
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that affects 36 million people worldwide, but currently has no effective treatment options. One of the original hallmarks of AD are plaques comprised of beta amyloid (Aβ) and neurofibrillary tangles comprised of phosphorylated Tau protein. However, it is soluble oligomeric Aβ which is more closely correlated with cognitive decline and is therefore considered to be the neurotoxic species. Oligomeric Aβ has recently been shown to form complexes with the glycosylphosphatidylinositol (GPI)-anchored membrane protein, cellular prion protein (PrPc), and these complexes are believed to play an important role in the progression of AD pathogenesis. Glutamate, the major excitatory neurotransmitter is responsible for mediating learning and memory under normal physiological conditions. However, the dysregulation of glutamatergic signaling has also been implicated in a number of neurodegenerative diseases including AD. Glutamate acts via both ionotropic glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluR), each of which have been implicated in AD. There is now growing evidence to suggest that mGluR5 may contribute the AD pathogenesis by acting as scaffolds for the PrPc/Aβ oligomer complex, enabling the propagation of neurotoxic signaling in AD. In addition, PrPc and Aβ oligomer signaling via NMDARs may also contribute to AD pathology. The current review overviews our current understanding of the role of PrPc and Aβ oligomers in regulating glutamate receptor signaling, as well as highlights the importance of understanding these signaling complexes to develop more effective therapeutic strategies to treat AD.
Similar content being viewed by others
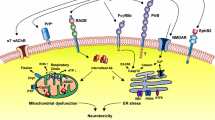
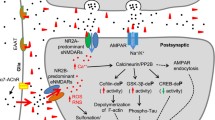

Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that is the leading cause of dementia affecting people over 50 years of age. There are approximately 36 million people worldwide with AD, and this number is expected to double by 2050, as the consequence of an aging world population [1]. Patients with AD suffer from memory loss and cognitive decline, which increases in severity as the disease progresses. Despite the rapidly growing incidence of AD there is no cure, and current therapeutic strategies have limited efficacy [1]. Pathophysiologically, the AD brain undergoes severe shrinkage that is caused by extensive synaptic and neuronal loss [2]. The original pathological features of AD were characterised by Alois Alzheimer in post mortem examination of patients suffering from severe memory loss [3,4]. These hallmarks include fibrillar plaques primarily composed of the protein beta amyloid (Aβ) and neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein [2,5]. However, additional AD markers include inflammation, characterized by activated microglia, which have been shown to cluster in brain regions showing substantial neurodegeneration [6] and oxidative stress, which results from an imbalance of reactive oxygen species (ROS) and antioxidants [7,8]. Recent studies have shown that the metabotropic glutamate receptor 5 (mGluR5) antagonist MTEP and mGluR5 knockout is protective in AD mice and that the N-methyl-D-aspartate (NMDA) receptor antagonist may be protective at low concentrations [9,10]. The present review will overview the contribution of β-amyloid and cellular prion protein (PrPC) in the regulation of mGluR5 and NMDA receptor signaling.
β-amyloid and PrPC
β-amyloid is a 39–43 amino acid peptide that is the primary component of the fibrillar plaques that characterize AD [11]. It is produced by the sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases, via what is known as either the amyloidogenic or β-amyloid pathway [12-14]. Often referred to as an overflow pathway, the amyloidogenic pathway, first cleaves APP extracellularly via a β-secretase producing a C99 peptide, which is subsequently cleaved by the γ-secretase complex resulting in Aβ oligomer production. However, the major pathway by which APP is cleaved is the non-amyloidogenic or α-secretase pathway, in which APP is cleaved by both α- and γ-secretases producing sAPPα and an APP intracellular domain. The products of the non-amyloidogenic pathway are considered to be beneficial and are associated with increased synaptic plasticity and neuronal survival [15]. Interestingly, β-amyloid is present in the brains of non-demented individuals, under normal physiological conditions, and is believed to act as part of a negative feedback loop in the regulation of synaptic plasticity and neuronal survival [14]. However, it is the over production of β-amyloid in AD that likely results in neurodegeneration and cognitive decline [11,16-19].
