
Abstract
A series of 1,3,4-oxadiazole bridged pyrazole/isoxazole bearing quinoline derivatives has been designed and synthesized by a clean and convenient method. Structures of the newly synthesized compounds have been confirmed by FTIR, 1H and 13C NMR, and HRMS spectral data. The titled compounds have been evaluated for their molecular docking guided antimicrobial and anti-inflammatory activity. One of 1,3,4-oxadiazole bridged quinolinyl-pyrazole derivatives has interacted efficiently with E. Coli protein (PDB file: 1KZN), and has been characterized by good antimicrobial activity against the majority of the tested pathogens. Another product has exhibited excellent anti-inflammatory activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Quinoline and its derivatives are considered as versatile prodrugs, among those are antimalarial quinine (A) [1], chloroquine (B), and hydroxychloroquine (C) (Fig. 1). The latter one is currently under clinical trials as a repurposed drug for COVID-19 [2].

Samples of promising quinoline alkaloids.
Currently, many quinoline-containing drugs are available on the market [3, 4]. Recently, three protein kinase inhibitors (Lenvatinib, Bosutinib, and Cabozantinib) and one Farnesyl transferase inhibitor (Tipifarnib) were presented as anticancer agents [5]. 1,3,4-Oxadiazole derivatives are well known for their antioxidant, anticancer, anticonvulsant, tyrosinase inhibitory, cathepsin K inhibitors, P-glycoprotein inhibitors, antimicrobial, antitumor, anti-inflammatory, and antiretroviral activities [6].
Combination of quinoline and pyrazole structural blocks in molecules makes pharmacophores with respect to anti-cancer, interleukin, A3 adenosine receptor antagonists, anti-ulcer, and anti-inflammatory to be of certain potential [7]. Similarly, combination of quinoline and isoxazole heterocycles can result in formation of anti-TB [8, 9], cytotoxic [10], antibacterial, antifungal [11], anti-inflammatory, and analgesic [12] compounds. Quinoline containing 1,3,4-oxadiazole derivatives exhibit pronounced biological activities [13, 14].
According to the earlier reports [15], the position 8 in quinoline was determined to be decisive for its biological activity. Generally, quinoline and its derivatives are considered as highly potent among derivatives of other five-membered heterocycles [16]. All the above inspired us to design and synthesize quinoline based compounds containing azoles combined by 1,3,4-oxadiazole bridge.
RESULTS AND DISCUSSION
The compounds 2 and 3 synthesized from benzaldehyde were subjected to cyclization reaction with ethyl acetoacetate in presence of the catalytic amount of anhydrous zinc chloride without using an additional solvent to give intermediates 4a and 4b, respectively (Scheme 1).

Synthesis of target compounds 11a–11l.
In the following step, esters 4a, 4b were treated with hydrazine hydrate in ethanol to afford 5a and 5b, respectively. Subsequently, esters 4a, 4b were hydrolyzed by NaOH (10%) in THF–water (1 : 1) medium to afford 6a and 6b, respectively. Compounds 8a–8f were synthesized by treatment of a variety of 8-hydroxyquinolines 7a–7f by ethyl 2-chloroacetate in acetone in presence of potassium carbonate. The isolated product 8a was subjected to hydrolysis by sodium hydroxide to afford acid 9a. Refluxing of 8a–8f with hydrazine hydrate in ethanol led to the key intermediates 10a–10f.
In the first approach to the target compounds, the compounds 11a and 11g were synthesized by refluxing 9a with 5a or 5b, respectively, in phosphorous oxychloride (Scheme 1, method A). Alternatively, 10a was treated with 6a or 6b with formation of the target compounds 11a and 11g under the similar reaction conditions (Scheme 1, method B).
Optimization of reaction conditions for both methods is presented in Table 1 and favored method B. Probably poor solubility of 5a could lead to lower rate of the reaction. Accordingly, method B was applied for synthesis of the other derivatives. It is noteworthy that higher yield was achieved in case of isoxazole (84 and 92%) than pyrazole (79 and 87%) due to absence of the bulky N-phenyl group in the former one which minimized the partial positive charge on the carbonyl group.
Structures of the synthesized compounds were confirmed by FTIR, 1H and 13C NMR, and HRMS spectral data. Two singlets at 2.41 ppm (CH3) and 5.35 ppm (O–CH2), and a doublet of doublets at 8.88– 8.90 ppm assigned to quinoline proton in 1H NMR spectrum of compound 11a indicated its formation. In IR spectra, presence of characteristic bands at 1065– 1089 cm–1 (N–N), 1618–1643 cm–1 (C=N), and 1229–1256 cm–1 (C–O–C) also supported formation of compounds 11a–11l.
