hereford and Charolais at first and later parities1. J Anim Sci. 2004;82:375–83. https://doi.org/10.2527/2004.822375x.
Cook BR, Tess MW, Kress DD. Effects of selection strategies using heifer pelvic area and sire birth weight expected progeny difference on dystocia in first-calf heifers. J Anim Sci. 1993;71:602–7. https://doi.org/10.2527/1993.713602x.
Bourdon RM, Brinks JS. Genetic, environmental and phenotypic relationships among gestation length, birth weight, growth traits and age at first calving in beef cattle. J Anim Sci. 1982;55:543–53. https://doi.org/10.2527/jas1982.553543x.
Boligon AA, Silva JAV, Sesana RC, Sesana JC, Junqueira JB, Albuquerque LG. Estimation of genetic parameters for body weights, scrotal circumference, and testicular volume measured at different ages in Nellore cattle1. J Anim Sci. 2010;88:1215–9. https://doi.org/10.2527/jas.2008-1719.
Utsunomiya YT, do Carmo AS, Carvalheiro R, HHHR N, Matos MC, Zavarez LB, et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013;14:52. https://doi.org/10.1186/1471-2156-14-52.
Spelman RJ, Huisman AE, Singireddy SR, Coppieters RJ, Arranz J, Georges M, et al. Quantitative trait loci analysis on 17 nonproduction traits in the New Zealand dairy population. J Dairy Sci. 2000;83:370. https://doi.org/10.3168/JDS.S0022-0302(00)74890-9.
Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, et al. Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci. 2004;82:3405–14. https://doi.org/10.2527/2004.82123405x.
Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A. Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein Jersey crossbred population. Anim Genet. 2008;40:27–34. https://doi.org/10.1111/j.1365-2052.2008.01793.x.
McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607. https://doi.org/10.1111/j.1365-2052.2010.02063.x.
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408. https://doi.org/10.1186/1471-2164-12-408.
Santiago GG, Siqueira F, Cardoso FF, Regitano LCA, Ventura R, Sollero BP, et al. Genomewide association study for production and meat quality traits in Canchim beef cattle. J Anim Sci. 2017;95:3381–90.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4:e5350. https://doi.org/10.1371/journal.pone.0005350.
Decker JE. Agricultural genomics: commercial applications bring increased basic research power. PLoS Genet. 2015;11:e1005621. https://doi.org/10.1371/journal.pgen.1005621.
Rincon G, Weber KL, Van Eenennaam AL, Golden BL, Medrano JF. Hot topic: performance of bovine high-density genotyping platforms in Holsteins and jerseys. J Dairy Sci. 2011;94:6116–21. https://doi.org/10.3168/JDS.2011-4764.
Garrick DJ. The nature, scope and impact of genomic prediction in beef cattle in the United States. Genet Sel Evol. 2011;43:17. https://doi.org/10.1186/1297-9686-43-17.
VanRaden PM, Null DJ, Sargolzaei M, Wiggans GR, Tooker ME, Cole JB, et al. Genomic imputation and evaluation using high-density Holstein genotypes. J Dairy Sci. 2013;96:668–78.
Wiggans GR, VanRaden PM, Cooper TA. The genomic evaluation system in the United States: past, present, future. J Dairy Sci. 2011;94:3202–11. https://doi.org/10.3168/jds.2010-3866.
Rowan TN, Hoff JL, Crum TE, Taylor JF, Schnabel RD, Decker JE. A multi-breed reference panel and additional rare variants maximize imputation accuracy in cattle. Genet Sel Evol. 2019;51:77. https://doi.org/10.1186/s12711-019-0519-x.
Smith JL, Wilson ML, Nilson SM, Rowan TN, Oldeschulte DL, Schnabel RD, et al. Genome-wide association and genotype by environment interactions for growth traits in U.S. Gelbvieh cattle. BMC Genomics. 2019;20:926. https://doi.org/10.1186/s12864-019-6231-y.
Saatchi M, McClure MC, McKay SD, Rolf MM, Kim J, Decker JE, et al. Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet Sel Evol. 2011;43:40.