In addition to its presence in the insoluble fibrillar plaques, which characterize AD, β-amyloid exists in the brain in the form of soluble Aβ oligomers [17]. While early AD research focused on Aβ plaques as the neurotoxic species [20], recent evidence suggests that plaques may actually be a physiological ‘end point’ of limited harmfulness, while soluble oligomeric Aβ isoforms, in particular the Aβ42 fragment, are the primary source of neurotoxicity [21,22]. The accumulation of Aβ oligomers predominantly occurs in brain regions associated with learning and memory, including the hippocampus, and binds to sites that are located at neuronal synapses to cause the disruption to neuronal signaling and ultimately neuronal cell death [23,24]. A number of Aβ oligomer binding sites have been proposed, encompassing, but not limited to, glutamate receptors (both ionotropic and metabotropic), insulin receptors, acetylcholine receptors (both muscarinic and nicotinic), as well as cellular prion protein (PrPc) which may function as a co-receptor for Aβ [9,25-28]. Although the precise binding site remains controversial, Aβ oligomers presumably act via multiple receptors at synapse, thereby contributing to the range of issues that characterize AD.
PrPc is a glycosylphosphatidylinositol (GPI)-anchored membrane protein, whose normal cellular function remains unclear [29]. However, PrPc can undergo a conformationally inappropriate folding resulting in scrapie prion protein (PrPSc) that is linked to transmittable spongiform encephalopathies that cause terminal neurodegenerative disorders in both human and animals [30]. Several ligands bind to PrPc including the laminin γ1-chain, Cu2+ ions and Aβ42 oligomers [28-30]. Aβ42 oligomers bind to PrPc with nM affinity [25]. PrPc is suggested to be required for the suppression of long-term potentiation (LTP) in hippocampal slices, but LTP has been reported in hippocampal slices derived from PrPc null mice [25].
Glutamate
Glutamate is the major excitatory neurotransmitter in the brain. Under normal physiological conditions glutamate mediates learning and memory, as well as other higher order integrative brain function, but pathological glutamate signaling is also known to contribute to neuronal cell death [31-33]. Glutamate is released into the synapse following the depolarization of pre-synaptic neurons and is cleared from the synapse by the GLT transporter into astrocytes. Under normal physiological conditions, the removal of glutamate is rapid and neuroprotective [34]. Glutamate exerts its effects via both ionotropic glutamate receptors (iGluR) and metabotropic glutamate receptors (mGluRs). Ionotropic GluRs are ligand-gated ion channels that include the NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainite receptors that function to mediate the rapid synaptic responses to glutamate [33]. In contrast, mGluRs encompass 8 subtypes and the activation of these receptors by glutamate results in slower, longer-lasting, modulatory alterations in synaptic activity [35].
The dysregulation of and/or pathological signaling of glutamate receptors is well established for a number of neurodegenerative diseases, including AD [35]. Glutamate becomes excitotoxic as the result of overproduction and impaired clearance from synapses [36]. The excess of glutamate appears to chronically activate both ionotropic and metabotropic glutamate receptors resulting in the elevation of intracellular Ca2+ associated with neurodegenerative disease that promotes neuronal injury and cell death [37]. In the present review, we will overview the hypothesis that mGluR5 and NMDAR function as extracellular molecular scaffolding proteins, which facilitate the cellular signaling of PrPc/Aβ oligomer complexes in the pathogenesis of AD. These PrPc/Aβ oligomer interactions with glutamate receptors represent a novel target for the prevention and/or attenuation of AD progression.
Metabotropic glutamate receptors
Metabotropic glutamate receptors are members of the G protein coupled receptor superfamily. There are eight mGluR subtypes, which are further divided into 3 groups based on sequence homology, G protein-coupling specificity and pharmacological profile [38]. The Group I mGluRs, mGluR1a and mGluR5a activate phospholipase C to generate both inositol 1,4,5 trisphosphate and diacylglycerol resulting in the release of intracellular Ca2+ stores and the activation of protein kinase C [38]. Group I mGluRs, in particular mGluR5, have been specifically implicated in neurodegenerative diseases such as AD, Parkinson’s disease and Huntington’s disease [35,39,40]. Interestingly, Group I mGluRs and their interacting proteins have the ability to function in both a neuroprotective and neurotoxic manner. For example, in a rat model of ischemia, mGluR5 antagonists and agonists, were found to have neuroprotective effects [41]. Group I mGluRs associate with and/or activate Homer, ERK1/2, AKT and mTOR signaling pathways [42-46]. These pathways are considered to be neuroprotective as they are important for cell survival, growth and proliferation, in addition to synaptic plasticity. For example, mGluR1a/Homer/PIKE protein interactions have been demonstrated to contribute to the activation of PI3K and functions to prevent neuronal apoptosis [46]. Furthermore, Group I mGluRs have been shown to modulate the activity of ligand-gated ion channels including the NMDAR and play an important role in regulating both long-term potentiation (LTP) and long-term depression (LTD), which represent cellular mechanisms involved in both the positive and negative regulation of synaptic strength, respectively [47,48]. Similarly, NMDAR activation increases mGluR5 activity [49,50]. Thus, mGluR5 activity is also associated with excitotoxic cell death, caused by elevated intracellular Ca2+ by virtue of the ability of mGluR5 to potentiate NMDAR function [48]. mGluR5 has also been linked to the RNA binding protein fragile X mental retardation protein (FMRP), which is known to repress protein synthesis at the synapse [51,52]. APP is among the proteins regulated by FMRP [51]. The activation of mGluR5 has been shown to increase FMRP mediated translation of APP, which could lead to neuroprotection under normal physiological conditions or excessive Aβ production in animal models of AD [53].