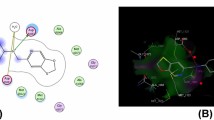
Biological activity. Molecular docking study. The molecular docking data are summarized in Table 2. The majority of target compounds demonstrated significant binding energy (–11.57 to –7.97 kJ/mol) with the target sites possessing H-bonds.
Antimicrobial activity. Results of antimicrobial activity (Table 3, Fig. 2) indicated that majority of products were of significant activity against most of the tested microbes. Compound 11d which contained the methyl group on quinoline ring was potent antimicrobial agent as well as 11a. The other compounds showed moderate activity. Compound 11b containing electron-withdrawing chlorine at C5 of quinoline displayed significant activity against gram-negative bacterial strain and demonstrated moderate activity against other pathogens. Compounds 11c and 11i that contained Cl at C5 and C7 on quinoline ring connected by pyrazole/isoxazole bridge with 1,3,4-oxadiazole ring exhibited excellent activity against gram-negative bacterial strains (E. coli and K. pneumonia) at high concentration (100 µg/mL). So, compounds containing electron-withdrawing substituents demonstrated excellent activity against majority of the gram-negative bacteria, while compounds containing electron-donating groups displayed significant activity against both gram-positive and gram-negative bacterial strains. Compounds 11g and 11h were highly active against S. flexneri gram-negative bacteria but showed moderate activity against other tested strains which could be attributed to the presence of Cl on quinoline cycle in addition to isoxazole heterocycle. 1,3,4-Oxadiazole bridged pyrazolyl-quinoline compounds were more potent than isoxazolyl-quinoline compounds. The compounds containing iodine (11f, 11l) exhibited the lowest antibacterial activity against all tested strains but showed good antifungal activity. Majority of the fungal strains were inhibited by compound 11a and 11d that contained the methyl group on quinoline ring while, other derivatives showed moderate antifungal activity.

Comparison of antimicrobial (MBC/MFC) activity of newly synthesized compounds 11a–11l.
Anti-inflammatory activity. Anti-inflammatory activity of the products were tested by inhibition of the albumin denaturation method according to Muzushima and Kabayashi with slight modification [17]. Diclofenac sodium was used as a standard (Table 4). The product 11c which contained Cl at C5 and C7 of quinoline ring demonstrated potent anti-inflammatory activity comparable with that of the standard drug. Compound 11b containing pyrazole and 1,3,4-oxadiazole and Cl at C5 demonstrated promising activity as well as 11h and 11i containing isoxazole ring. Compounds 11f and 11l carrying iodine at C5 and C7 of quinoline ring were of the lowest activity. According to the data summarized in Table 4, anti-inflammatory activity of products was influenced by electronegative Cl substituent and pyrazole ring attached to quinoline via 1,3,4-oxadiazole heterocycle.
All reagents were purchased from Sigma Aldrich. Melting points were determined on a Buchi oil melting point apparatus and are uncorrected. IR spectra (KBr discs) were recorded on a SHIMADZU 8400 S FTIR spectrophotometer. 1H and 13C NMR spectra were measured on a Bruker Avance spectrometer (400 MHz for proton and 100 MHz for carbon) respectively using CDCl3 as a solvent and TMS as an internal standard. High resolution mass spectra (HRMS) were measured by the direct infusion method on a MicroTOF-QII-ESI-Qq-TOF liquid chromatography mass spectrometer (Bruker Daltonics, Billerica, USA), electrospray ionization (ESI). Column chromatography was carried out using silica gel 60 (15–40 mm, Merck). TLC was performed on pre-coated (silica gel 60 F254, 0.2 mm, Merck) plates and visualized under UV light and/or by iodine vapor.
Synthesis of intermediates 10a–10f. Intermediate compounds 10a–10f were synthesized by the reported earlier methods. 1-Benzylidene-2-phenylhydrazine (2) and benzaldehyde oxime (3) were synthesized from benzaldehyde [18, 19]. Ethyl 5-methyl-1,3-diphenyl-1H-pyrazole-4-carboxylate (4a) and ethyl 5-methyl-3-phenylisoxazole-4-carboxylate (4b) were synthesized by cyclization of compounds 2 and 3 with ethyl acetoacetate, respectively [20, 21]. 5-Methyl-1,3-diphenyl-1H-pyrazole-4-carbohydrazide (5a) and 5-methyl-3-phenylisoxazole-4-carbohydrazide (5b) were synthesized by treating compounds 4a and 4b with hydrazine hydrate [22, 23]. 5-Methyl-1,3-diphenyl-1H-pyrazole-4-carboxylic acid (6a) and 5-methyl-3-phenylisoxazole-4-carboxylic acid (6b) were synthesized by hydrolysis in aqueous methanol [24, 25]. Ethyl 2-(quinolin-8-yloxy)acetates 8a–8f and 2-(quinolin-8-yloxy)acetohydrazides 10a–10f were synthesized by treating the corresponding 8-hydoxyquinoline 7a–7f with ethyl 2-chloroacetate followed by hydrazine hydrate [26, 27]. 2-(Quinolin-8-yloxy)acetic acid 9a was obtained via hydrolysis of 8a in aqueous methanol.
