
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease that inevitably results in dementia and death. Currently, there are no pathogenetically grounded methods for the prevention and treatment of AD, and all current treatment regimens are symptomatic and unable to significantly delay the development of dementia. The accumulation of β-amyloid peptide (Aβ), which is a spontaneous, aggregation-prone, and neurotoxic product of the processing of signaling protein APP (Amyloid Precursor Protein), in brain tissues, primarily in the hippocampus and the frontal cortex, was for a long time considered the main cause of neurodegenerative changes in AD. However, attempts to treat AD based on decreasing Aβ production and aggregation did not bring significant clinical results. More and more arguments are arising in favor of the fact that the overproduction of Aβ in most cases of AD is not the initial cause, but a concomitant event of pathological processes in the course of the development of sporadic AD. The concept of neuroinflammation has come to the fore, suggesting that inflammatory responses play the leading role in the initiation and development of AD, both in brain tissue and in the periphery. The hypothesis about the key role of neuroinflammation in the pathogenesis of AD opens up new opportunities in the search for ways to treat and prevent this socially significant disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The term proteinopathies integrates a number of diseases in which the aggregation of certain proteins as a result of overproduction, mutations that result in primary structure changes, or misfolding, are observed [1–5]. Proteinopathies of the nervous system, or neurodegenerative diseases, are characterized by gradually progressing loss of nerve cells with cognitive and/or motor impairments. Among neurodegenerative diseases, Alzheimer’s disease (AD) is of the highest socio-economic significance [6, 7]. The clinical course of AD is prolonged with steady, lasting for years, worsening of the patients’ condition. In AD, progressive memory loss, cognitive impairments, and the development of dementia, are observed. The inevitable outcome of AD is the death of the patient (as a rule, as a result of concomitant complications such as pneumonia or bedsores).
The typical form of AD (sporadic senile form) usually develops in individuals over 65. As of 2000, AD occurs in 1.6% of the population at the age of 65–74, in the 75–84 years old group the number of patients reaches 16%, and, in individuals older than 84, it increases to 42%. An inevitable increase in morbidity is predicted in developed countries, as a consequence of increasing lifespan [6–8]. Besides the senile AD form, there is the familial (pre-senile) form, which is associated with genetic predisposition and is characterized by early development (at 45–50) and aggressive disease course [9–12]. Diagnostics, treatment, and nursing of AD patients is a serious economic problem for developed counties [7, 13], and at present there are no effective ways to treat AD [14–16].
The accumulation of insoluble Aβ peptide (β-amyloid, further Аβ) in the brain is believed to be the main cause of AD development. Today, alternative hypotheses have also been proposed, where the key factors of AD development are believed to be the aggregation of hyperphosphorylated tau protein (tauopathy), chronic inflammation of brain tissue (neuroinflammation), or the accumulation of mtDNA mutations, resulting in the dysfunction of the mitochondrial respiratory chain and the development of oxidative stress [10, 17, 18]. A combination of several hypotheses (β amyloid, tauopathy, and where the leading role of neuroinflammation is postulated) into a single concept seems to be most promising.
β-AMYLOID HYPOTHESIS: THE FIRST ATTEMPT TO UNDERSTAND THE CAUSE OF ALZHEIMER’S DISEASE
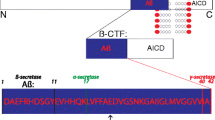
The leading pathomorphological symptom, occurring in 100% of AD cases, was described in 1907 by A. Alzheimer, after who the disease was later named, he observed the formation of so-called senile plaques, revealed via staining postmortem histological brain slices. Later on, it was shown that the plaques consist of fibrillar aggregates of Аβ, 40–42 amino acid residues in length, with a mass of 4 kDa. Β-amyloid is formed as a result of proteolysis of АРР (amyloid precursor protein), a transmembrane protein, which’s functions are yet studied insufficiently for the complete understanding of its role. АРР is involved in intercellular communications during nervous system development and synapse formation [19]. There are two alternative pathways for АРР processing with the participation of three endoproteases: α-, β-, and γ‑secretases. In the first, non-amyloidogenic, pathway the secreted form of APP (ectodomain) and the short peptide, indicated as р3, are generated via cleavage by α- and γ-secretases. In the the amyloidogenic pathway, the secreted form of АРР and a set of peptide of 30 to 51 amino acid residues in length are formed. Both mechanisms of АРР proteolysis generate an AICD (Amyloid precursor protein Intracellular Cytoplasmic/C-terminal Domain) intracellular fragment, which is involved in various signal pathways and resembles the intracellular domain, which is removed in the processing of Notch protein [20]. In healthy people (without AD), three forms of Аβ peptides are most widely expressed: Аβ40 (50%), Аβ38 (16%), and Аβ42 (10%) [21]. β- and γ-secretases are responsible for the proteolysis of APP via the amyloidogenic pathway [19]. The functions of Аβ were unknown for a long time; Аβ peptides were mainly considered “molecular garbage”, a by-product of АРР processing, were the main role is played by the splitting of the precursor protein into extracellular and intracellular fragments, involved in intercellular communications. Now there are data on the antimicrobial activity of Аβ peptides, making it possible to consider them one of the components of the innate immune system [22, 23].
The toxicity of Aβ for neurons has been shown in vitro and in vivo [24, 25]. Аβ peptides, mainly Аβ42 and, to a lesser degree, Аβ40, are prone to aggregation and the formation of insoluble fibrils [10, 26]. Senile plaques, detected in the brain of patients who died of AD, consist mainly of Аβ fibrillary deposits. It is known today that the formation of Аβ fibrils is preceded by the oligomerization of soluble amyloid β peptides,and the soluble oligomers are more toxic to neurons than large insoluble fibrils [25, 27, 28]. The key significance of accumulation and aggregation of Аβ was confirmed by data, obtained on patients with presenile (familial) AD. This form of the disease constitutes of up to 5–10% of the total number of AD cases, is familial, and is determined by mutations in the genes of АРР and presenilins (components of the γ-secretase complex), resulting in the overproduction of Аβ or the formation of Аβ peptides with increased predisposition to aggregation [29–32]. The location of the АРР gene on chromosome 21 determines the increased frequency and early development of AD in patients with Down’s syndrome (trisomy 21) as a consequence of the increase in the copy number of the АРР gene [33]. Mutations in the АРР gene, providing the opposite effect and statistically significantly decreasing the risk of AD development, have also been found [34]. AD is also associated with some epigenetic changes, for example, it is associated with a decrease in the DNA methylation level in the promotor area of the APP gene [35, 36].
The “amyloid cascade” hypothesis (also the amyloid hypothesis), which still remains popular today, was formulated based on study of the hereditary forms of AD and Down’s syndrome [37–39]. It was suggested that β amyloid, accumulating mainly in the fibrillary form, itself exerts a toxic effect leading to damage and loss of neurons [10, 37]. Later, it was found that soluble monomers and oligomers of Аβ have far higher toxicity [25, 27, 28, 40–42]. According to contemporary assumptions, the toxic effect of excessive Аβ peptide is due to its direct interaction with a number of receptor proteins both on the surface and in the cytoplasm of neurons. It was shown that the monomeric form of Аβ interacts with the α1β1 complex of Na,K-АТРase, inhibiting its activity, which results in the disturbance of the neuronal transmembrane potential [41, 42]. Аβ oligomers interact with adrenergic receptor α2А, increasing its activity, which leads to the activation of 3β-glycogen synthase (GSK-3β) and hyperphosphorylation of tau protein (see below) [43]. The increased concentration of Аβ results in the hyperactivation of NMDA receptors (excitotoxicity), inducing the disturbance of cell calcium homeostasis [44]. Disturbances of cholinergic system, oxidative stress etc. are also considered toxic effects of Аβ. All these and other changes result in the disturbance of long-term potentiation, synapse impairment, and neurodegeneration via apoptosis or necrosis [45].