In the human AD brain, mGluR1 expression has been found to be reduced, with the reduced level of mGluR expression being correlated to the severity of the disease, whereas mGluR5 protein expression was unchanged [54]. Interestingly, there is some discrepancy over changes to mGluR5 expression, with some research suggesting that mGluR5 mRNA expression is up-regulated [55] and mGluR5 cell surface expression is increased in a APPswePS1ΔE9 mouse model of AD [10]. Additionally, elevated expression of mGluR5 has been observed in astrocytes, most notably with this elevation is observed in astrocytes found clustered around Aβ plaques [54]. However, mGluR5 represents a candidate receptor contributing to the underlying pathogenesis associated with AD [9,10,27]. Differences in mGluR1 and mGluR5 expression, in the AD brain, present the possibility that these receptors play functionally distinct roles in the progression of AD pathology. This has suggested that mGluR1 may play a protective role in AD, whereas altered mGluR5 signaling in AD may be neurotoxic [55]. However, this hypothesis remains to be clearly determined.
Role of group I mGluRs as scaffolds for the binding of Aβ oligomers and PrPc
Numerous studies have implicated Group I mGluR signaling in the regulation of Aβ42 toxicity in neurons and in AD mouse models [9,10,27] and Aβ42 oligomers and PrPC increase mGluR5a-dependent LTD in vivo [56]. Aβ42 oligomers also induce changes in the subcellular localization of CaMKII resulting in a reduction of AMPARs in the synaptic membrane [57]. Aβ42 peptide is also reported to uncouple mGluR5-dependent activation of PKC, but not ERK1/2 activity [58]. Oligomeric forms of Aβ42 also bind to PrPc to alter synaptic plasticity and recent work clearly supports the concept that PrPc is required for Aβ42 oligomer-mediated neuronal toxicity [25,59-65]. PrPc has been shown to stimulate Ca2+ release from neuronal intracellular stores in response to laminin γ1-chain peptide-dependent activation of endogenous PrPc, as well as stimulate PKC translocation to the plasma membrane in response to either mGluR1a or mGluR5a activation [28]. Laminin γ1-chain peptide-mediated Ca2+ release from neurons is antagonized by either mGluR1a or mGluR5a antagonists and is not observed in primary neurons derived from PrPc null mice [28]. More recently, mGluR5, but not mGluR1, has been suggested to be the primary co-receptor for both PrPc and Aβ oligomers [9] (Figure 1). Specifically, convincing data from the Stritmatter laboratory indicates that mGluR5, but not an extensively investigated battery of other GPCRs (including mGluR1), functions as the extracellular scaffolding protein receptor that is essential for the regulation of Aβ oligomer and PrPc signaling in AD [9]. However, a potential role for mGluR1 cannot be dismissed given its role in laminin γ1-chain peptide-mediated Ca2+ release [28]. The report that mGluR5 functions as a receptor for both Aβ oligomers and PrPc is consistent with a previous study showing that Aβ oligomers stimulate the lateral diffusion and clustering of mGluR5 at synapses [27]. This ultimately results in increased Ca2+ release from intracellular stores ultimately resulting in synaptic deterioration [27]. Whether this Aβ oligomer-stimulated clustering contributes to mGluR5- and/or mGluR1-dependent alterations in LTP and LTD remains to be determined. Nevertheless, the seminal study by Stritmatter and colleagues provides evidence that mGluR5 plays an integral role in regulated Aβ oligomer pathology, as the deletion of mGluR5 expression reduced Aβ oligomer-mediated synapse loss and mGluR5 antagonist (MTEP) treatment results in the amelioration of cognitive deficits normally observed APPswePS1ΔE9 mice at 9 months of age [9]. Consistent with these observations, we have recently shown that the genetic deletion of mGluR5 in APPswePS1ΔE9 mice also results in the reversal of the cognitive deficits associated with this mouse model of AD at both 9 and 12 months of age, and that soluble Aβ oligomer levels and Aβ plaques in APPswePS1ΔE9 mice are reduced in the absence of mGluR5 expression at 12 month of age [12]. More recently, it has been shown that mGluR5 allosteric modulators can disrupt Aβ oligomer-stimulated interactions between mGluR5 and PrPC suggesting that it may be possible to develop new compounds to interrupt the pathogenesis observed in AD [66]. The observation that mGluR5 activation increases FMRP-mediated translation of APP, suggests the possibility that Aβ oligomer stimulation of mGluR5 may leaded to increased Aβ production resulting in a positive feedback loop the exacerbates AD pathology [53] (Figure 1).