Typical procedure for synthesis of 2-(5-methyl-1,3-diphenyl-1H-pyrazol-4-yl)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (11a) and 2-(5-methyl-3-phenylisoxazol-4-yl)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (11g). Method A. 5-Methyl-1,3-diphenyl-1H-pyrazole-4-carbohydrazide (5a, 0.29 g, 1.00 mmol) and 5-methyl-3-phenylisoxazole-4-carbohydrazide (5b, 0.21 g, 1.00 mmol) were placed in round bottom flasks fitted with a water cool condenser. To the flasks 2-(quinolin-8-yloxy)acetic acid (9a, 0.21 g, 1.00 mmol) was added followed by 10 mL of phosphorous oxychloride.
Method B. 5-Methyl-1,3-diphenyl-1H-pyrazole-4-carboxylic acid (6a, 0.27 g, 1.00 mmol) or 5-methyl-3-phenylisoxazole-4-carboxylic acid (6b, 0.21 g, 1.00 mmol), 2-(quinolin-8-yloxy)acetohydrazide (10a, 0.22 g, 1.00 mmol) and 10 mL of phosphorous oxychloride were mixed together.
The other products were synthesized from the corresponding compounds following the method B.
The above reaction mixtures were refluxed on a water bath at 80°C upon monitoring by TLC, then reaction mixture was cooled down to 25°C and poured into crushed ice slowly with constant stirring. The resultant solution was neutralized by 20% KOH and extracted by dichloromethane (2×25 mL). The organic layer was washed with 5% hydrochloric acid (2× 25 mL), 5% sodium hydroxide (2×25 mL), brine solution (1×25 mL), and finally with water (25 mL). The organic layer obtained was dried by anhydrous Na2SO4 and the solvent was removed under reduced pressure. The product was purified by column chromatography using hexane : ethyl acetate (8 : 2) as an eluent to afford the corresponding pure product as a white amorphous solid.
2-(5-Methyl-1,3-diphenyl-1H-pyrazol-4-yl)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (11a). IR spectrum, ν, cm–1: 3261 (C–H), 1622 (C=N), 1241 (Ar–O–C), 1068 (N–N). 1H NMR spectrum, δ, ppm: 2.41 s (3H, CH3), 5.35 s (2H, CH2), 7.40–7.60 m (14H, ArH), 8.79–8.80 d (1H, ArH), 8.88–8.90 d. d (1H, ArH, J = 1.6, 6.4 Hz). 13C NMR spectrum, δC, ppm: 9.88 (C8), 71.03 (C19), 103.54 (C2), 106.53 (C21), 117.55 (C23), 120.24 (C26), 124.11 (C9, C13), 126.80 (C11), 127.25 (C22), 127.59 (C14, C18), 128.57 (C16), 129.14 (C15, C17), 129.52 (C24), 129.92 (C10, C12), 132.82 (C7), 135.13 (C25), 137.72 (C3), 139.34 (C6), 140.68 (C28), 147.48 (C1), 148.65 (C27), 153.41 (C20), 163.40 (C5), 165.10 (C4). HRMS: m/z: 460.1227. C28H21N5O2.
2-{[(5-Chloroquinolin-8-yl)oxy]methyl}-5-(5-methyl-1,3-diphenyl-1H-pyrazol-4-yl)-1,3,4-oxadiazole (11b). Yield 79%. IR spectrum, ν, cm–1: 3251 (C–H), 1635 (C=N), 1256 (Ar–O–C), 1064 (N–N). 1H NMR spectrum, δ, ppm: 2.44 s (3H, CH3), 5.31 s (2H, CH2), 7.39–7.59 m (13H, ArH), 8.53–8.54 d (1H, ArH), 8.98–8.99 d. d (1H, ArH, J = 1.2, 5.2 Hz). 13C NMR spectrum, δC, ppm: 9.90 (C8), 71.32 (C19), 103.33 (C2), 106.83 (C21), 118.90 (C23), 122.37 (C26), 122.77 (C9, C13), 126.26 (C11), 127.39 (C14, C18), 127.82 (C22), 128.41 (C16), 129.02 (C15, C17), 129.39 (C24), 129.60 (C10, C12), 131.94 (C7), 132.73 (C25), 134.00 (C3), 137.56 (C6), 139.79 (C28), 147.41 (C1), 152.72 (C27), 154.16 (C20), 162.86 (C5), 164.25 (C4). HRMS: m/z: 494.0671. C28H20ClN5O2.