HyPERPHOSPHORYLATION AND TAU PROTEIN AGGREGATION: THE SECOND KEY EVENT IN ALZHEIMER’S DISEASE
The second typical morphologic symptom of AD is neurofibrillary tangles (NFT), which are formed in neuronal cytoplasm (especially in axoplasm) and are insoluble filaments, consisting mainly of hyperphosphorylated tau protein (which is related to the microtubule-associated protein group) [46–48]. Impairments of tau protein functions result in destabilization and depolymerization of microtubules; the degradation of microtubules causes a disconnection of intracellular transport in the cytoplasm, loss of synapses, and, ultimately, nerve cell death. The formation of Аβ aggregates is accompanied by tau protein aggregation and precedes the development of structural impairments in brain tissues at later stages of the diseases. The question of interconnection between the accumulation of Аβ oligomers and aggregates and the hyperphosphorylation of tau followed by aggregation remains open, since Аβ is a secreted peptide, which accumulates mainly in intercellular space, while tau is an intracellular protein. One model considers tau protein hyperphosphorylation as the main trigger mechanism for its aggregation. It was shown that the following protein kinases are involved in tau phosphorylation: Ca2+/calmodulin-dependent protein kinase (CaMKII), protein kinase C (РКС), сAMP-dependent protein kinase (PKA), GSK-3β, and MAPK family kinases: ERK2, JNK, and р38/SAPK (stress-activated protein kinases) [20, 46, 49–51]. The activity of these protein kinases increases in the development of AD, probably as a consequence of the response to the formation of Аβ oligomers, or in response to neuroinflammation in brain tissues. Hyperactivation of glutamate receptors, induced by direct interaction with Аβ42, is considered a possible pathway of activation of protein kinases involved in tau phosphorylation. Phospholipase С activation, release of Ca2+ ions from endoplasmic reticulum, and activation of PKC are observed as a result of that [52]. In the course of AD development, GSK-3β is activated via adrenaline receptor α2А after its interaction with Аβ [43]. Thus, the cause of the synaptic activity loss and apoptosis of neurons may be the continuous stagewise process of induction of signal cascades, resulting in hyperphosphorylation and gradual aggregation of tau, which induces axonal transport impairment followed by degradation of synapses and nerve cell death, rather than the damaging effect of Аβ aggregates.
An alternative hypothesis postulates the prionic properties of some forms of Аβ (mainly Аβ42, with certain specificities in tertiary structure) and tau protein with impaired three-dimensional structure, which can induce pathological misfolding and the aggregation of other Аβ molecules and tau-protein, and some other proteins as well (in particular, PrPC) [53, 54]. The Аβ42 peptide, isomerized by the asparagine acid residue (Asp7), has the highest predisposition to the induction of amyloid deposits [55]. It was shown that some amount of Аβ can penetrate through membranes or be captured by neurons via endocytosis and, apparently, in that way interact with tau. In vitro experiments, the interaction of Аβ and tau, accompanied by tau oligomerization, was shown [56–58]. Injections of Aβ into hippocampus or right brain hemisphere of transgenic male rats expressing human mutant APP, accelerated the aggregation of Aβ of transgenic origin. Аβ aggregation was observed initially in the immediate proximity of the injection site, and, then the deposits of Aβ propagate along axons to brain regions connected to the hippocampus. The administration of mutant tau protein, with increased predisposition to aggregation, into the brain, caused the propagation of NFT formation in the cortex of mice expressing human wild-type tau protein which does not form aggregates spontaneously [59–61]. There is convincing evidence that β amyloid and tau protein micro-aggregations are capable of inducing the development of encephalopathies as a result of iatrogenic transition during brain surgery, when using contaminated tools or donor materials with admixtures of β-amyloid or tau aggregates [62–67]. We emphasize that, although in the described cases of hypothetic iatrogenic transmission of β-amyloid, Аβ aggregation was observed in the patients’ brain, and, in some cases the aggregation of tau was observed, it resulted in the development of Creutzfeldt-Jakob disease or Cerebral amyloid angiopathy (CAA), where β‑amyloid is deposited in the walls of small cerebral vesicles, rather than classic AD [62, 64]. Thus, the initial increase in the level of Аβ may play the role of a trigger inducing the cascade of interactions resulting in massive accumulation of aggregates, at first, of Aβ-itself, and then those of tau protein, which, in turn, leads to neuronal loss and the loss of function of central nervous system regions. The study of the mechanism of interaction between β‑amyloid and tau protein, including their identification in neuronal cells, opens new opportunities for the search for pharmaceutical agents, that can prevent accumulation of these toxic products in cells and the intercellular space [62].
β-AMYLOID HYPOTHESIS: PRO AND CONTRA ARGUMENTS
The data, provided above, convincingly enough indicate the important role of various forms of Аβ in the development of AD. At the same time, the amyloid hypothesis itself does not explain the causes of the increase in Аβ production in the sporadic form of AD, which is not associated with genetic predisposition. It is clear, however, that the main factor promoting the development of AD, is advanced age. Perhaps, the increased content of various forms of β-amyloid in the brain of senior people is less associated with overproduction than with age-related decline of effectiveness of different systems for Аβ removal. Age-related impairments of protein folding systems (in particular, the decrease in expression of Hsp70 family chaperon proteins) [69], the decrease in efficiency of Аβ-peptide removal involving microglia [70], and the age-related decrease in the brain content of neprilysin, a metalloproteinase providing proteolysis of Аβ [71, 72], are considered factors that promote an increase in Aβ42 concentration. There are data that the development of sporadic AD can be accompanied by accumulation of the toxic Аβ42 isomer, containing isoasparagine-7, which increases due to the age-related decrease in catalytic activity of the protein L-isoaspartyl-(D-aspartyl)-methyl transferase (PIMT), responsible for repair of isoasparagine residues [73, 74]. There are also hypotheses about the leading role of the increase in Аβ production in the development of AD as a result of inflammatory processes in brain tissues or the age-related accumulation of mtDNA mutations, which leads to the disturbance of electron transport by the respiratory chain, an increase in the production of reactive oxygen species (ROS) and oxidative stress, which triggers many signal cascades, resulting in the increase in APP processing via the amyloid pathway [18]. However, although the important role of β-amyloid in AD pathogenesis is beyond any doubt, at present there are numerous results calling into question the thesis about the increase in Аβ level as the initial cause of sporadic AD [75]. First, the form of β-amyloid (Аβ42) most predisposed to aggregation is found in healthy individuals as well those with AD. So, brain tissues of up to 40% aged individuals not suffering from AD, contain β-amyloid plaques, which is no less than the number of plaques in individuals with clinical manifestations of AD [76, 77]. Second, β-amyloid inclusions can be found in all brain regions, but only strictly determined structures are subjected to degeneration in AD, in particular, certain areas of the cortex and hippocampus. Third, the amyloid hypothesis does not consider the initial cause of Aβ overproduction in the sporadic form of AD in individuals without mutations in genes involved in Aβ production. A long time period lies between the beginning of the formation of β-amyloid aggregates and the appearance of the first symptoms of the disease (years or decades) [8, 78]. The direct toxic effect of Аβ on neurons is shown in vitro experiments in neuronal cultures and in vivo via injections to the brain of experimental animals. However, the toxic effect was observed only when Аβ was used at a concentration of about 1 mkM, i.e., exceeding the normal content of β-amyloid in patients’ brain 1000-fold [75]. Finally, a number of therapeutic approaches, aimed at β-amyloid as the target, for example, immunization of patients with Аβ42 and using monoclonal antibodies to different forms of Аβ, failed at the clinical trial stage. Some clinical studies have shown a decrease in the level of Аβ in patients’ brains, but, at the same time, have not observed recovery of cognitive functions and improvement of condition. The application of β- and γ-secretase inhibitors, aimed at decreasing Аβ production, has failed as well. In a number of cases, clinical trials were ceased ahead of schedule because of the detection of serious side effects. Direct inhibitors of Аβ oligomerization and aggregation also turned out to be ineffective [14, 76, 77, 79, 80]. On the other hand, failure to apply the “anti-β-amyloid” strategy in AD can be explained by the fact that, as a rule, the therapy begins after the development of clinical symptoms, when the degeneration has gone so far that the changes are irreversible. Data pro and contra the classic β-amyloid hypothesis are summarized in Table 1.