mGluR5 function as scaffolds for the formation of A β oligomer and PrP C signaling complexes. The extracellular domain of mGluR5 interacts with both PrPc and Aβ42 which results in the activation of Ca2+ release from intracellular stores, PKC translocation and ERK1/2 phosphorylation. Aβ42 and PrPc also activate mGluR5 to stimulate Fyn kinase-mediated and mGluR5 activation has been linked to the regulation of APP protein translation. APP, amyloid precursor protein; DAG, diacylglycerol; ER, endoplasmic reticulum; ERK, extracellular regulated kinase; PLCβ, phospholipase Cβ; PrPc, cellular prion protein.
Dysregulation of Group I mGluRs may also further exacerbate AD progression, as a consequence of bidirectional cross talk with NMDARs, as NMDAR activation may increase mGluR5 signaling [49,50]. This cross activity between mGluR5 and NMDARs involves reduced mGluR5 desensitization in response to GRK2 and is also due to NMDAR-stimulated dephosphorylation of mGluR5 and Homer protein regulated cross-regulation of AMPAR activity [49,55,67]. This likely contributes to Group I mediated regulation of NMDAR-dependent LTP and LTD [40,68].
NMDARS
In the previous sections, we have discussed mGluRs as scaffolds for PrPc and β-amyloid in AD. However, glutamate also plays a central role in regulating synaptic transmission by activating ionotropic receptors, namely AMPA, kainate and NMDA receptors [69]. There is new evidence that NMDARs are regulated by both PrPc and Aβ oligomers [70]. NMDARs are activated by glutamate, as well as the synthetic agonist NMDA, and the co-agonists glycine and D-serine which function to modulate glutamate-dependent activation of NMDARs resulting in the opening of a cation channel that is permeant to both Ca2+ and Na+ [71]. This has two major consequences, first, it creates an electrogenic response that induces an excitatory postsynaptic potential, and second, Ca2+ entering via NMDARs triggers cytoplasmic signaling cascades that eventually culminate in altered cellular activity, such as changes in gene expression. While NMDARs are important for learning and memory [72,73], their dysfunction is linked to pathophysiological signaling that is associated with neurodegenerative diseases such as AD [74-76]. Glutamate binding to NMDARs mediates NMDAR channel opening, whereas the dissociation of the ligand causes channel closure (or deactivation) [77]. NMDARs also undergo a process called desensitization in the prolonged presence of receptor agonist [78], which is designed to protect cells from Ca2+ overload. This desensitization process is slowed in a dose-dependent manner by the co-agonist glycine [78,79]. NMDARs are potently regulated by extracellular Mg2+ ions, which occlude the channel pore at hyperpolarized potentials, and this block is relieved following membrane depolarization of the post-synaptic neuron [80,81]. Other metal ions that regulate NMDARs include Zn2+ and Cu2+ [82-85].
PrPc interactions with NMDARs
NMDARs are functionally modulated by both PrPc and PrPSc, both in vivo and in vitro [70]. For example, PrPc null mice exhibit depressive-like behaviours [86] and reduced pain thresholds [87], which are reversed by delivery of the NMDA receptor antagonist MK-801. Moreover, cell death of neuronal cultures infected with PrPSc is partially abrogated by MK801 treatment [88]. NMDAR activity is also enhanced in PrPc null neurons [89]. Field potential recordings from hippocampal slices exhibit an increased number of population spikes that are exacerbated by the removal of extracellular Mg2+, and are prevented by the NMDAR blocker APV. In PrPc null neurons there is also an increase in the amplitude and duration of the miniature synaptic NMDA currents. Also, in PrPc null neurons there is a drastic slowing of the deactivation kinetics of NMDAR currents and direct delivery of NMDA into the brains of PrPc null mice increases lesion size compared to WT animals [89]. This suggests a mechanism by which PrPc regulates subunit stoichiometry between GluN1 and various types of GluN2 subunits to account for divergent effects on NMDAR function [70]. In this regard, it is noteworthy that NMDARs can be co-immunoprecipitated with PrPc from brain homogenate [85,89], suggesting that PrPc forms a molecular complex with NMDARs and may function as a NMDAR ligand, rather than regulating NMDAR function solely via second-messenger signal transduction pathways.