2-{[(5,7-Dichloroquinolin-8-yl)oxy]methyl}-5-(5-methyl-1,3-diphenyl-1H-pyrazol-4-yl)-1,3,4-oxadiazole (11c). Yield 73%. IR spectrum, ν, cm–1: 3238 (C–H), 1630 (C=N), 1251 (Ar–O–C), 1065 (N–N). 1H NMR spectrum, δ, ppm: 2.53 s (3H, CH3), 5.45 s (2H, CH2), 7.41–7.61 m (12H, ArH), 8.54–8.55 d (1H, ArH), 8.99–9.01 d. d (1H, ArH, J = 1.2, 5.2 Hz). 13C NMR spectrum, δC, ppm: 10.01 (C8), 71.40 (C19), 103.78 (C2), 107.11 (C21), 120.85 (C23), 122.48 (C26), 122.88 (C9, C13), 126.37 (C11), 127.47 (C14, C18), 127.90 (C22), 128.52 (C16), 129.11 (C15, C17), 129.54 (C24), 129.72 (C10, C12), 132.04 (C7), 132.92 (C25), 134.24 (C3), 137.73 (C6), 139.80 (C28), 147.63 (C1), 152.92 (C27), 154.28 (C20), 162.90 (C5), 164.45 (C4). HRMS: m/z: 527.9884. C28H19Cl2N5O2.
2-(5-Methyl-1,3-diphenyl-1H-pyrazol-4-yl)-5-{[(2-methylquinolin-8-yl)oxy]methyl}-1,3,4-oxadiazole (11d). Yield 74%. IR spectrum, ν, cm–1: 3241 (C–H), 1628 (C=N), 1242 (Ar–O–C), 1089 (N–N). 1H NMR spectrum, δ, ppm: 2.36 s (3H, CH3), 2.68 s (3H, CH3), 5.42 s (2H, CH2), 7.41–7.62 m (13H, ArH), 8.79–8.80 d (1H, ArH), 8.87–8.88 d. d (1H, ArH, J = 1.6, 6.4 Hz). 13C NMR spectrum, δC, ppm: 9.96 (C8), 25.32 (C29), 71.12 (C19), 103.63 (C2), 106.73 (C21), 117.81 (C23), 120.37 (C26), 124.31 (C9, C13), 126.92 (C11), 127.36 (C22), 127.64 (C14, C18), 128.68 (C16), 129.35 (C15, C17), 129.74 (C24), 130.04 (C10, C12), 133.05 (C7), 135.35 (C25), 137.67 (C3), 139.42 (C6), 140.71 (C28), 147.57 (C1), 148.81 (C27), 153.62 (C20), 163.58 (C5), 165.27 (C4). HRMS: m/z: 474.0633. C29H23N5O2.
2-{[(5,7-Dichloro-2-methylquinolin-8-yl)oxy]methyl}-5-(5-methyl-1,3-diphenyl-1H-pyrazol-4-yl)-1,3,4-oxadiazole (11e). Yield 70%. IR spectrum, ν, cm–1: 3252 (C–H), 1623 (C=N), 1233 (Ar–O–C), 1073 (N–N). 1H NMR spectrum, δ, ppm: 2.54 s (3H, CH3), 2.86 s (3H, CH3), 5.56 s (2H, CH2), 7.53–7.73 m (12H, ArH), 8.55–8.56 d (1H, ArH), 8.99–9.00 d. d (1H, ArH, J = 1.2, 5.2 Hz). 13C NMR spectrum, δC, ppm: 10.11 (C8), 24.52 (C29), 71.52 (C19), 103.69 (C2), 107.23 (C21), 120.79 (C23), 122.46 (C26), 123.08 (C9, C13), 126.42 (C11), 127.58 (C14, C18), 127.83 (C22), 128.63 (C16), 129.24 (C15, C17), 129.63 (C24), 129.95 (C10, C12), 132.13 (C7), 132.78 (C25), 134.68 (C3), 138.06 (C6), 140.05 (C28), 147.73 (C1), 153.07 (C27), 154.34 (C20), 162.87 (C5), 164.36 (C4). HRMS: m/z: 542.0048. C29H21Cl2N5O2.