THE ROLE OF NEUROINFLAMMATION IN THE PATHOGENESIS OF ALZHEIMER’S DISEASE IS THE POSSIBLE INITIAL CAUSE OF β-AMYLOID ACCUMULATION
In the last two decades, a lot of attention was given to inflammatory processes in the CNS, whose development, as was shown independently by many authors, is connected with AD pathogenesis. Chronic inflammation in the form of activation of microglia and astrocytes, secretion of a number of pro-inflammatory cytokines and nitric oxide (NO), an increase in the level of ROS, and exerting cytotoxic effect on neurons are typical of AD. Also, in the brain, in AD, the expression level of receptors involved in the development of the inflammatory response, is increased. In AD patients, the increase in TNF-α, IL-1, IL-6, and complement system components occurs both in the brain and in the periphery (in the blood plasma), and the number of mRNA and TLR4 receptors, NF-κB activity, and ROS level grow as well [17, 81–85]. Similar data have been obtained with transgenic mice expressing mutant forms of АРР and presenilins and predisposed to the accumulation of Аβ aggregates and the development of cognitive disorders corresponding to human AD symptoms [86]. It has also been found that the probability of the development of sporadic AD correlates with some allele variants of genes involved in the immune response: TLR4, CD14 (a coreceptor of TLR4), CD33, TREM2 (triggering receptor expressed on myeloid cells 2), CR1 (complement receptor 1), and IL-6 [83, 84, 87, 88]. The suppression of TNF-α synthesis with the help of an inhibitor, 3,6′-ditiothalidomide, or RNAi lentivirus constructions slows down neurodegenerative processes and recovers cognitive functions (first of all, memory and learning ability) in model animals [82, 89]. In mice with genetically determined overproduction of IL-6 by astrocytes, starting from the age of six months, progressive neurodegeneration is developed [90], which proves the cytotoxic effect on neurons during neuroinflammation. Besides, a number of studies (retrospective studies and direct clinical trials) shows that long-term application of non-steroidal anti-inflammatory drugs (NSAID) slows down the development of AD in patients and exerts a positive effect on model animals [91–94]. Inflammation in CNS can induce many effects ascribed to β-amyloid: activation of protein kinase cascades which lead to tau protein hyperphosphorylation; increased activity of NMDA receptors; oxidative stress etc. [95].

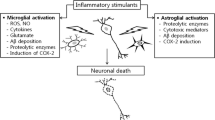
Hypothetical mechanism of the development of Alzheimer’s disease subject to the effects of neuroinflammation. Overproduction of Аβ can be induced by inflammatory responses in the sporadic form or be a consequence of mutations of genes involved in АРР processing, in the senile form of AD. The formation of a high number of Аβ oligomers, in turn, induces inflammatory response in the brain, forming a vicious cycle. Inflammation leads to neuronal loss both directly (for example, via oxidative stress) and indirectly via tau hyperphosphorylation and other effects.
There are convincing data about interconnections between the development of neuroinflammation and the accumulation of β-amyloid. It was shown that oligomers and fibrillar aggregates of Аβ in the brain are able to induce neuroinflammation via microglia activation and secretion of pro-inflammatory cytokines and prostaglandins [96]. Thus, Аβ accumulation can promote neuron apoptosis not only via cascades involving tau protein, but also via initiation of chronic inflammation in brain tissues as well. It was shown that TLR2/4 receptors, which are able to act as inductors of expression of pro-inflammatory cytokines, are responsible for the recognition of the monomeric form of Аβ [84, 97]. Many experimental and epidemiological works suggest that inflammatory responses are not only determined by Аβ accumulation but can act as the initial trigger mechanism in sporadic AD as well. It was shown that AD can develop as a long-term effect of brain trauma, including surgeries [98–100]. In some cases, not only primary neuroinflammation is involved, but also chronic inflammatory processes on the periphery as a result of persistant infections or metabolic disorders. Positive data in this direction were obtained in experiments on mice. For example, regular intraperitoneal injection of lipopolysaccharides (LPS) or poly(I:C) (as an analog of virus RNA) induced Аβ42 accumulation in the hippocampus and cortex of mice, astrocyte activation, and cognitive decline [101–103]. Epidemiological studies reveal a correlation between AD and preceding infections which cause chronic inflammatory responses [8]. It was shown that herpes virus infection (especially caused by type 1 and 2 herpesviruses) is a substantial risk factor of AD development. Herpesvirus DNA is found in the content of β-amyloid plaques with high frequency, and the reproduction of the virus in neuronal culture induces Аβ accumulation [104–106]. The development of HIV infection is often accompanied with so-called HIV-associated neurocognitive disorder (HAND). In the brain, Аβ synthesis increases and β-amyloid plaques are formed. HIV persists in microglia and astrocytes of the brain and induces an increase in the production of pro-inflammatory cytokines: IL-6, IL-1, TNF-α, and IL-8 [107]. Reports about the possible connection of cytomegalovirus and hepatitis С virus with AD development have also been published [108]. There is also association of AD with periodontitis, chlamydial infections, and some other inflammatory diseases, caused by Chlamydia pneumoniae, Porphyromonas gingivalis, and Treponema denticola [16, 109–112]. Finally, metabolic disorders, resulting in the development of inflammatory responses (diabetes is a classic example) substantially increase the risk of AD [45, 113]. Data are accumulating about the fact that in chronic inflammatory processes in the periphery, there is a response of the brain in the form of activation of microglial cells and astrocytes and the secretion of pro-inflammatory cytokines. On the one hand, the brain, separated from the circulatory system by the blood-brain barrier (BBB), is an immunoprivileged organ. On the other hand, the BBB is permeable to peripheral monocytes and prostaglandins. Besides, there are areas in the brain (the circumventricular organs, for instance, epiphysis), where the BBB has increased permeability [114]. Thus, chronic inflammation on the periphery or the persistence of slow infection in CNS (like in the case of herpesviruses and HIV) can induce inflammatory responses in brain tissues as the first cause of Аβ overproduction, development of the pathological cascade in the form of tauopathy and neurodegeneration and, eventually sporadic AD. It was shown that inflammation promotes proliferation and activation of microglia, following which, activated astrocytes begin to secrete increased amounts of both Аβ40 and Аβ42 [101–103, 107]. Inflammation also accelerates Аβ oligomerization due to the formation and secretion of complexes acting as oligomerization centers, for example, inflammasome, containing NLRP3 protein (cryopyrin) [8]. The increased concentration of Аβ oligomers henceforth can serve as an additional inductor of neuroinflammation. As a result, inflammation and accumulation of Аβ form a vicious cycle, promoting disease progression [115]. At the same time, deposits of fibrillar aggregates of β-amyloid (including those in healthy people) can be considered not a pathological process, but one of the mechanisims to remove Аβ peptides. In hereditary AD, neither chronic infections nor impairment of microglial activity regulation lead to inflammation, but the overloading of nervous system tissues with Аβ oligomers, whose overproduction is caused by mutations in the genes of АРР and/or presenilins.