Saatchi M, Schnabel RD, Rolf MM, Taylor JF, Garrick DJ. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet Sel Evol. 2012;44:38.
Saatchi M, Garrick DJ, Tait RG, Mayes MS, Drewnoski M, Schoonmaker J, et al. Genome-wide association and prediction of direct genomic breeding values for composition of fatty acids in Angus beef cattle. BMC Genomics. 2013;14:730.
Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442.
Ryu J, Lee C. Genomic heritability of bovine growth using a mixed model. Asian Australasian J Anim Sci. 2014;27:1521–5. https://doi.org/10.5713/ajas.2014.14287.
Pryce JE, Hayes BJ, Bolormaa S, Goddard ME. Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011;187:981–4.
Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–15.
Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83.
Pausch H, Flisikowski K, Jung S, Emmerling R, Edel C, Götz KU, et al. Genome-wide association study identifies two major loci affecting calving ease and growth-related traits in cattle. Genetics. 2011;187:289–97.
Abo-Ismail MK, Brito LF, Miller SP, Sargolzaei M, Grossi DA, Moore SS, et al. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet Sel Evol. 2017;49:82.
Seabury CM, Oldeschulte DL, Saatchi M, Beever JE, Decker JE, Halley YA, et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genomics. 2017;18:386. https://doi.org/10.1186/s12864-017-3754-y.
Rowan TN, Durbin HJ, Seabury CM, Schnabel RD, Decker JE. Powerful detection of polygenic selection and evidence of environmental adaptation in US beef cattle. PLoS Genet. 2021;17:e1009652. https://doi.org/10.1371/journal.pgen.1009652.
Eltaher S, Baenziger PS, Belamkar V, Emara HA, Nower AA, Salem KFM, et al. GWAS revealed effect of genotype × environment interactions for grain yield of Nebraska winter wheat. BMC Genomics. 2021;22:1–14. https://doi.org/10.1186/s12864-020-07308-0.
Spindel JE, Begum H, Akdemir D, Collard B, Redoña E, Jannink JL, et al. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity (Edinb). 2016;116:395–408. https://doi.org/10.1038/hdy.2015.113.
Zheng JS, Lai CQ, Parnell LD, Lee YC, Shen J, Smith CE, et al. Genome-wide interaction of genotype by erythrocyte n-3 fatty acids contributes to phenotypic variance of diabetes-related traits. BMC Genomics. 2014;15:1–13. https://doi.org/10.1186/1471-2164-15-781.
Boldt RJ, Speidel SE, Thomas MG, Enns RM. Genetic parameters for fertility and production traits in red Angus cattle. J Anim Sci. 2018;96:4100–11. https://doi.org/10.1093/jas/sky294.
Xia J, Fan H, Chang T, Xu L, Zhang W, Song Y, et al. Searching for new loci and candidate genes for economically important traits through gene-based association analysis of Simmental cattle. Sci Rep. 2017;7:1–9.
Jiang J, Ma L, Prakapenka D, VanRaden PM, Cole JB, Da Y. A large-scale genome-wide association study in U.S. Holstein cattle. Front Genet. 2019;10:412.
Olsen HG, Hayes BJ, Kent MP, Nome T, Svendsen M, Lien S. A genome wide association study for QTL affecting direct and maternal effects of stillbirth and dystocia in cattle. Anim Genet. 2010;41:273–80. https://doi.org/10.1111/j.1365-2052.2009.01998.x.
Zhang F, Wang Y, Mukiibi R, Chen L, Vinsky M, Plastow G, et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: feed efficiency and component traits. BMC Genomics. 2020;21:36. https://doi.org/10.1186/s12864-019-6362-1.
An B, Xia J, Chang T, Wang X, Xu L, Zhang L, et al. Genome-wide association study reveals candidate genes associated with body measurement traits in Chinese wagyu beef cattle. Anim Genet. 2019;50:386–90. https://doi.org/10.1111/age.12805.
Akanno EC, Chen L, Abo-Ismail MK, Crowley JJ, Wang Z, Li C, et al. Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle. Genet Sel Evol. 2018;50:48. https://doi.org/10.1186/s12711-018-0405-y.
Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, et al. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) gene. Genetics. 2009;183:951–64.
Liu Y, Duan X, Chen S, He H, Liu X. NCAPG is differentially expressed during longissimus muscle development and is associated with growth traits in Chinese Qinchuan beef cattle. Genet Mol Biol. 2015;38:450–6.
Lindholm-Perry AK, Kuehn LA, Oliver WT, Sexten AK, Miles JR, Rempel LA, et al. Adipose and muscle tissue gene expression of two genes (NCAPG and LCORL) located in a chromosomal region associated with cattle feed intake and gain. PLoS One. 2013;8:e80882. https://doi.org/10.1371/journal.pone.0080882.
Meredith BK, Kearney FJ, Finlay EK, Bradley DG, Fahey AG, Berry DP, et al. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012;13:21. https://doi.org/10.1186/1471-2156-13-21.
Bouwman AC, Daetwyler HD, Chamberlain AJ, Ponce CH, Sargolzaei M, Schenkel FS, et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat Genet. 2018;50:362–7. https://doi.org/10.1038/s41588-018-0056-5.
Crispim AC, Kelly MJ, Guimarães SEF, FF ES, MRS F, Wenceslau RR, et al. Multi-trait GWAS and new candidate genes annotation for growth curve parameters in Brahman cattle. PLoS One. 2015;10:e0139906. https://doi.org/10.1371/journal.pone.0139906.
Kiser JN, Keuter EM, Seabury CM, Neupane M, Moraes JGN, Dalton J, et al. Validation of 46 loci associated with female fertility traits in cattle. BMC Genomics. 2019;20:576. https://doi.org/10.1186/s12864-019-5935-3.
Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ, Everts-Van Der Wind A, Lee JH, et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–44.
Do DN, Bissonnette N, Lacasse P, Miglior F, Sargolzaei M, Zhao X, et al. Genome-wide association analysis and pathways enrichment for lactation persistency in Canadian Holstein cattle. J Dairy Sci. 2017;100:1955–70.
Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, et al. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-met substitution in NCAPG as a positional candidate. BMC Genet. 2009;10:43.
Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, et al. A multi-trait, Meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 2014;10:e1004198.
Pryce JE, Bolormaa S, Chamberlain AJ, Bowman PJ, Savin K, Goddard ME, et al. A validated genome-wide association study in 2 dairy cattle breeds for milk production and fertility traits using variable length haplotypes. J Dairy Sci. 2010;93:3331–45. https://doi.org/10.3168/jds.2009-2893.
Uzbekova S, Arlot-Bonnemains Y, Dupont J, Dalbiès-Tran R, Papillier P, Pennetier S, et al. Spatio-temporal expression patterns of Aurora kinases a, B, and C and cytoplasmic polyadenylation-element-binding protein in bovine oocytes during meiotic Maturation1. Biol Reprod. 2008;78:218–33. https://doi.org/10.1095/biolreprod.107.061036.
Buzanskas ME, Grossi DA, Ventura RV, Schenkel FS, Sargolzaei M, Meirelles SLC, et al. Genome-wide association for growth traits in canchim beef cattle. PLoS One. 2014;9:e94802.
Hancock AM, Witonsky DB, Gordon AS, Eshel G, Pritchard JK, Coop G, et al. Adaptations to climate in candidate genes for common metabolic disorders. PLoS Genet. 2008;4:e32. https://doi.org/10.1371/journal.pgen.0040032.
Yurchenko AA, Deniskova TE, Yudin NS, Dotsev AV, Khamiruev TN, Selionova MI, et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genomics. 2019;20:1–19.
Bolormaa S, Pryce JE, Kemper KE, Hayes BJ, Zhang Y, Tier B, et al. Detection of quantitative trait loci in Bos indicus and Bos taurus cattle using genome-wide association studies. Genet Sel Evol. 2013;45:1–12. https://doi.org/10.1186/1297-9686-45-43.
Saunders J, Wisidagama DR, Morford T, Malone CS. Maximal expression of the evolutionarily conserved Slit2 gene promoter requires Sp1. Cell Mol Neurobiol. 2016;36:955–64.