2-{[(5,7-Diiodoquinolin-8-yl)oxy]methyl}-5-(5-methyl-1,3-diphenyl-1H-pyrazol-4-yl)-1,3,4-oxadiazole (11f). Yield 64%. IR spectrum, ν, cm–1: 3241 (C–H), 1634 (C=N), 1245 (Ar–O–C), 1066 (N–N). 1H NMR spectrum, δ, ppm: 2.64 s (3H, CH3), 5.47 s (2H, CH2), 7.55–7.75 m (12H, ArH), 8.60–8.61 d (1H, ArH), 8.98–9.99 d. d (1H, ArH, J = 1.2, 5.2 Hz). 13C NMR spectrum, δC, ppm: 9.82 (C8), 71.53 (C19), 76.34 (C21), 85.78 (C23), 102.54 (C2), 121.62 (C26), 123.11 (C9, C13), 126.44 (C11), 127.69 (C14, C18), 128.79 (C22), 128.78 (C16), 129.47 (C15, C17), 129.68 (C24), 129.93 (C10, C12), 132.11 (C7), 132.86 (C25), 134.56 (C3), 137.88 (C6), 139.92 (C28), 147.77 (C1), 153.05 (C27), 154.35 (C20), 162.83 (C5), 164.67 (C4). HRMS: m/z: 711.8631. C28H19I2N5O2.
2-(5-Methyl-3-phenylisoxazol-4-yl)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (11g). IR spectrum, ν, cm–1: 3266 (C–H), 1631 (C=N), 1251 (Ar–O–C), 1069 (N–N). 1H NMR spectrum, δ, ppm: 2.40 s (3H, CH3), 5.13 s (2H, CH2), 7.45–7.55 m (9H, ArH), 8.56–8.58 d. d (1H, ArH, J = 1.2, 5.2 Hz), 8.96–8.97 d. d (1H, ArH, J = 1.6, 5.6 Hz). 13C NMR spectrum, δC, ppm: 14.81 (C6), 73.96 (C13), 106.83 (C15), 110.96 (C2), 113.41 (C17), 120.33 (C20), 124.64 (C16), 124.88 (C8, C12), 128.46 (C10), 129.02 (C9, C11), 129.62, (C18), 132.41 (C7), 140.48 (C19), 148.39 (C22), 150.24 (C21), 153.30 (C1), 154.91 (C14), 162.36 (C3), 162.50 (C5), 163.28 (C4). HRMS: m/z: 385.0467. C22H16N4O3.
2-{[(5-Chloroquinolin-8-yl)oxy]methyl}-5-(5-methyl-3-phenylisoxazol-4-yl)-1,3,4-oxadiazole (11h). Yield 87% yield. IR spectrum, ν, cm–1: 3258 (C–H), 1623 (C=N), 1243 (Ar–O–C), 1067 (N–N). 1H NMR spectrum, δ, ppm: 2.13 s (3H, CH3), 5.45 s (2H, CH2), 7.46– 7.59 m (8H, ArH), 8.57–8.59 d. d (1H, ArH, J = 1.6, 6.4 Hz), 9.00–9.01 d. d (1H, ArH, J = 1.6, 5.6 Hz). 13C NMR spectrum, δC, ppm: 13.72 (C6), 72.57 (C13), 106.38 (C15), 110.38 (C2), 118.46 (C17), 122.34 (C20), 126.16 (C16), 127.61 (C8, C12), 127.88 (C10), 128.48 (C9, C11), 128.72 (C18), 131.75 (C7), 132.87 (C19), 139.84 (C22), 152.80 (C21), 155.19 (C1), 155.44 (C14), 163.29 (C3), 164.34 (C5), 165.59 (C4). HRMS: m/z: 419.0205. C22H15ClN4O3.
2-{[(5,7-Dichloroquinolin-8-yl)oxy]methyl}-5-(5-methyl-3-phenylisoxazol-4-yl)-1,3,4-oxadiazole (11i). Yield 87%. IR spectrum, ν, cm–1: 3259 (C–H), 1633 (C=N), 1238 (Ar–O–C), 1066 (N–N). 1H NMR spectrum, δ, ppm: 2.20 s (3H, CH3), 5.46 s (2H, CH2), 7.47–7.59 m (7H, ArH), 8.61–8.62 d. d (1H, ArH, J = 1.6, 5.6 Hz), 9.03–9.05 d. d (1H, ArH, J = 1.6, 5.6 Hz). 13C NMR spectrum, δC, ppm: 14.29 (C6), 73.07 (C13), 110.38 (C15), 110.44 (C2), 119.57 (C17), 122.54 (C20), 127.16 (C16), 127.83 (C8, C12), 127.93 (C10), 128.85 (C9, C11), 128.93 (C18), 131.80 (C7), 133.17 (C19), 140.14 (C22), 153.00 (C21), 155.34 (C1), 155.53 (C14), 164.03 (C3), 164.54 (C5), 166.12 (C4). HRMS: m/z: 452.5274. C22H14Cl2N4O3.