АΒ PEPTIDE AS AN ELEMENT OF INNATE IMMUNITY
For a long time, Аβ peptides were exclusively considered by-products of АРР processing. Recent data make it possible to suggest that Аβ production may be due to non-specific defense mechanisms, which protect the brain from infiltration by infection agents. It was shown that the addition of various forms of Аβ peptides to a cultural medium suppresses replication of many bacteria including Escherichia coli, Staphylococcus, Streptococcus, Listeria [22]. In a study by D.K. Kumar et al. [25], it was shown that transgenic mice expressing human Аβ are less susceptible to meningitis caused by Salmonella enterica administrated directly to the brain than wild-type mice. Also, the survival rate of transgenic Caenorhabditis elegans nematodes and neuroglioma cell culture, expressing Аβ peptide, was compared with the survival rate of wild-type nematodes and neuroglioma culture, infected with Candida albicans. It turned out that transgenic cultures and nematodes were more resistant. It was revealed that Аβ peptides form fibrils on the surface of cells of the infection agent, preventing its replication. In this respect, the activity of Аβ peptide resembles classic mammal antibacterial peptides, for example, LL-37 [22]. Antiviral properties of β-amyloid have also been described. In glioma cell culture expressing Аβ peptide, the replication of human herpes simplex virus 1 (HSV-1) is suppressed [116]. In 5XFAD transgenic mice, expressing mutant human Аβ peptide, and in a 3D-culture of human neurons, it was shown that Аβ oligomers directly interact with surface glycoprotein HSV-1, slowing down the infection process. On the other hand, herpesvirus infection induces Аβ overproduction in in 5XFAD mice and in 3D-cultures [117]. Thus, Аβ peptide turns out to be not “molecular garbage”, but a component of innate (non-specific) immunity [118]. The brain, as an immunoprivileged organ, where the work of the adaptive component of immunity is absent or restricted, needs a high activity of innate immunity mechanisms for protection against infections. Accordingly, inflammatory responses in the central nervous system can trigger Аβ overexpression as a protective mechanism. As a result, sporadic AD can be considered a consequence of a difficult choice between possible infection damage to the brain “now” and neurodegeneration “in 20 years”. At the same time, the ability of Аβ, at excessive accumulation, to lead (according to the amyloid hypothesis) to neurodegeneration in the form of long-term effects is not subjected to the influence of natural selection, since the clinical development of AD begins after a human has passed childbearing age. The diagram of possible development of AD with involvement of neuroinflammatory processes is shown in Fig. 1.
The hypothesis about the leading role of neuroinflammatory responses in the pathogenesis of AD opens up new opportunities for the search of strategies of therapy and/or prevention of AD. There are a high number of meta-analyses, showing that long-term application of NSAIDs decreases the risk of AD onset and slows down the development of symptoms, [91–94, 119, 120]. According to some data, application of NSAIDs after the appearance of AD symptoms did not have a positive outcome, but in people without AD symptoms, it statistically significantly decreased the risk of development of symptoms for the following 2–3 years [121–123]. The positive effect of NSAIDs was also shown in experiments on AD models (rodents) [94]. The data make it possible consider timely treatment or prevention or inflammatory disease prevention as an approach to the prevention of AD.
TLR4 is a key receptor, playing a role both in the development of inflammatory responses and the utilization of Аβ. TLR4, along with some other receptors, recognizes LPS (endotoxins of gram-negative bacteria), at the same time triggering the signal cascades Akt–GSK-3β and NF-kB, leading to overproduction of pro-inflammatory cytokines and ROS by macrophages and neutrophils [124]. TLR4 is able to recognize not only LPS, but many other ligands as well, however, only some of them cause hyperactivation of the signal pathways, leading to an inflammatory response [125]. In the nervous system, TLR4 is expressed by glial macrophages (microglia) and astrocytes [126]. Besides other ligands, TLR4 recognizes the oligomeric form of Аβ. The response to interaction between TLR4 and Аβ can be implemented in two ways. In health, TLR4 plays and important role in the breakdown of of Аβ via phagocytosis [70], while in the development of AD, the activation of TLR4 by β‑amyloid leads to the overproduction of pro-inflammatory cytokines [127, 128]. TLR4 can be considered a possible therapeutical target in AD.
One of the natural ligands of TLR4 is the secreted from of Hsp70, a key chaperone protein. Like some cytokines, Hsp70 can be secreted from cells via the neoclassic mechanism [129–131]. Recombinant Hsp70 has an anti-inflammatory effect and inhibits some pro-inflammatory regulatory cascades. Administration of recombinant Hsp70 blocks the production of TNF-α and ROS by neutrophils and macrophages in response to LPS [132–138]. The interaction between Hsp70 and TLR4 results in fast phagocytosis of the formed complex [139].
Recently, new data about the effectiveness of exogenous (extracellular) Hsp70 as a neuroprotective factor have appeared. In our work [139–141], we have shown that recombinant Hsp70, labeled with Alexa-fluor or radioactive iodine, easily infiltrates the brain when administered intranasally. Subchronic intranasal administration of recombinant Hsp70 resulted in a decrease in Аβ concentration, neurogenesis activation, and cognitive function recovery in mice modelling AD: bulbectomized mice and transgenic 5XFAD mice [140, 141]. At the same time, the analysis of the hippocampal transcriptome of 5XFAD transgenic mice, which received recombinant Hsp70, revealed a substantial decrease in the expression of genes responsible for the development of neuroinflammation [141], and increased expression of genes responsible for the presentation of antigens, particularly of MHC I and II genes [142]. The transcriptome changes also indicate the increase in the level of neurorestoration markers and the activity of neurotransmitter synthesis systems after the administration of exogenous Hsp70 [142]. In independent studies transgenic mice and drosophilae secreting Hsp70 into the intercellular media, have been obtained. The expressed neuroprotective effect of extracellular Hsp70 was observed in lines obtained via crossing producents of extracellular Hsp70 with transgenes expressing Аβ42 and predisposed to development of neurodegeneration [143]. Perhaps, Hsp70, being a ligand of TLR4, switches it from pro-inflammatory activity to phagocytosis of Аβ. Thus, in the development of new means of therapy or prevention of AD, Hsp70 and other proteins with pro-inflammatory activity can be considered prospective neuroprotectors.
CONCLUSION
We can say with confidence that Аβ peptide is a necessary, but insufficient, participant of the process that ultimately leads to the development of AD [144]. Accumulation of Аβ is observed in many cases in clinically healthy individuals and cannot be considered the sole cause of the disease. Hyperphosphorylation and aggregation of tau, oxidative stress, and neuroinflammation play a no less important roles in the pathogenesis of AD. Furthermore, according to recent data, it is the neuroinflammation of various etiology that can be the initial stimulus, leading to Аβ accumulation and the development of sporadic AD. Besides, perhaps, the regulatory cascades triggered by neuroinflammation, and not the accumulation of Аβ, lead to the hyperphosphorylation of tau and the induction of other mechanisms of neurodegeneration. The identification of neuroinflammation as an important factor of AD pathogenesis opens new directions in the search for means of therapy and/or prevention of the disease.
REFERENCES
Ganguly G., Chakrabarti S., Chatterjeeт U., Saso L. 2017. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Design, Dev. Ther. 11, 797–810.
Briston T., Hicks A.R. 2018. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Transact. 46 (4), 829–842.
Ciccocioppo F., Bologna G., Ercolino E., Pierdomenico L., Simeone P., Lanuti P., Pieragostino D., Del Boccio P., Marchisio M., Miscia S. 2020. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural. Regen. Res. 15 (5), 850–856.