Iqbal Z, Cejudo-Martin P, de Brouwer A, van der Zwaag B, Ruiz-Lozano P, Scimia MC, et al. Disruption of the Podosome adaptor protein TKS4 (SH3PXD2B) causes the skeletal dysplasia, eye, and cardiac abnormalities of frank-Ter Haar syndrome. Am J Hum Genet. 2010;86:254–61.
Gebreyesus G, Buitenhuis AJ, Poulsen NA, Visker MHPW, Zhang Q, Van Valenberg HJF, et al. Multi-population GWAS and enrichment analyses reveal novel genomic regions and promising candidate genes underlying bovine milk fatty acid composition. BMC Genomics. 2019;20:178. https://doi.org/10.1186/s12864-019-5573-9.
Mosley JD, Shaffer CM, Van Driest SL, Weeke PE, Wells QS, Karnes JH, et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenomics J. 2016;16:231–7. https://doi.org/10.1038/tpj.2015.51.
Hu W, Zuo J, De Jager PL, Heintz N. The human glutamate receptor δ2 gene (GRID2) maps to chromosome 4q22. Genomics. 1998;47:143–5.
Patel K, Scrimieri F, Ghosh S, Zhong J, Kim M-S, Ren YR, et al. FAM190A deficiency creates a cell division defect. Am J Pathol. 2013;183:296–303. https://doi.org/10.1016/j.ajpath.2013.03.020.
Wang Y, Zhang F, Mukiibi R, Chen L, Vinsky M, Plastow G, et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: II: carcass merit traits. BMC Genomics. 2020;21:36. https://doi.org/10.1186/s12864-019-6273-1.
Qrafli M, Asekkaj I, Bourkadi JE, El Aouad R, Sadki K. New variant identified in major susceptibility locus to tuberculosis on chromosomal region 8q12-q13 in Moroccan population: a case control study. BMC Infect Dis. 2017;17:712.
Rolf MM, Taylor JF, Schnabel RD, McKay SD, McClure MC, Northcutt SL, et al. Genome-wide association analysis for feed efficiency in Angus cattle. Anim Genet. 2012;43:367–74. https://doi.org/10.1111/j.1365-2052.2011.02273.x.
Hernandez MC, Andres-Barquin PJ, Martinez S, Bulfone A, Rubenstein JLR, Israel MA. ENC-1: a novel mammalian kelch-related gene specifically expressed in the nervous system encodes an actin-binding protein. J Neurosci. 1997;17:3038–51.
Moriwaki Y, Yoshikawa K, Fukuda H, Fujii YX, Misawa H, Kawashima K. Immune system expression of SLURP-1 and SLURP-2, two endogenous nicotinic acetylcholine receptor ligands. Life Sci. 2007;80:2365–8.
Wellcome Trust Case Control Consortium TWTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. https://doi.org/10.1038/nature05911.
Braz CU, Rowan TN, Schnabel RD, Decker JE. Genome-wide association analyses identify genotype-by-environment interactions of growth traits in Simmental cattle. Sci Reports. 2021;11:1–15. https://doi.org/10.1038/s41598-021-92455-x.
Chander P, Kennedy MJ, Winckler B, Weick JP. Neuron-specific gene 2 (NSG2) encodes an AMPA receptor interacting protein that modulates excitatory neurotransmission. eNeuro. 2019;6:292–310. https://doi.org/10.1523/ENEURO.0292-18.2018.
Rosen H, Krichevsky A, Polakiewicz RD, Benzakine S, Bar-Shavit Z. Developmental regulation of proenkephalin gene expression in osteoblasts. Mol Endocrinol. 1995;9:1621–31. https://doi.org/10.1210/mend.9.11.8584038.
Zhen Y, Xinghui Z, Chao W, Yi Z, Jinwen C, Ruifang G, et al. Several microRNAs could predict survival in patients with hepatitis B-related liver cancer. Sci Rep. 2017;7:45195.
Goda N, Murase H, Kasezawa N, Goda T, Yamakawa-Kobayashi K. Polymorphism in microRNA-binding site in HNF1B influences the susceptibility of type 2 diabetes mellitus: a population based case-control study. BMC Med Genet. 2015;16:75.