More recently, we have described a type of PrPc-mediated modulation of NMDAR activity that is dependent upon Cu2+ ions [85]. PrPc is a high-affinity Cu2+ binding protein that contains five Cu2+ binding sites that exhibit varying Cu2+ binding-affinity [90-92]. When Cu2+ ions are chelated by the exogenous application of either cuprizone or bathocuproinesulfonate (BCS), NMDAR current amplitudes are increased and receptor desensitization is slowed, leading to a persistent current [85]. A similar slowing of NMDAR desensitization is also observed in neurons either derived from PrPc null mice or upon acute enzymatic cleavage of PrPc. Importantly, BCS has no further effect on NMDAR desensitization in PrPc null neurons. Compared with wild type neurons, the glycine concentration-dependence of the magnitude of the persistent NMDAR currents is shifted leftward in PrPc null neurons. Hence, over a wide range of glycine concentrations, there is an increase in the non-desensitizing current component. Based on these findings, we conclude that in the presence of PrPc, glycine affinity for the NMDAR is reduced and thus the receptor desensitizes more rapidly. Thus, BCS-mediated chelation of copper appears to weaken the biochemical interactions between PrPc and the NMDAR complex [85] suggesting that this PrPc/NMDAR complex is copper-ligand regulated.
Aβ-mediated regulation of NMDA receptor function
Numerous studies have linked NMDARs to AD and the clinically utilized AD drug, memantine, is a known allosteric blocker of NMDARs [93-96]. In cultured neurons Aβ42 oligomers appear to initiate spontaneous NMDAR currents and NMDAR internalization [97,98] and a number of studies have shown that the molecular mechanisms underlying Aβ42-dependent alterations in NMDAR activity and trafficking are complex [97,99]. It has been reported that Cu2+ binds to Aβ42 with an attomolar affinity-constant [100]. In line with what we have described above, the application of either nM concentrations of Aβ42 oligomers, or application of μM concentrations of Aβ42 monomers, mimics the effect of BCS on NMDAR function [70], yielding NMDA currents that exhibit reduced desensitization. Based on our findings, we propose a mechanism by which binding of Aβ42 oligomers at nM concentrations bind directly to PrPc and alter PrPc-mediated NMDAR desensitization, whereas Aβ42 monomers, when present in the low μM range, might in manner analogous to BCS, simply chelate Cu2+, thus leading to PrPc dissociation from the NMDAR [70]. This mechanism may also help to account for the disparate findings by various groups who have investigated the role of Aβ oligomer/PrPc complexes, in regulating reduction in LTP mediated by both mGluRs and NMDARs in wild-type mice and AD mouse models [25,40,59,60,65].
It is important to note that one of the consequences of slowed NMDAR desensitization is the potential for persistent calcium entry following prolonged glutamate activation resulting in neuronal cell death [70]. However, if the elimination of PrPc expression results in slowed NMDAR desensitization kinetics that are analogous to that seen in the presence of Aβ42 oligomers, then one would also expect to observe AD-like pathology in PrPc null mice, but this is not the case. They key to answering this conundrum likely lies in the fact that a slowing of desensitization kinetics manifests itself pathologically only when there is prolonged excess of glutamate agonist. In PrPc null mice, synaptic glutamate is likely cleared quickly by reuptake GLT-1 transport mechanisms on astrocytes, and thus NMDARs are only briefly activated at synapses. In contrast, in mouse models of AD, it is likely that glutamate reuptake mechanisms are perturbed by Aβ42 oligomers, leading to the buildup of synaptic and extra-synaptic glutamate that triggers prolonged activation of both NMDAR- and mGluR-mediated calcium entry resulting in cell damage that may contribute to synaptic loss in AD [101]. In this context it is important to note that extrasynaptic GluN2B containing NMDA receptors are thought to be the primary drivers of excitotoxicity, whereas synaptic GluN2A receptors were found to be neuroprotective [102-104]. On the other hand a recent study has linked the degenerative effects of Aβ to a dysregulation of GluN2A containing receptors [105]. Precisely how the interactions with the NMDAR complex occur in a NMDAR subtype specific manner remains to be determined. Taken together, there is increasing body of experimental evidence suggesting that PrPc is an important regulator of NMDAR functions that are regulated by Cu2+ ions. Aβ peptides (in both their monomeric and oligomeric forms) appear to mediate dysregulation of NMDAR-mediated synaptic function, thereby potentially contributing to AD pathology. These processes may also be further complicated by hyperphosphorylated tau protein and Fyn kinase, which are also closely linked to NMDARs and PrPc activity, but beyond the scope of the present review [105-109].