Bayer T.A. 2015. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 25 (5), 713–724.
Shelkovnikova T.A., Kulikova A.A., Tsvetkov F.O., Peters O., Bachurin S.O., Bukhman V.L., Ninkina N.N. 2012. Proteinopathies, neurodegenerative disorders with protein aggregation-based pathology. Mol. Biol. (Moscow). 46 (3), 362–374.
Dubois B., Feldman H.H., Jacova C., Hampel H., Molinuevo J.L., Blennow K., Dekosky S.T., Gauthier S., Selkoe D., Bateman R., Cappa S., Crutch S., Engelborghs S., Frisoni G.B., Fox N.C., et al. 2014. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 13 (6), 614–629.
Kochanek D.K., Murphy S.L., Xu J., Arias E. 2019. Deaths: Final data for 2017. Nat. Vital Statistics Repts. 68 (9), 1–15.
Leng F., Edison P. 2020. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 17 (3), 157–172.
Haass C. 1996. Presenile because of presenilin: The presenilin genes and early onset Alzheimer’s disease. Curr. Opin. Neurol. 9 (4), 254–259.
Reisberg B., Saeed M.U. 2004. Alzheimer’s disease. In: Comprehensive Textbook of Geriatric Psychiatry, 3rd ed. Eds Sadavoy J., Jarvik L.F., Grossberg G.T., Meyers B.S. New York: Norton Professional Books, pp. 449–509.
Waring S.C., Rosenberg R.N. 2008. Genome-wide association studies in Alzheimer disease. Arch. Neurol. 65 (3), 329–334.
Wong T.H., Seelaar H., Melhem S., Rozemuller A.J.M., van Swieten J.C. 2019. Genetic screening in early-onset Alzheimer’s disease identified three novel presenilin mutations. Neurobiol. Aging. 86, 201.e9–201.e14.
Meek P.D., McKeithan K., Schumock G.T. 1998. Economic considerations in Alzheimer’s disease. Pharmacotherapy. 18 (2, Pt. 2), 68–73. https://pubmed.ncbi.nlm.nih.gov/9543467/
Hung S.Y., Fu W.M. 2017. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 24 (1), 47.
Weller J., Budson A. 2018. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 7, 1161.
Panza F., Lozupone M., Solfrizzi V., Watling M., Imbimbo B.P. 2019. Time to test antibacterial therapy in Alzheimer’s disease. Brain: J. Neurol. 142 (10), 2905–2929.
Heppner F.L., Ransohoff R.M., Becher B. 2015. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16 (6), 358–372.
Swerdlow R.H., Burns J.M., Khan S.M. 2010. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 20, 265–279.
Müller U.C., Deller T., Korte M. 2017. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18 (5), 281–298.
Sulistio Y.A., Heese K. 2016. The ubiquitin-proteasome system and molecular chaperone deregulation in Alzheimer’s disease. Mol. Neurobiol. 53 (2), 905–931.
Bibl M., Gallus M., Welge V., Esselmann H., Wolf S., Rüther E., Wiltfang J. 2012. Cerebrospinal fluid amyloid-β 2–42 is decreased in Alzheimer’s, but not in frontotemporal dementia. J. Neural. Transm. 119 (7), 805–813.
Soscia S.J., Kirby J.E., Washicosky K.J., Tucker S.M., Ingelsson M., Hyman B., Burton M.A., Goldstein L.E., Duong S., Tanzi R.E., Moir R.D. 2010. Alzheimer’s disease-associated amyloid b-protein is an antimicrobial peptide. PLoS One. 5 (3), e9505.
Kumar D.K., Choi S.H., Washicosky K.J., Eimer W.A., Tucker S., Ghofrani J., Lefkowitz A., McColl G., Goldstein L.E., Tanzi R.E., Moir R.D. 2016. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 8, 340ra72.
Puzzo D., Arancio O. 2013. Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimer’s Dis. 33 (Suppl. 1), S111–S120.
Walsh D.M., Selkoe D.J. 2007. A beta oligomers – a decade of discovery. J. Neurochem. 101 (5), 1172–1184.
Musiek E.S., Holtzman D.M. 2015. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 18 (6), 800–806.
Haass C., Selkoe D.J. 2007. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 8, 101–112.
He Y., Zheng M.M., Ma Y., Han X.J., Ma X.Q., Qu C.Q., Du Y.F. 2012. Soluble oligomers and fibrillar species of amyloid β-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem. Biophys. Res. Commun. 429 (3–4), 125–130.
St George-Hyslop P.H. 2000. Molecular genetics of Alzheimer’s disease. Biol. Psychiatry. 47 (3), 183–199.
Brouwers N., Sleegers K., Van Broeckhoven C. 2008. Molecular genetics of Alzheimer’s disease: An update. Ann. Med. 40 (8), 562–583.
Scheltens P., Blennow K., Breteler M.M., de Strooper B., Frisoni G.B., Salloway S., Van der Flier W.M. 2016. Alzheimer’s disease. Lancet. 388, 505–517.
Pimenova A.A., Raj T., Goate A.M. 2017. Untangling genetic risk for Alzheimer’s disease. Biol. Psychiatry. 83 (4), 300–310.
Hartley D., Blumenthal T., Carrillo M., DiPaolo G., Esralew L., Gardiner K., Granholm AC., Iqbal K., Krams M., Lemere C., Lott I., Mobley W., Ness S., Nixon R., Potter H., et al. 2015. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 11 (6), 700–709.
Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R.R., et al. 2012. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 488, 96–99.
West R.L., Lee J.M., Maroun L.E. 1995. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 6 (2), 141–146.
Stoccoro A., Coppedè F. 2018. Role of epigenetics in Alzheimer’s disease pathogenesis. Neurodegenerative Dis. Manag. 8 (3), 181–193.
Hardy J.A., Higgins G.A. 1992. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 256 (5054), 184–185.
Armstrong R.A. 2011. The pathogenesis of Alzheimer’s disease: A reevaluation of the “amyloid cascade hypothesis”. Internat. J. Alzheimer’s Dis. 2011, 630865.
Kozin S.A., Makarov A.A. 2019. The Convergence of Alzheimer’s disease pathogenesis concepts. Mol. Biol. (Moscow). 53 (6), 896–904.
Selkoe D.J. 2008. Soluble oligomers of the amyloid β‑protein impair synaptic plasticity and behavior. Behav. Brain Res. 192 (1), 106–113.
Petrushanko I.Y., Mitkevich V.A., Anashkina A.A., Adzhubei A.A., Burnysheva K.M., Lakunina V.A., Kamanina Y.V., Dergousova E.A., Lopina O.D., Ogunshola O.O., Bogdanova A.Y., Makarov A.A. 2016. Direct interaction of beta-amyloid with Na,K-ATPase as a putative regulator of the enzyme function. Sci. Repts. 6, 27738.
DiChiara T., DiNunno N., Clark J., Bu R.L., Cline E.N., Rollins M.G., Gong Y., Brody D.L., Sligar S.G., Velasco P.T., Viola K.L., Klein W.L. 2017. Alzheimer’s toxic amyloid beta oligomers: Unwelcome visitors to the Na/K ATPase alpha3 docking station. Yale J. Biol. Med. 90 (1), 45–61.
Zhang F., Gannon M., Chen Y., Yan S., Zhang S., Feng W., Tao J., Sha B., Liu Z., Saito T., Saido T., Keene C.D., Jiao K., Roberson E.D., Xu H., Wang Q. 2020. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci. Translat. Med. 12 (526), 6931.