Ying G, Frederick JM, Baehr W. Deletion of both centrin 2 (CETN2) and CETN3 destabilizes the distal connecting cilium of mouse photoreceptors. J Biol Chem. 2019;294:3957–73.
Néant I, Mellström B, Gonzalez P, Naranjo JR, Moreau M, Leclerc C. Kcnip1 a Ca2+−dependent transcriptional repressor regulates the size of the neural plate in Xenopus. Biochim Biophys Acta - Mol Cell Res. 2014;1853:2077–85.
Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9:1–13.
Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–12.
Bouwman AC, Bovenhuis H, Visker MHPW, van Arendonk JAM. Genome-wide association of milk fatty acids in Dutch dairy cattle. BMC Genet. 2011;12:43. https://doi.org/10.1186/1471-2156-12-43.
Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci. 2006;11:356–67. https://doi.org/10.2741/1803.
Ibeagha-Awemu EM, Peters SO, Akwanji KA, Imumorin IG, Zhao X. High density genome wide genotyping-by-sequencing and association identifies common and low frequency SNPs, and novel candidate genes influencing cow milk traits. Sci Rep. 2016;6:1–18. https://doi.org/10.1038/srep31109.
Hardie LC, VandeHaar MJ, Tempelman RJ, Weigel KA, Armentano LE, Wiggans GR, et al. The genetic and biological basis of feed efficiency in mid-lactation Holstein dairy cows. J Dairy Sci. 2017;100:9061–75. https://doi.org/10.3168/jds.2017-12604.
Lu Z, Zhou L, Killela P, Rasheed AB, Di C, Poe WE, et al. Glioblastoma proto-oncogene SEC61γ is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009;69:9105–11. https://doi.org/10.1158/0008-5472.CAN-09-2775.
Moniz LS, Stambolic V. Nek10 mediates G2/M cell cycle arrest and MEK autoactivation in response to UV irradiation. Mol Cell Biol. 2011;31:30–42. https://doi.org/10.1128/mcb.00648-10.
Hilgendorf KI, Johnson CT, Jackson PK. The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol. 2016;39:84–92. https://doi.org/10.1016/j.ceb.2016.02.008.
Höglund JK, Sahana G, Guldbrandtsen B, Lund MS. Validation of associations for female fertility traits in Nordic Holstein, Nordic red and Jersey dairy cattle. BMC Genet. 2014;15:8. https://doi.org/10.1186/1471-2156-15-8.
Alings F, Sarin LP, Fufezan C, Drexler HCA, Leidel SA. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA. 2015;21:202–12. https://doi.org/10.1261/rna.048199.114.
Uno Y, Kanda M, Miwa T, Umeda S, Tanaka H, Tanaka C, et al. Increased expression of DNAJC12 is associated with aggressive phenotype of gastric Cancer. Ann Surg Oncol. 2019;26:836–44.
Gui L, Wang H, Wei S, Zhang Y, Zan L. Molecular characterization, expression profiles, and analysis of Qinchuan cattle SIRT1 gene association with meat quality and body measurement traits (Bos taurus). Mol Biol Rep. 2014;41:5237–46. https://doi.org/10.1007/s11033-014-3393-1.
Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2005;2:67–76.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, et al. A whole-genome scan to map quantitative trait loci for conformation and functional traits in Canadian Holstein bulls. J Dairy Sci. 2008;91:2844–56.
Lu D, Miller S, Sargolzaei M, Kelly M, Vander Voort G, Caldwell T, et al. Genome-wide association analyses for growth and feed efficiency traits in beef cattle. J Anim Sci. 2013;91:3612–33. https://doi.org/10.2527/jas.2012-5716.
Mateescu RG, Garrick DJ, Reecy JM. Network analysis reveals putative genes affecting meat quality in Angus cattle. Front Genet. 2017;8:171. https://doi.org/10.3389/fgene.2017.00171.