Summary
In summary, mGluR5 and NMDARs represent potential therapeutic targets for the treatment of AD. Particularly encouraging, are recent studies in which mouse models of AD were treated with the mGluR5 antagonist MTEP, and memory was rescued in the Morris water maze (MWM) a spatial memory behavioural paradigm [10]. Additionally, in an AD mouse model, in which mGluR5 was genetically deleted, mice showed improved memory in the MWM, as well as a substantial reduction in Aβ plaques and oligomers [12]. Of the drugs available for the treatment of AD, memantine, is a known allosteric blocker of NMDARs [93-96]. Although memantine is of limited therapeutic value, the modulation of NMDAR function may nonetheless offer the possibility for effective target for the treatment of AD. It is likely that the effects of Aβ42 oligomers and PrPc on mGluR5 and NMDAR signaling do not occur in isolation as Aβ42 oligomer binding to PrPc and mGluR5 results in the activation of Fyn kinase to regulate signaling downstream of mGluR5 [10] (Figure 1). This activation of Fyn results in the phosphorylation of the GluN2B subunit of the NMDAR that ultimately results in a loss of cell surface NMDARs [10]. Of particular interest will be to determine whether the antagonism of Aβ42 oligomer and PrPc interactions with both mGluR5 and NMDARs will serve as an effective treatment for AD.
Abbreviations
- AD:
-
Alzheimer’s disease
- NFT:
-
Neurofibrillary tangles
- Aβ:
-
Beta amyloid
- ROS:
-
Reactive oxygen species
- APP:
-
Amyloid precursor protein
- PrPc :
-
Cellular prion protein
- LTP:
-
Long term potentiation
- LTD:
-
Long term depression
- mGluR:
-
Metabotropic glutamate receptor
- iGluR:
-
Ionotropic glutamate receptor
- NMDA:
-
N-methyl-D-aspartate
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- GPCR:
-
g protein coupled receptor
- PLC:
-
Phospholipase C
- DAG:
-
Diacylglycerol
- mTOR:
-
Mammalian target of rapamycin
- CNS:
-
Central nervous system
- PS1:
-
Presenilin 1
- FMRP:
-
Fragile X mental retardation protein
References
Association A. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2013;2013(9):208–45.
Dubois B, Feldman H, Jacova C, Hampel H, Molinuevo J, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29.
Alzheimer A. Ueber eine eigenartige Erkrankung der Hirnrinde. Zeitschrift fuer Psychiatr. 1907;64:146–9.
Alzheimer A, Stelzmann RA, Schnitzlen HN, Murthagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429–31.
Braak E, Braak H. Alzheimer’s disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon's horn. Acta Neuropathol. 1997;93:323–5.
Glass C, Saijo K, Winner B, Marchetto M, Gage F. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34.
Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208.
Guglielmotto M, Giliberto L, Tamagno E, Tabaton M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front Aging Neurosci. 2010;2(February):3.
Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25.
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron. 2013;79:887–902.
Zhang Y, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4:3.
Hamilton A, Esseltine JL, DeVries RA, Cregan SP, Ferguson SSG. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol Brain. 2014;7:40.
Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998;8:447–53.
Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55.
Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12:1–12.
Ma H, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, et al. Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–72.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP Processing and Synaptic Function State University of New York at Stony Brook. Neuron. 2003;37:925–37.
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12.
Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proc Natl Acad Sci U S A. 1996;93:13170–5.
Gordon MN, King DL, Diamond DM, Jantzen PT, Boyett KV, Hope CE, et al. Correlation between cognitive deficits and Abeta deposits in transgenic APP + PS1 mice. Neurobiol Aging. 2001;22:377–85.
McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2006;47:191–9.
Lesne S, Koh M, Kotilinek L, Kayed R, Glabe C, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7.
Gong Y, Chang L, Viola K, Lacor P, Lambert M, Finch C, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–22.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–200.
Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Stephen M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-B oligomers. Nature. 2009;457:1128–32.
De Felice FG, Vieira MNN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–6.
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–54.
Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 2011;25:265–79.
Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–4.
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728.
Albasanz JL, Dalfó E, Ferrer I, Martín M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol Dis. 2005;20:685–93.
Piani D, Frei K, Do K, Cuenod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci let. 1991;133:159–62.
Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol. 1992;9:2429–36.
Rao V, Bowen K, Dempsey R. Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem Res. 2001;26:497–502.
Ribeiro FM, Paquet M, Cregan SP, Ferguson SSG. Group 1 metabotropic glutamate receptor signaling and its implication in neurological disease. CNS Neurol Discord Drug Targets. 2010;9:574–95.
Balazs R, Miller S, Romano C, de Vries A, Chun Y, Cotman CW. Metabotropic glutamate receptor mGluR5 in astrocytes: pharmacological properties and agonist regulation. J Neurochem. 1997;69:151–63.
Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;8:623–34.
Dhami GK, Ferguson SSG. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther. 2006;111:260–71.
Bordi F, Ugolini A. Group 1 metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol. 1999;59:55–79.
Bruno V, Battaglia G, Copani A, Cespédes VM, Galindo MF, Ceña V, et al. An activity-dependent switch from facilitation to inhibition in the control of excitotoxicity by group I metabotropic glutamate receptors. Eur J Neurosci. 2001;13:1469–78.
Opitz T, Reymann K. (1S, 3R)-ACPD protects synaptic transmission from hypoxia in hippocampal slices. Neuropharmacology. 1993;32:103–4.
Choe ES, Wang JQ. Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci Lett. 2001;313:129–32.
Nicodemo AA, Pampillo M, Ferreira LT, Dale LB, Cregan T, Ribeiro FM, et al. Pyk2 uncouples metabotropic glutamate receptor G protein signaling but facilitates ERK1/2 activation. Mol Brain. 2010;3:4.
Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–61.
Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci. 2005;25:2741–52.
Rong R, Ahn J-Y, Huang H, Nagata E, Kalman D, Kapp JA, et al. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–61.
Wu J, Rush A, Rowan MJ, Anwyl R. NMDA receptor and metabotropic glutamate receptor-dependent synaptic plasticity induced by high frequency stimulation in the rat dentate gyrus in vitro. J Physiol. 2001;533:745–55.
Doherty AJ, Palmer MJ, Henley JM, Collingridge GL, Jane DE. Chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5 but not mGlu1 receptors expressed in CHO cells and potentiates NMDAA responses in the hippocampus. Neuropharmacology. 1997;36:265–7.
Alagarsamy S, Saugstad J, Warren L, Mansuy IM, Gereau RWI, Conn PJ. NMDA-induced potentiation of mGluR5 is mediated by activation of protein phosphatase 2B/calcineurin. Neuropharmacology. 2005;49:135–45.
Ribeiro FM, Paquet M, Ferreira LT, Cregan T, Swan P, Cregan SP, et al. Metabotropic glutamate receptor-mediated cell signaling pathways are altered in a mouse model of Huntington’s disease. J Neurosci. 2010;30:316–24.
Bear M, Huber K, Warren S. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–7.
Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52.
Sokol DK, Mahoney B, Long JM, Ray B, Lahiri DK. Autism, Alzheimer’s disease, and fragile X. APP, FMRP and mGluR5 are molecular links. Neurology. 2011;76:1344–52.
Casley CS, Lakics V, Lee H-G, Broad LM, Day TA, Cluett T, et al. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-beta peptide. Brain Res. 2009;1260:65–75.
Caraci F, Battaglia G, Sortino MA, Spampinato S, Molinaro G, Copani A, et al. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: still a hot topic? Neurochem Int. 2012;61:559–65.
Hu NW, Nicoll AJ, Zhang D, Mably AJ, O’Malley T, Purro SA, et al. mGlu5 receptors and cellular prion protein mediate amyloid-β-facilitated synaptic long-term depression in vivo. Nat Commun. 2014;5:3374.
Gu Z, Liu W, Yan Z. β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284:1063–10649.
Tyszkiewicz JP, Yan Z. β-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J Neurophysiol. 2005;93:3102–11.
Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–74.
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, et al. Alzheimer’s disease brain-derived Amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31:7259–63.
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–300.
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, et al. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–14.
Cissé M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci. 2011;31:10427–31.
Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–70.
Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336.
Haas LT, Kostylev MA, Strittmatter SM. Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J Biol Chem. 2014;289:28460–77.