Yamin G. 2009. NMDA receptor-dependent signaling pathways that underlie amyloid β protein disruption of LTP in the hippocampus. J. Neurosci. Res. 87 (8), 1729–1736.
Mroczko B., Groblewska M., Litman-Zawadzka A., Kornhuber J., Lewczuk P. 2018. Cellular receptors of amyloid β oligomers (AβOs. in Alzheimer’s disease. Int. J. Mol. Sci. 19 (7), 1884.
Iqbal Kh., del Alonso C.A., Chen S., Chohan M.O., El-Akkad E., Gong C.-X., Khatoon S., Li B., Liu F., Rahman A., Tanimukai H., Grundke-Iqbal I. 2005. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta. 1739 (2–3), 198–210.
Gao Y., Tan L., Yu J.T., Tan L. 2018. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 15 (3), 283–300.
Neumann K., Farias G., Slachevsky A., Perez P., Maccioni R.B. 2011. Human platelets tau: A potential peripheral marker for Alzheimer’s disease. J. Alzheimer’s Dis. 25 (1), 103–109.
Avila J. 2006. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 580 (12), 2922–2927.
Domise M., Didier S., Marinangeli C., Zhao H., Chandakkar P., Buée L., Viollet B., Davies P., Marambaud P., Vingtdeux V. 2016. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Repts. 6, 26758.
Qi H., Prabakaran S., Cantrelle F.X., Chambraud B., Gunawardena J., Lippens G., Landrieu I. 2016. Characterization of neuronal tau protein as a target of extracellular signal-regulated kinase. J. Biol. Chem. 291 (14), 7742–7753.
Hamilton A., Zamponi G.W., Ferguson S.S. 2015. Glutamate receptors function as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol. Brain. 8, 18.
Kong C., Xie H., Gao Z., Shao M., Li H., Shi R., Cai L., Gao S., Sun T., Li C. 2019. Binding between prion protein and Aβ oligomers contributes to the pathogenesis of Alzheimer’s disease. Virol. Sinica. 34 (5), 475–488.
Schwarzman A.L., Sarantseva S.V. 2017. Transmission of pathogenic protein aggregates in Alzheimer’s disease. Mol. Biol. (Moscow). 51(3), 368–378.
Kozin S.A., Cheglakov I.B., Ovsepyan A.A., Telegin G.B., Tsvetkov P.O., Lisitsa A.V., Makarov A.A. 2013. Peripherally applied synthetic peptide isoAsp7-Aβ(1–42) triggers cerebral β-amyloidosis. Neurotox. Res. 24 (3), 370–376.
Guo J., Arai T., Miklossy J., McGeer P. 2006. Abeta and tau form soluble complexes that may promote self-aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 103 (6), 1953–1958.
Lasagna-Reeves C., Castillo-Carranza D., Guerrero-Muoz M., Jackson G., Kayed R. 2010. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 49 (47), 10039–10041.
Zhao L.N., Long H.W., Mu Y., Chew L.Y. 2012. The toxicity of amyloid oligomers. Int. J. Mol. Sci. 13 (6), 7303–7327.
Eisele Y.S., Obermüller U., Heilbronner G., Baumann F., Kaeser S.A., Wolburg H., Walker L.C., Staufenbiel M., Heikenwalder M., Jucker M. 2010. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science. 330 (6006), 980–982.
Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A.L., Vigouret J.-M., Paganetti P., Walsh D.M., Mathews P.M., Ghiso J., et al. 2006. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 313 (5794), 1781–784.
Walker L.C., Callahan M.J., Bian F., Durham R.A., Roher A.E., Lipinski W.J. 2002. Exogenous induction of cerebral β-amyloidosis in βAPP-transgenic mice. Peptides. 23 (7), 1241–1247.
Jaunmuktane Z., Quaegebeur A., Taipa R., Viana-Baptista M., Barbosa R., Koriath C., Sciot R., Mead S., Brandner S. 2018. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 135 (5), 671–679.
Jaunmuktane Z., Mead S., Ellis M., Wadsworth J.D., Nicoll A.J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A.S., Rudge P., Collinge J., Brandner S. 2015. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 525, 247–250.
Swerdlow A.J., Higgins C.D., Adlard P., Jones M.E., Preece M.A. 2003. Creutzfeldt-Jakob disease in United Kingdom patients treated with human pituitary growth hormone. Neurology. 61 (6), 783–791.
Purro S.A., Farrow M.A., Linehan J., Nazari T., Thomas D.X., Chen Z., Mengel D., Saito T., Saido T., Rudge P., Brandner S., Walsh D.M., Collinge J. 2018. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 564 (7736), 415–419.
Lauwers E., Lalli G., Brandner S., Collinge J., Compernolle V., Duyckaerts C., Edgren G., Haik S., Hardy J., Helmy A., Ivinson A.J., Jaunmuktane Z., Jucker M., Knight R., Lemmens R., et al. 2020. Potential human transmission of amyloid β pathology: Surveillance and risks. Lancet Neurol. 19 (10), 872–878.
Cali I., Cohen M.L., Haïk S., Parchi P., Giaccone G., Collins S.J., Kofskey D., Wang H., McLean C.A., Brandel J., Privat N., Sazdovitch V., Duyckaerts C., Kitamoto T., Belay E.D., et al. 2018. Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: An international study. Acta Neuropathol. Commun. 6(1), 5. https://pubmed.ncbi.nlm.nih.gov/29310723/
Tatarnikova O.G., Orlov M.A., Bobkova N.V. 2015. Beta-amyloid an Tau protein: Structure, interaction, and prion-like properties. Usp. Biol. Khim. 55, 351–390.
Leak R.K. 2014. Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 8, 293–310.
Tahara K., Kim H.D., Jin J.J., Maxwell J.A., Li L., Fukuchi K. 2006. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 129 (11), 3006–3019.
Nalivaeva N.N., Turner A.J. 2019. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 176 (18), 3447–3463.
Nalivaeva N.N., Zhuravin I.A., Turner A.J. 2020. Neprilysin expression and functions in development, ageing and disease. Mech. Ageing Dev. 192, 111363.
Desrosiers R.R., Fanélus I. 2011. Damaged proteins bearing L-isoaspartyl residues and aging: A dynamic equilibrium between generation of isomerized forms and repair by PIMT. Curr. Aging Sci. 4 (1), 8–18.
Mitkevich V.A., Petrushanko I.Y., Yegorov Y.E., Simonenko O.V., Vishnyakova K.S., Kulikova A.A., Tsvetkov P.O., Makarov A.A., Kozin S.A. 2013. Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 4 (11), e939.
Kepp K.P. 2017. Ten challenges of the amyloid hypothesis of Alzheimer’s disease. J. Alzheimer’s. Dis. 55 (2), 447–457.
Holmes C., Boche D., Wilkinson D., Yadegarfar G., Hopkins V., Bayer A., Jones R.W., Bullock R., Love S., Neal J.W., Zotova E., Nicoll J.A.R. 2008. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet. 372 (9634), 216–223.
Aisen P.S., Gauthier S., Ferris S.H., Saumier D., Haine D., Garceau D., Duong A., Suhy J., Oh J., Lau W.C., Sampalis J. 2011. Tramiprosate in mild-to-moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 7 (1), 102–111.
Bateman R., Xiong C., Benzinger T., Fagan A., Goate A., Fox N., Marcus D., Cairns N., Xie X., Blazey T., Holtzman D., Santacruz A., Buckles V., Oliver A., Moulder K., et al. 2012. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804.
Li Y., Liu Y., Wang Z., Jiang Y. 2013. Clinical trials of amyloid-based immunotherapy for Alzheimer’s disease: End of beginning or beginning of end? Expert Opin. Biol. Therapy. 13 (11), 1515–1522.