Kasahara T, Kubota-Sakashita M, Nagatsuka Y, Hirabayashi Y, Hanasaka T, Tohyama K, et al. Cardiolipin is essential for early embryonic viability and mitochondrial integrity of neurons in mammals. FASEB J. 2020;34:1465–80. https://doi.org/10.1096/fj.201901598R.
Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–65.
Zhou C, Li C, Cai W, Liu S, Yin H, Shi S, et al. Genome-wide association study for milk protein composition traits in a chiniese Holstein population using a single-step approach. Front Genet. 2019;10:72. https://doi.org/10.3389/fgene.2019.00072.
Lemos MVA, Chiaia HLJ, Berton MP, Feitosa FLB, Aboujaoud C, Camargo GMF, et al. Genome-wide association between single nucleotide polymorphisms with beef fatty acid profile in Nellore cattle using the single step procedure. BMC Genomics. 2016;17:213. https://doi.org/10.1186/s12864-016-2511-y.
Jiang J, Cole JB, Freebern E, Da Y, VanRaden PM, Ma L. Functional annotation and Bayesian fine-mapping reveals candidate genes for important agronomic traits in Holstein bulls. Commun Biol. 2019;2:1–12. https://doi.org/10.1038/s42003-019-0454-y.
Raven LA, Cocks BG, Hayes BJ. Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genomics. 2014;15:62. https://doi.org/10.1186/1471-2164-15-62.
Yan Z, Wang Z, Zhang Q, Yue S, Yin B, Jiang Y, et al. Identification of whole-genome significant single nucleotide polymorphisms in candidate genes associated with body conformation traits in Chinese Holstein cattle. Anim Genet. 2020;51:141–6. https://doi.org/10.1111/age.12865.
Bourens M, Barrientos A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J Biol Chem. 2017;292:7774–83. https://doi.org/10.1074/jbc.M117.778514.
Gaisne M, Bonnefoy N. The COX18 gene, involved in mitochondrial biogenesis, is functionally conserved and tightly regulated in humans and fission yeast. FEMS Yeast Res. 2006;6:869–82. https://doi.org/10.1111/j.1567-1364.2006.00083.x.
Timón-Gómez A, Bartley-Dier EL, Fontanesi F, Barrientos A. HIGD-driven regulation of cytochrome c oxidase biogenesis and function. Cells. 2020;9:2620. https://doi.org/10.3390/cells9122620.
Magalhães AFB, De Camargo GMF, Junior Fernandes GA, Gordo DGM, Tonussi RL, Costa RB, et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS One. 2016;11:e0157845. https://doi.org/10.1371/journal.pone.0157845.
Oliveira Júnior GA, Perez BC, Cole JB, Santana MHA, Silveira J, Mazzoni G, et al. Genomic study and medical subject headings enrichment analysis of early pregnancy rate and antral follicle numbers in Nelore heifers. J Anim Sci. 2017;95:4796–812. https://doi.org/10.2527/jas2017.1752.
Barreto Amaral Teixeira D, Alves Fernandes Júnior G, Beraldo dos Santos Silva D, Bermal Costa R, Takada L, Gustavo Mansan Gordo D, et al. Genomic analysis of stayability in Nellore cattle. PLoS One. 2017;12:e0179076. https://doi.org/10.1371/journal.pone.0179076.
Gebreyesus G, Buitenhuis AJ, Poulsen NA, Visker MHPW, Zhang Q, van Valenberg HJF, et al. Combining multi-population datasets for joint genome-wide association and meta-analyses: the case of bovine milk fat composition traits. J Dairy Sci. 2019;102:11124–41.
Maskell LJ, Qamar K, Babakr AA, Hawkins TA, Heads RJ, Budhram-Mahadeo VS. Essential but partially redundant roles for POU4F1/Brn-3a and POU4F2/Brn-3b transcription factors in the developing heart. Cell Death Dis. 2017;8:e2861. https://doi.org/10.1038/cddis.2017.185.
Shin MK, Levorse JM, Ingram RS, Tilghman SM. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature. 1999;402:496–501. https://doi.org/10.1038/990040.
Lahav R, Ziller C, Dupin E, Le Douarin NM. Endothelin 3 promotes neural crest cell proliferation and mediates a vast increase in melanocyte number in culture. Proc Natl Acad Sci U S A. 1996;93(9):3892–7.