2-(5-Methyl-3-phenylisoxazol-4-yl)-5-{[(2-methylquinolin-8-yl)oxy]methyl}-1,3,4-oxadiazole (11j). Yield 84%. IR spectrum, ν, cm–1: 3251 (C–H), 1639 (C=N), 1243 (Ar–O–C), 1085 (N–N). 1H NMR spectrum, δ, ppm: 2.22 s (3H, CH3), 2.53 s (3H, CH3), 5.51 s (2H, CH2), 7.16–7.17 d (1H, ArH), 7.53–7.65 m (8H, ArH), 8.42–8.44 d. d (1H, ArH, J = 1.6, 5.6 Hz). 13C NMR spectrum, δC, ppm: 12.00 (C6), 24.42 (C23), 71.89 (C13), 107.12 (C15), 111.13 (C2), 113.76 (C17), 120.13 (C20), 124.00 (C16), 124.97 (C8, C12), 128.56 (C10), 128.89 (C9, C11), 132.76 (C18), 136.57 (C7), 140.63 (C19), 148.68 (C22), 150.00 (C21), 153.52 (C1), 155.06 (C14), 162.44 (C3), 162.86 (C5), 163.47 (C4). HRMS: m/z: 399.1336. C23H18N4O3.
2-{[(5,7-Dichloro-2-methylquinolin-8-yl)oxy]methyl}-5-(5-methyl-3-phenylisoxazol-4-yl)-1,3,4-oxadiazole (11k). Yield 82%. IR spectrum, ν, cm–1: 3263 (C–H), 1643 (C=N), 1229 (Ar–O–C), 1077 (N–N). 1H NMR spectrum, δ, ppm: 2.23 s (3H, CH3), 2.45 s (3H, CH3), 5.55 s (2H, CH2), 7.21–7.22 d (1H, ArH), 7.46– 7.55 m (6H, ArH), 8.61–8.63 d. d (1H, ArH, J = 1.6, 5.6 Hz). 13C NMR spectrum, δC, ppm: 14.37 (C6), 25.26 (C23), 73.12 (C13), 110.47 (C15), 111.14 (C2), 118.76 (C17), 122.72 (C20), 127.34 (C16), 128.02 (C8, C12), 128.01 (C10), 128.98 (C9, C11), 128.79 (C18), 131.91 (C7), 133.27 (C19), 140.42 (C22), 153.13 (C21), 155.47 (C1), 155.67 (C14), 164.42 (C3), 164.97 (C5), 166.32 (C4). HRMS: m/z: 466.8572. C23H16Cl2N4O3.
2-{[(5,7-Diiodoquinolin-8-yl)oxy]methyl}-5-(5-methyl-3-phenylisoxazol-4-yl)-1,3,4-oxadiazole (11l). Yield 77%. IR spectrum, ν, cm–1: 3249 (C–H), 1639 (C=N), 1243 (Ar–O–C), 1072 (N–N). 1H NMR spectrum, δ, ppm: 2.32 s (3H, CH3), 5.39 s (2H, CH2), 7.44– 7.56 m (7H, ArH), 8.61–8.62 d. d (1H, ArH, J = 1.6, 5.6 Hz), 8.89–8.91 d. d (1H, ArH, J = 1.6, 5.2 Hz). 13C NMR spectrum, δC, ppm: 13.81 (C6), 73.23 (C13), 76.97 (C15), 86.78 (C17), 110.56 (C2), 122.76 (C20), 127.86 (C8, C12), 127.26 (C10), 128.90 (C9, C11), 128.87 (C18), 131.77 (C7), 133.47 (C19), 140.38 (C22), 147.42 (C16), 153.15 (C21), 155.47 (C1), 155.68 (C14), 164.17 (C3), 164.46 (C5), 166.23 (C4). HRMS: m/z: 636.2477. C22H14I2N4O3: 635.9155.
Molecular docking study. Docking study was performed using Autodock-4.2. For evaluating the best binding sites the study was carried out for the crystal structure of E. coli (complex with Clorobiocin, PDB file 1KZN) by the Lamarckian Genetic Algorithm computational method. To avoid interactions of solvent molecules, the software was calibrated by eliminating water and lower occupancy residues. Hydrogen bond with other docking constants represented the possible proximity of ligands with the receptor.