Herrup K. 2015. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18 (6), 794–799.
Dursun E., Gezen-Ak D., Hanağası H., Bilgiç B., Lohmann E., Ertan S., Atasoy IL., Alaylıoğlu M., Araz ÖS., Önal B., Gündüz A., Apaydın H., Kızıltan G., Ulutin T., Gürvit H., Yılmazer S. 2015. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 283, 50–57.
Zhang F., Jiang L. 2015. Neuroinflammation in Alzheimer’s disease. Neuropsychiatric Dis. Treatment. 11, 243–256.
Heneka M.T., Carson M.J., Khoury J.E., Landreth G.E., Brosseron F., Feinstein D.L., Jacobs A.H., Wyss-Coray T., Vitorica J., Ransohoff R.M., Herrup K., Frautschy S.A., Finsen B., Brown G.C., Verkhratsky A., et al. 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14 (4), 388–405.
Heneka M.T., Golenbock D.T., Latz E. 2015. Innate immunity in Alzheimer’s disease. Nat. Immunol. 16 (3), 229–236.
Bolós M., Perea J.R., Avila J. 2017. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts. 8 (1), 37–43.
Nazem A., Sankowski R., Bacher M., Al-Abed Y. 2015. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflammation. 12, 74.
Balistreri C.R., Grimaldi M.P., Chiappelli M., Licastro F., Castiglia L., Listì F., Vasto S., Lio D., Caruso C., Candore G. 2008. Association between the polymorphisms of TLR4 and CD14 genes and Alzheimer’s disease. Curr. Pharm. Des. 14 (26), 2672–2677.
Chen Y., Yip P., Huang Y., Sun Y., Wen L., Chu Y., Chen T. 2012. Sequence variants of Toll like receptor 4 and late-onset Alzheimer’s disease. PLoS One. 7 (2), e50771.
McAlpine F.E., Lee J.K., Harms A.S., Ruhn K.A., Blurton-Jones M., Hong J., Das P., Golde T.E., LaFerla F.M., Oddo S., Blesch A., Tansey M.G. 2009. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 34 (1), 163–177.
Millington C., Sonego S., Karunaweera N., Rangel A., Aldrich-Wright J.R., Campbell I.L., Gyengesi E., Münch G. 2014. Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. Biomed. Res. Int. 2014, 309129.
Cakala M., Malik A.R., Strosznajder J.B. 2007. Inhibitor of cyclooxygenase-2 protects against amyloid peptide-evoked memory impairment in mice. Pharmacol. Repts. 59 (2), 164–172.
Moore A.H., Bigbee M.J., Boynton G.E., Wakeham C.M., Rosenheim H.M., Staral C.J., Morrissey J.L., Hund A.K. 2010. Non-steroidal anti-inflammatory drugs in Alzheimer’s disease and Parkinson’s disease: Reconsidering the role of neuroinflammation. Pharmaceuticals. 3 (6), 1812–1841.
Rubio-Perez M.J., Morillas-Ruiz J.M. 2012. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 756357.
Daniels M.J., Rivers-Auty J., Schilling T., Spencer N.G., Watremez W., Fasolino V., Booth S.J., White C.S., Baldwin A.G., Freeman S., Wong R., Latta C., Yu S., Jackson J., Fischer N., Koziel V., et al. 2016. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 7, 12504–12504.
Wenk G.L. 2006. Neuropathologic changes in Alzheimer’s disease: Potential targets for treatment. J. Clin. Psychiatry. 67(Suppl. 3), 3–7.
Carrero I., Gonzalo M.R., Martin B., Sanz-Anquela J.M., Arévalo-Serrano J., Gonzalo-Ruiz A. 2012. Oligomers of beta-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 236 (2), 215–227.
Go M., Kou J., Lim J., Yang J., Fukuchi K. 2016. Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer’s mouse model: Implication of TLR4 signaling in disease progression. Biochem. Biophys. Res. Commun. 479 (2), 331–337.
Lye T.C., Shores E.A. 2000. Traumatic brain injury as a risk factor for Alzheimer’s disease: A review. Neuropsychol. Rev. 10 (2), 115–129.
Szczygielski J., Mautes A., Steudel W.I., Falkai P., Bayer T.A., Wirths O. 2005. Traumatic brain injury: Cause or risk of Alzheimer’s disease? A review of experimental studies. J. Neural. Transm. (Vienna). 112 (11), 1547–1564.
Van Den Heuvel C., Thornton E., Vink R. 2007. Traumatic brain injury and Alzheimer’s disease: A review. Prog. Brain Res. 161, 303–316.
Lee J.W., Lee Y.K., Yuk D.Y., Choi D.Y., Ban S.B., Oh K.W., Hong J.T. 2014. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 5, 37.
Weintraub M.K., Kranjac D., Eimerbrink M.J., Pearson S.J., Vinson B.T. 2014. Peripheral administration of polyI:C leads to increased hippocampal amyloid-beta and cognitive deficits in a non-transgenic mouse. Behav. Brain Res. 266, 183–187.
Catorce M.N., Gevorkian G. 2016. LPS-induced murine neuroinflammation model: Main features and suitability for pre-clinical assessment of nutraceuticals. Curr. Neuropharmacol. 14 (2), 155–164.
Lin W.R., Wozniak M.A., Cooper R.J., Wilcock G.K., Itzhaki R.F. 2002. Herpesviruses in brain and Alzheimer’s disease. J. Pathol. 197 (3), 395–402.
Harris S.A., Harris E.A. 2015. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 48 (2), 319–353.
Piacentini R., Li Puma D.D., Ripoli C., Marcocci M.E., De Chiara G., Garaci E., Palamara A.T., Grassi C. 2015. Herpes Simplex Virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-β protein accumulation. Sci. Rep. 5, 15444.
Fulop T., Witkowski J.M., Larbi A., Khalil A., Herbein G., Frost E.H. 2019. Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J. Neurovirol. 25 (5), 634–647.
Sochocka M., Zwolinska K., Leszek J. 2017. The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 15 (7), 996–1009.
Olsen I., Singhrao S.K. 2015. Can oral infection be a risk factor for Alzheimer’s disease? J. Oral Microbiol. 7, 29143.
Gaur S., Agnihotri R. 2015. Alzheimer’s disease and chronic periodontitis: Is there an association? Geriatr. Gerontol. Int. 15 (4), 391–404.
Kamer A.R., Pirraglia E., Tsui W., Rusinek H., Vallabhajosula S., Mosconi L., Yi L., McHugh P., Craig R.G., Svetcov S., Linker R., Shi C., Glodzik L., Williams S., Corby P., Saxena D., de Leon M.J. 2015. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging. 36, 627–633.
Abbott A. 2020. Are infections seeding some cases of Alzheimer’s disease? Nature. 587, 22–25.
Rad S.K., Arya A., Karimian H., Madhavan P., Rizwan F., Koshy S., Prabhu G. 2018. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Design, Dev. Therapy. 12, 3999–4021.
Duvernoy H.M., Risold P.Y. 2007. The circumventricular organs: An atlas of comparative anatomy and vascularization. Brain Res. Rev. 56 (1), 119–147.
Frost G.R., Li Y.M. 2017. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 7 (12), 170228.
Bourgade K., Le Page A., Bocti C., Witkowski J.M., Dupuis G., Frost E.H., Fulop Jr.T. 2016. Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimer’s Dis. 50 (4), 1227–1241.
Eimer W.A., Vijaya Kumar D.K., Navalpur Shanmugam N.K., Rodriguez A.S., Mitchell T., Washicosky K.J., György B., Breakefield X.O., Tanzi R.E., Moir R.D. 2018. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 99 (1), 56–63.