Gautier M, Flori L, Riebler A, Jaffrézic F, Laloé D, Gut I, et al. A whole genome Bayesian scan for adaptive genetic divergence in west African cattle. BMC Genomics. 2009;10:550. https://doi.org/10.1186/1471-2164-10-550.
Strachan FE, Spratt JC, Wilkinson IB, Johnston NR, Gray GA, Webb DJ. Systemic blockade of the endothelin-B receptor increases peripheral vascular resistance in healthy men. Hypertension. 1999;33:581–5. https://doi.org/10.1161/01.hyp.33.1.581.
Huang EJ, Liu W, Fritzsch B, Bianchi LM, Reichardt LF, Xiang M. Brn3a is a transcriptional regulator of soma size, target field innervation and axon pathfinding of inner ear sensory neurons. Dev (Cambridge)2. 2001;128:2421–32.
Liu WA, Chen S, Li Z, Lee CH, Mirzaa G, Dobyns WB, et al. PARD3 dysfunction in conjunction with dynamic HIPPO signaling drives cortical enlargement with massive heterotopia. Genes Dev. 2018;32:763–80. https://doi.org/10.1101/gad.313171.118.
Takekuni K, Ikeda W, Fujito T, Morimoto K, Takeuchi M, Monden M, et al. Direct binding of cell polarity protein PAR-3 to cell-cell adhesion molecule nectin at neuroepithelial cells of developing mouse. J Biol Chem. 2003;278:5497–500. https://doi.org/10.1074/jbc.C200707200.
Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–37. https://doi.org/10.1038/ncb1368.
Chen H, Bagri A, Zupicich JA, Zou Y, Stoeckli E, Pleasure SJ, et al. Neuropilin-2 regulates the development of select cranial and sensory nerves and hippocampal mossy fiber projections. Neuron. 2000;25:43–56.
Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, et al. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41.
Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129:4797–806.
Liu G, Clement LC, Kanwar YS, Avila-Casado C, Chugh SS. ZHX proteins regulate podocyte gene expression during the development of nephrotic syndrome. J Biol Chem. 2006;281:39681–92. https://doi.org/10.1074/jbc.M606664200.
Yamada K, Kawata H, Shou Z, Hirano S, Mizutani T, Yazawa T, et al. Analysis of zinc-fingers and homeoboxes (ZHX)-1-interacting proteins: molecular cloning and characterization of a member of the ZHX family, ZHX3. Biochem J. 2003;373:167–78. https://doi.org/10.1042/BJ20021866.
Zhao C, Tian F, Yu Y, Luo J, Mitra A, Zhan F, et al. Functional genomic analysis of variation on beef tenderness induced by acute stress in Angus cattle. Comp Funct Genomics. 2012;2012:1–11. https://doi.org/10.1155/2012/756284.
Buzanskas ME, Grossi D do A, Ventura RV, Schenkel FS, TCS C, Stafuzza NB, et al. Candidate genes for male and female reproductive traits in Canchim beef cattle. J Anim Sci Biotechnol. 2017;8:67. https://doi.org/10.1186/s40104-017-0199-8.
Covarrubias-Pazaran G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS One. 2016;11:1–15.
R Core Team. R: The R Project for Statistical Computing. R Foundation for Statistical 844 Computing. https://www.r-project.org/.
PRISM Climate Group. PRISM. 2011. http://prism.oregonstate.edu/.
Hennig C. CRAN - package fpc. 2013. https://cran.r-project.org/web/packages/fpc/index.html.
Leutner B, Horning N. Tools for Remote Sensing Data Analysis [R package RStoolbox version 0.1.10]. 2017. https://cran.r-project.org/web/packages/RStoolbox/index.html.
Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience. 2020;9:1–9. https://doi.org/10.1093/gigascience/giaa021.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. https://doi.org/10.1086/519795.
Loh P-R, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, et al. Reference-based phasing using the haplotype reference consortium panel. Nat Genet. 2016;48:1443–8. https://doi.org/10.1038/ng.3679.
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;48:955–9.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4. https://doi.org/10.1038/ng.2310.