In vitro antibacterial and antifungal activity. The antimicrobial assay was performed by well-known methods [28]. The target compounds were tested for their antimicrobial activity against Gram-positive (Staphylococcus aureus (MTCC 96) and Bacillus cereus (MTCC 8372)) and Gram-negative (Escherichia coli (MTCC 724), Klebsiella pneumonia (MTCC 3384) , and Shigella flexneri (MTCC 1457)) bacterial strains using nutrient agar medium by utilizing the paper disc diffusion method. Antifungal activity of synthesized compounds was tested on Candida albicans (MTCC 183) and Aspergillus niger (MTCC 281) fungal strains by the disc-diffusion method. Chloramphenicol and Fluconazole were used as standard drugs for antimicrobial and antifungal activity respectively. All experiments were done in triplicates and the results were represented in terms of diameter of the zone of inhibition. The MIC values were determined by the liquid dilution method against the same organisms, and absorbance of the test solutions was recorded on a spectrophotometer at 555 nm.
In vitro anti-inflammatory activity. Anti-inflammatory study was carried out by Protein denaturation by bovine serum albumin method [29]. The procedure described by Mamata D N and coworkers was used for preparing the solutions with slight modification [30]. The bovine serum albumin solution was obtained using tris buffer saline and pH adjusted to 6.8 by acetic acid. Series of test samples were prepared in 0.5 mL DMSO by treating different concentrations of target compounds with 0.5 mL of bovine serum albumin. A standard solution containing Diclofenac sodium (0.05 mL) and bovine serum albumin (0.05 mL) were used for comparison. The sample solutions were incubated at 37°C (20 min) and then at 57°C (3 min), then cooled down to room temperature and 2.5 mL of phosphate buffer were added. The mixtures were shaken thoroughly, and absorbance was measured on a UV-Vis spectrophotometer at 620 nm. Protein denaturation in terms of percentage inhibition (I, %) of precipitation was calculate using the following formula:
where Vc—absorbance of control and Vs—absorbance of sample solution.
CONCLUSIONS
The new 1,3,4-oxadiazole bridged quinoline linked pyrazole/isoxazole heterocyclic compounds have been synthesized. The molecular docking study of the final compounds against E. coli protein has indicated that those containing pyrazole heterocycle with methyl (CH3) group at quinoline moiety have high binding energy and demonstrate excellent antimicrobial activity against the majority of the tested strains. Hence, the above templates can be considered as a useful lead for future development of potent drugs.
REFERENCES
Singh, S.K., and Singh, S., Int. J. Pharm. Sci. Rev. Res., 2014, vol. 25, p. 295.
Awadhesh, K.S., Akriti, S., Altamash, S., Ritu, S., and Anoop, M., Diabetes Metab. Syndr., 2020, vol. 14, p. 241-246. https://doi.org/10.1016/j.dsx.2020.03.011
Arezoo, R.P., and Azar, T., Iran. J. Med Sci., 2017, vol. 42, p. 115.
Ramadan, A.M., Mariam, A.A.S., Hanadi, Y.M., and Kamal, U.S., RSC Adv., 2020, vol. 10 p. 19867. https://doi.org/10.1039/D0RA02786C
Obaid, A., Suresh, K., and Rafi, M.H., Eur. J. Med. Chem., 2015, vol. 97, p. 871. https://doi.org/10.1016/j.ejmech.2014.07.044
Khalilullah, H., Ahsan, J.M., Hedaitullah, M., Khan, S., and Ahmed, B., Mini Rev. Med. Chem., 2012, vol. 12, p. 789. https://doi.org/10.2174/138955712801264800
Basavanna, V., Ningaiah, S., Chandramouli, M., Sobha, A., and Doddamani, S., J. Iran. Chem. Soc., 2021, vol. 18, p. 1479. https://doi.org/10.1007/s13738-020-02152-1
Annamaria, L., Jialin, M., Baojie, W., Yuehong, W., Scott, G.F., and Alan, P.K., J. Med. Chem., 2009, vol. 52, p. 2109. https://doi.org/10.1021/jm900003c