Degos V, Peineau S, Nijboer C, Kaindl AM, Sigaut S, Favrais G, et al. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann Neurol. 2013;73:667–78.
Rammes G, Hasenjäger A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of β-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60:982–90.
McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–60.
Stys PK, You H, Zamponi GW. Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J Physiol. 2012;590:1357–68.
Shleper M, Kartvelishvily E, Wolosker H. D-serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. J Neurosci. 2005;25:9413–7.
Herron CE, Lester RA, Coan EJ, Collingridge GL. Frequency-dependent involvement of NMDA receptors in the hippocampus: a novel synaptic mechanism. Nature. 1986;322:265–8.
MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84.
Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Eng J Med. 1994;330:613–22.
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–50.
Loftis JM, Janowsky A. The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003;97:55–85.
Vance KM, Simorowski N, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat Commun. 2011;2:294.
Mayer ML, Vyklicky LJ, Clements J. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature. 1989;338:425–7.
Vyklicky LJ, Benveniste M, Mayer ML. Modulation of N-methyl-D-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. J Physiol. 1990;428:313–31.
Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–5.
Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–3.
Peters S, Koh J, Choi DW. Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science. 1987;236:589–93.
Rachline J, Perin-Dureau F, Le Goff A, Neyton J, Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J Neurosci. 2005;25:308–17.
Vlachová V, Zemková H, Vyklický Jr L. Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur J Neurosci. 1996;8:2257–64.
You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, et al. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–42.
Gadotti VM, Bonfield SP, Zamponi GW. Depressive-like behaviour of mice lacking cellular prion protein. Behav Brain Res. 2012;227:319–23.
Gadotti VM, Zamponi GW. Cellular prion protein protects from inflammatory and neuropathic pain. Mol Pain. 2011;7:59.
Müller WE, Ushijima H, Schröder HC, Forrest JM, Schatton WF, Rytik PG, et al. Cytoprotective effect of NMDA receptor antagonists on prion protein (PrionSc)-induced toxicity in rat cortical cell cultures. Eur J Pharmacol. 1993;246:261–7.
Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–65.
Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, et al. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–7.
Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, et al. Frequency-dependent involvement of NMDA receptors in the hippocampus: a novel synaptic mechanism. Proc Natl Acad Sci U S A. 2001;98:8531–5.
Brown DR, Sassoon J. Copper-dependent functions for the prion protein. Mol Biotechnol. 2002;22:165–78.
Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–70.
Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–55.
Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci. 2009;29:2774–9.
Di Stefano A, Iannitelli A, Laserra S, Sozio P. Drug delivery strategies for Alzheimer’s disease treatment. Expert Opin Drug Deliv. 2011;8:581–603.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8.
Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49:184–90.
Rush T, Buisson A. Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res. 2014;356:279–86.
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, et al. Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neuroschem. 2000;75:1219–33.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801.
Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33.
Kaufman AM, Milnerwood AJ, Sepers MD, Coquinco A, She K, Wang L, et al. Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. J Neurosci. 2012;32:3992–4003.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–2527.
Tackenberg C, Grinschgl S, Trutzel A, Santuccione AC, Frey MC, Konietzko U, et al. NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 2013;4:e608.
Wang H, Ren CH, Gunawardana CG, Schmitt-Ulms G. Overcoming barriers and thresholds - signaling of oligomeric Aβ through the prion protein to Fyn. Mol Neurodegener. 2013;8:24.
Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci. 2008;28:1989–2002.
Vergara C, Ordonez-Gutierrez L, Wandosell F, Ferrer I, Del Rio JA, Gavin R. Role of PrPC Expression in Tau Protein Levels and Phosphorylation in Alzheimer’s Disease Evolution. Mol Neurobiol. 2014. [Epub ahead of print]
Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A. 2006;103:2892–7.
Acknowledgements
S. S. G. F. holds a Tier I Canada Research Chair in Molecular Neurobiology and is a Career Investigator of the Heart and Stroke Foundation of Ontario. G. W. Z. also holds a Tier I Canada Research Chair.
Funding
This work was supported by the Canadian Institutes of Health Research [grant number MOP-119437] to S. S. G. F.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AH: contributed review of AD and mGluRs. GZ: contributed review of NMDARs. SF: senior author who edited manuscript and provided figures. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hamilton, A., Zamponi, G.W. & Ferguson, S.S.G. Glutamate receptors function as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol Brain 8, 18 (2015). https://doi.org/10.1186/s13041-015-0107-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-015-0107-0