Ueki M, Fujii M, Tamiya G. Quick assessment for systematic test statistic inflation/deflation due to null model misspecifications in genome-wide environment interaction studies. PLoS One. 2019;14:e0219825. https://doi.org/10.1371/journal.pone.0219825.
Turner SD. Qqman: an R package for visualizing GWAS results using Q-Q and Manhattan plots. J Open Source Softw. 2018;3:731. https://doi.org/10.21105/joss.00731.
Team RC. R: The R project for statistical computing. R Found Stat 844 Comput 2013. https://www.r-project.org/.
Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. https://doi.org/10.1111/j.0006-341X.1999.00997.x.
Shim H, Chasman DI, Smith JD, Mora S, Ridker PM, Nickerson DA, et al. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS One. 2015;10:e0120758.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9. https://doi.org/10.1038/nmeth.2848.
Pirinen M, Benner C, Marttinen P, Järvelin MR, Rivas MA, Ripatti S. biMM: efficient estimation of genetic variances and covariances for cohorts with high-dimensional phenotype measurements. Bioinformatics. 2017;33(15):2405–7. https://doi.org/10.1093/bioinformatics/btx166.
Acknowledgements
We sincerely appreciate the Red Angus Association of America and the Red Angus breeders for sharing the genotypes and phenotypes used in this research. The computation for this work completed at the University of Missouri was performed on the high-performance computing infrastructure provided by Research Computing Support Services and in part by the National Science Foundation under grant number CNS-1429294 at the University of Missouri, Columbia MO, DOI: https://doi.org/10.32469/10355/69802.
Funding
This project was supported by Agriculture and Food Research Initiative Competitive Grant no. 2016–68004-24827 from the USDA National Institute of Food and Agriculture to JED and CMS.
Author information
Authors and Affiliations
Contributions
JED and CMS conceptualized and designed the research. RDS managed data acquisition, storage, and retrieval. TNR performed initial quality control, phasing and imputation. SMN pre-adjusted phenotypes. JLS and MLW performed GWAA. JLS, JED, and CMS interpreted results. JLS, CMS, and JED wrote the initial version of the manuscript, which was edited by all authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Prior to the planning and onset of the current study, data were collected under typical production practices and reported to the Red Angus Association of America, for inclusion in an existing data repository; thus an animal care and use protocol is not necessary. Staff at the Red Angus Association of America have read and approved the manuscript.
Consent for publication
NA
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Description and acronym definitions for summary data. Summary data for all genome-wide association analyses (GWAA) and genotype-by-environment genome-wide association analyses (GxE GWAA) performed for bovine birth weight, weaning weight, and yearling weight in tabular format.
Additional file 2: Table S1.
Summary of QTL supporting pleiotropy detected for birth weight, weaning weight, and yearling weight GWAA and GxE GWAA in U.S. Red Angus cattle. Table S2. Summary of QTL with 2 to 4 supporting SNPs detected for birth weight in U.S. Red Angus cattle. Table S3. Genomic inflation factors (λ) calculated using observed P-values and expected P-values for GWAA for growth traits in U.S. Red Angus beef cattle. Table S4. Summary of QTL with 2 to 4 supporting SNPs detected for weaning weight in U.S. Red Angus cattle. Table S5. Summary of QTL with 2 to 4 supporting SNPs detected for yearling weight in U.S. Red Angus cattle. Table S6. Summary of GxE interactions with 2 to 4 supporting SNPs detected for birth weight in U.S. Red Angus cattle. Table S7. Genomic inflation factors (λ) calculated using observed P-values and expected P-values for GxE GWAA for growth traits in U.S. Red Angus beef cattle. Table S8. Summary of GxE interactions with 2 to 4 supporting SNPs detected for weaning weight in U.S. Red Angus cattle. Table S9. Summary of GxE interactions with 2 to 4 supporting SNPs detected for yearling weight in U.S. Red Angus cattle.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Smith, J.L., Wilson, M.L., Nilson, S.M. et al. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genomics 23, 517 (2022). https://doi.org/10.1186/s12864-022-08667-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08667-6