Jialin, M., Hai, Y., Yuehong, W., Baojie, W., Dennis, P., Rong, H., and Scott, G.F., Bioorg. Med. Chem. Lett., 2010, vol. 20, p. 1263. https://doi.org/10.1016/j.bmcl.2009.11.105
Sambasiva, R.P., Kurumurthy, C., Veeraswamy, B., Poornachandra, Y., Ganesh, K.C., and Narsaiah, B., Bioorg. Med. Chem. Lett., 2014, vol. 24, p. 1349. https://doi.org/10.1016/j.bmcl.2014.01.038
Mariappan, B., Kasi, P., and Penugonda, R., Eur. J. Med. Chem., 2012, vol. 47, p. 608. https://doi.org/10.1016/j.ejmech.2011.10.045
Rajanarendar, E., Nagi, R.M., Rama, K.S., Rama, M.K., Reddy, Y.N., and Rajam, M.V., Eur. J. Med. Chem., 2012, vol. 55, p. 273. https://doi.org/10.1016/j.ejmech.2012.07.029
Rania, H., Samia, A.E., Noha, I.Z., Arwyn, T.J., and Andrew, D.W., Molecules., 2019, vol. 24, p. 1274. https://doi.org/10.3390/molecules24071274
Sudeesh, K., and Gururaja, R., Organic Chem. Curr. Res., 2017, vol. 6, p. 1. https://doi.org/10.4172/2161-0401.1000183
Damoder, R.M., Dilipkumar, U., and Blake, W.E., Chem. Sci., 2018, vol. 9, p. 1782. https://doi.org/10.1039/C7SC04107A
Ibrahim, C., Ola, H.R., Tamer, M.I., Shery, S.H., El-Sayeda, M.E.K., Aida, E.B., Ibrahim, M.E.A., and Hisham, A.N., Bioorg. Chem., 2018, vol. 78, p. 220. https://doi.org/10.1016/j.bioorg.2018.03.023
Muzushima, Y. and Kobayashi, M., J. Pharm. Pharmacol., 1968, vol. 20, p. 169. https://doi.org/10.1111/j.2042-7158.1968.tb09718.x
Mohammadizadeh, M.R. and Basti, F., J. Iran. Chem. Soc., 2015, vol. 12, p. 1171. https://doi.org/10.1007/s13738-014-0578-4
Zhang, J.W., Yang, W.W., Chen, L.L., Chen, P., Wang, Y.B., and Chen, D.Y., Org. Biomol. Chem., 2019, vol. 17, p. 7461. https://doi.org/10.1039/C9OB01387C
Katayama, H. and Oshiyama, T., Can. J. Chem., 1997, vol. 75, p. 913. https://doi.org/10.1139/v97-109
Srikantamurthy, N., Vishalakshi, G.J., Jeyaseelan, S., Umesha, K.B., and Mahendra, M., Acta Cryst. E, 2013, vol. 69, p. 897. https://doi.org/10.1107/S1600536813011410
Umesha, K.B., Rai, K.M., and Nayaka, M.H., Int. J. Biomed. Sci., 2009, vol. 4, p. 359-368.
Jagadish, S., Rajeev, N., Naveen Kumar, S.K., Kumar, K.S., Paul, M., Hegde, M., Sadashiva, M.P., Girish, K.S., and Rangappa, K.S., Mol. Cell. Biochem., 2016, vol. 414, p. 137. https://doi.org/10.1007/s11010-016-2667-4
Shubakar, K., Umesha, K.B., Srikantamurthy, N., and Chethan, J., Bulg. Chem. Commun., 2013, vol. 45, p. 274.
Ningaiah, S., Bhadraiah, U.K., Doddaramappa, S.D., Keshavamurthy, S., and Javarasetty, C., Bioorg. Med. Chem., 2014, vol. 24, p. 245. https://doi.org/10.1016/j.bmcl.2013.11.029
Soliman, R. and Hammouda, N.A., J. Pharm. Sci., 1979, vol. 68, p. 1377. https://doi.org/10.1002/jps.2600681110
Naik, R.N., Pramod, H., Patil, S.C., Shridharamurthy, S., and Satyanarayana, S.B., Inventi Rapid: Med. Chem. 2013, vol. 2013, p. 29.
Khandelwal, N., Gautam, N., and Gautam, D.C., Int. J. Chem. Sci., 2013, vol. 125, p. 85. https://doi.org/10.1007/s12039-013-0363-4
Rahman, H., Eswaraiah, M.C., and Dutta, A.M., Am. Eur. J. Agri. Environ. Sci., 2015, vol. 15, p. 115. https://doi.org/10.5829/idosi.aejaes.2015.115.121
Naik, M.D., Bodke, Y.D., Prashantha, J., and Naik, J.K., Int. J. Chem. Sci., 2021, vol. 133, p. 1. https://doi.org/10.1007/s12039-020-01875-1
ACKNOWLEDGMENTS
The authors acknowledge the Management, Vidyavardhaka College of Engineering, Mysuru for the support to carry out research activity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Supplementary information
Rights and permissions
About this article

Cite this article
Basavanna, V., Chandramouli, M., Kempaiah, C. et al. A New Series of 1,3,4-Oxadiazole Linked Quinolinyl-Pyrazole/Isoxazole Derivatives: Synthesis and Biological Activity Evaluation. Russ J Gen Chem 91, 2257–2266 (2021). https://doi.org/10.1134/S1070363221110128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363221110128