Moir R.D., Lathe R., Tanzi R.E. 2018). The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dementia. 14 (12), 1602–1614.
Schneider L.S., Mangialasche F., Andreasen N., Feldman H., Giacobini E., Jones R., Mantua V., Mecocci P., Pani L., Winblad B., Kivipelto M. 2014. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 275 (3), 251–283.
Benito-León J., Contador I., Vega S., Villarejo-Galende A., Bermejo-Pareja F. 2019. Non-steroidal anti-inflammatory drugs use in older adults decreases risk of Alzheimer’s disease mortality. PLoS One. 14 (9), e0222505.
Szekely C.A., Town T., Zandi P.P. 2007. NSAIDs for the chemoprevention of Alzheimer’s disease. Subcell. Biochem. 42, 229–248.
Breitner J.C., Baker L.D., Montine T.J., Meinert C.L., Lyketsos C.G., Ashe K.H., Brandt J., Craft S., Evans D.E., Green R.C., Ismail M.S., Martin B.K., Mullan M.J., Sabbagh M., Tariot P.N. 2011. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimer’s Dementia. 7 (4), 402–411.
Hoozemans J.J., Veerhuis R., Rozemuller J.M., Eikelenboom P. 2011). Soothing the inflamed brain: Effect of non-steroidal anti-inflammatory drugs on Alzheimer’s disease pathology. CNS Neurol. Disord. Drug Targets. 10 (1), 57–67.
Medzhitov R., Preston-Hurlburt P., Janeway C.A. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388 (6640), 394–397.
Peri F., Calabrese V. 2014. Toll-like receptor 4 (TLR4. modulation by synthetic and natural compounds: An update. J. Med. Chem. 57 (9), 3612–3622.
Fellner L., Irschick R., Schanda K., Reindl M., Klimaschewski L., Poewe W., Wenning G.K., Stefanova N. 2013. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 61 (3), 349–360.
Jin J.J., Kim H.D., Maxwell J.A., Li L., Fukuchi K. 2008. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation. 5, 23.
Tang S.C., Lathia J.D., Selvaraj P.K., Jo D.G., Mughal M.R., Cheng A., Siler D.A., Markesbery W.R., Arumugam T.V., Mattson M.P. 2008. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid β-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 213 (1), 114–121.
Prudovsky I., Mandinova A., Soldi R., Bagala C., Graziani I., Landriscina M., Tarantini F., Duarte M., Bellum S., Doherty H., Maciag T. 2003. The non-classical export routes: FGF1 and IL-1alpha point the way. J. Cell. Sci. 116 (Pt. 24), 4871–4881.
Ferrari D., Pizzirani C., Adinolfi E., Lemoli R.M., Curti A., Idzko M., Panther E., Di Virgilio F. 2006. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 176 (7), 3877–3883.
Mambula S.S., Stevenson M.A., Ogawa K., Calderwood S.K. 2007. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods. 43 (3), 168–175.
Aneja R., Odoms K., Dunsmore K., Shanley T.P., Wong H.R. 2006. Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J. Immunol. 177 (10), 7184–7192.
Kustanova G., Murashev A., Karpov V., Margulis B., Guzhova I.V., Prokhorenko I.R., Grachev S.V., Evgen’ev M.B. 2006. Exogenous heat shock protein 70 mediates sepsis manifestations and decreases the mortality rate in rats. Cell Stress Chaperones. 11 (3), 276–286.
Rozhkova E., Yurinskaya M., Zatsepina O., Garbuz D., Murashev A., Ostrov V., Margulis B., Evgenev M., Vinokurov M. 2010. Exogenous mammalian extracellular HSP70 reduces endotoxin manifestations at the cellular and organism levels. Ann. N.Y. Acad. Sci. 1197, 94–107.
Vinokurov M., Ostrov V., Yurinskaya M., Garbuz D., Murashev A., Antonova O., Evgen’ev M. 2012. Recombinant human Hsp70 protects against lipoteichoic acid-induced inflammation manifestations at the cellular and organismal levels. Cell Stress Chaperones. 17 (1), 89–101.
Borges T.J., Lopes R.L., Pinho N.G., Machado F.D., Souza A.P., Bonorino C. 2013. Extracellular Hsp70 inhibits pro-inflammatory cytokine production by IL-10 driven down-regulation of C/EBPβ and C/EBPδ. Int. J. Hyperthermia. 29 (5), 455–463.
Hsu J.H., Yang R.C., Lin S.J., Liou S.F., Dai Z.K., Yeh J.L., Wu J.R. 2014. Exogenous heat shock cognate protein 70 pretreatment attenuates cardiac and hepatic dysfunction with associated anti-inflammatory responses in experimental septic shock. Shock. 42 (6), 540–547.
Ghosh A.K., Sinha D., Mukherjee S., Biswas R., Biswas T. 2015. LPS stimulates and Hsp70 down-regulates TLR4 to orchestrate differential cytokine response of culture-differentiated innate memory CD8+ T cells. Cytokine. 73 (1), 44–52.
Yurinskaya M., Zatsepina O.G., Vinokurov M.G., Bobkova N.V., Garbuz D.G., Morozov A.V., Kulikova D.A., Mitkevich V.A., Makarov A.A., Funikov S.Y., Evgen’ev M.B. 2015. The fate of exogenous human HSP70 introduced into animal cells by different means. Curr. Drug. Deliv. 12 (5), 524–532.
Bobkova N.V., Garbuz D.G., Nesterova I., Medvinskaya N., Samokhin A., Alexandrova I., Yashin V., Karpov V., Kukharsky M.S., Ninkina N.N., Smir-nov A.A., Nudler E., Evgen’ev M. 2014. Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer’s disease. J. Alzheimer’s Dis. 38, 425–435.
Evgen’ev M.B., Krasnov G.S., Nesterova I.V., Garbuz D.G., Karpov V.L., Morozov A.V., Snezhkina A.V., Samokhin A.N., Sergeev A., Kulikov A.M., Bobkova N.V. 2017. Molecular mechanisms underlying neuroprotective effect of intranasal administration of human Hsp70 in mouse model of Alzheimer’s disease. J. Alzheimers Dis. 59 (4), 1415–1426.
Evgen’ev M., Bobkova N., Krasnov G., Garbuz D., Funikov S., Kudryavtseva A., Kulikov A., Samokhin A., Maltsev A., Nesterova I. 2019. The effect of human Hsp70 administration on a mouse model of Alzheimer’s disease strongly depends on transgenicity and age. J. Alzheimer’s Dis. 67 (4), 1391–1404.
de Mena L., Chhangani D., Fernandez-Funez P., Rincon-Limas D.E. 2017. secHsp70 as a tool to approach amyloid-β42 and other extracellular amyloids. Fly. 11 (3), 179–184.
Emelin A.Yu. 2011. New criteria for the diagnosis of Alzheimer’s disease. Nevrol. Neiropsikhiatr. Psikhosomat. 4, 5–8.
Funding
The work is supported by the Russian Science Foundation (grant no. 19-14-00167 (DG)).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Statement on the welfare of animals. This paper does not content any studies with participation of animals as research subjects.
Statement of compliance with standards of research involving humans. This paper does not content any studies with participation of humans as research subjects.
Conflict of interests. The authors declare no conflict of interests.
Additional information
Translated by A. Khaitin
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garbuz, D.G., Zatsepina, O.G. & Evgen’ev, M.B. Beta Amyloid, Tau Protein, and Neuroinflammation: An Attempt to Integrate Different Hypotheses of Alzheimer’s Disease Pathogenesis. Mol Biol 55, 670–682 (2021). https://doi.org/10.1134/S002689332104004X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002689332104004X