
Abstract
2-Oxoacids are involved in a number of important metabolic processes and can be used as biomarkers in some human diseases. A new optimized method for quantification of 2,4-dinitrophenylhydrazine derivatives of 2-oxoacids using high-performance liquid chromatography was developed based on available techniques for quantification of 2-oxoacids in mammalian brain. The use of the 2,4-dinitrophenylhydrazine derivatives of 2-oxoacids was shown to be more advantageous in comparison with the previously used phenylhydrazine derivatives, due to a high chemical stability of the former. Here, we determined the concentrations of pyruvate, glyoxylate, 2-oxoglutarate, 2-oxomalonate, and 4-methylthio-2-oxobutyrate in the methanol/acetic acid extracts of the rat brain using the developed method, as well discussed the procedures for the sample preparation in analysis of mammalian brain extracts. The validation parameters of the method demonstrated that the quantification limits for each of the analyzed of 2-oxoacids was 2 nmol/mg tissue. The developed method facilitates identification of subtle changes in the tissue and cellular content of 2-oxoacids as (patho)physiological biomarkers of metabolism in mammalian tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
2-Oxoacids (or α-ketoacids) are organic compounds containing keto group in the α-position to the carboxyl group. 2-Oxoacids participate in numerous reactions of various metabolic networks. In particular, pyruvate (Pyr), 2-oxoglutarate (2-OG), and oxaloacetate form the nodes of metabolic pathways mediating interactions between glycolysis, Krebs cycle, gluconeogenesis, and catabolism of some amino acids [1]. Other 2-oxoacids participate in the metabolism of certain amino acids as intermediates [2-oxo-isovalerate, 2-oxo-isocaproate, 3-methyl-2-oxovalerate, 2-oxoadipate, 2-oxobutyrate, and 4-methylthio-2-oxobutyrate (MTOB)] or side products [phenylpyruvate, glyoxylate (Glx)]. That is why they can serve as prognostic markers in the diagnostics and monitoring of disease course. For example, an altered content (deficiency or excess) of 2-oxoacids has been observed in hypovitaminosis of group B vitamins, hypoxia, cancer, starvation, diabetes, various hereditary diseases, models of neurodegenerative diseases, and other disorders [2-8].
Recently developed high-throughput mass spectrometry techniques for analysis of low molecular weight metabolites (e.g., LC-MS/MS/MS) allow to analyze several hundred compounds (forming the so-called metabolomes) in order to elucidate organism’s response to various types of stress, as well as to investigate manifestations of various diseases such as cancer, diabetes, and neurodegeneration. In particular, in the studies of the Huntington’s disease in Drosophila melanogaster model, changes in the content of Pyr and 2-OG were observed at the symptomatic stage, which indicated significant disease-associated changes in metabolism, considering positions of these metabolites in the metabolic pathway nodes [7]. Changes in the blood plasma levels of 2-OG during starvation are believed to act as signaling events, as 2-OG is capable of transcription modulation. In particular, it activates 2-OG-dependent prolyl hydroxylases, resulting in the destabilization of transcription factors followed by the activation of global catabolic and anabolic processes during long-term starvation [8]. Recently, the role of some 2-oxoacids in cancer has been extensively investigated because these compounds can regulate gene expression and affect modification of DNA and histones through epigenetic mechanisms. Hence, assessing the cellular content of 2-oxoacids can help in elucidating possible epigenetic changes [9].
The majority of methods developed for the quantification of 2-oxoacids include the use of analytical HPLC in combination with derivatization of analyzed compounds. For example, the use of phenylhydrazine as a derivatization agent has allowed spectrophotometric detection of formed phenylhydrazone derivatives, which can be used for quantitative analysis of Glx, Pyr, and 2-OG in the urine [10, 11]. Outside the chromatographic techniques, one of the earlier suggested agents for quantitative detection of 2-oxoacids was 2,4-dinitrophenyl hydrazine (DNPH), a derivative of phenylhydrazine [12]. Considering that DNPH derivatives of 2-oxoacids have different absorption maxima within the 380-520 nm range, DNPH was suggested, for example, for the use in the transaminase activity assay [13]. Due to the high stability of DNPH derivatives, DNPH derivatization followed by HPLC and spectrophotometric detection of the resulting compounds was used for quantitative determination of aldehydes and ketones [14, 15]. So far, derivatization of 2-oxoacids with DNPH and their chromatographic separation was used only for the comparison of detection sensitivity with another derivatization agent – o-phenylenediamine [16].
One of the serious drawbacks of earlier studies on derivatization of 2-oxoacids was that they were aimed solely solving at practical issues in the analysis of a limited number of 2-oxoacids. At the same time, no validated methods for simultaneous quantification of several biologically relevant 2-oxoacids have been developed. In addition, even that o-phenylenediamine was successfully used as a derivatizing agent for the HPLC-based detection of 2-oxoacids in urine and blood plasma [17], the used technique required preliminary purification of 2-oxoacids on a hydrazide gel column and extraction of derivatives with ethyl acetate.
Later studies reported some method validation parameters, such as limit of detection (LOD), limit of quantification (LOQ), and signal-to-noise ratio. When o-phenylenediamine was used as a derivatization agent with the following fluorometric detection, LOQ was 1.2 µM [18, 19] and LOD was ~0.25 µM [11] and 0.39 µM [19], while LOD for 1,2-diamino-4,5-methylenebenzene was lower (0.014 µM) at a signal-to-noise ratio of 5 : 1 [6]. The use of DNPH as a derivatization agent followed by spectrophotometric detection of the formed derivatives was validated for the quantification of aldehydes, but not 2-oxoacids, in the animal feed [15] with reported sensitivity of ~40 µM.
The most common method used in the studies employing chromatographic techniques for determination of 2-oxoacids is reversed-phase HPLC (RP-HPLC). Furthermore, in all the studies without exception, chromatographic separation is preceded by 2-oxoacid derivatization. The most often used derivatization agents are phenylhydrazine and its 2,4-dinitro derivative (spectrophotometric detection) and o-phenylenediamine and its 4,5-methylenedioxy derivative (fluorometric detection).
In this work, we proposed a method for detection of 2-oxoacids in the animal brain extracts using DNPH derivatization and RP-HPLC with the following spectrophotometric detection. Among the analyzed 2-oxoacids, Glx, 2-OG, and Pyr have been previously quantified in the human and animal blood and urine samples, while the data on the HPLC detection of 2-oxomalonate (2-OM) and MTOB in the animal sample were absent. Similarly to the quantitative determination of formaldehyde [15], the reactions of DNPH with 2-oxoacids produce compounds (hydrazones) that absorb light in the visible range (380-520 nm), which might ensure more sensitive detection in comparison with the detection based on the adsorption of derivatives produced in the reaction with phenylhydrazine in the UV range (324 nm) [10, 11]. Moreover, the DNPH derivatives of carbonyl compounds are chemically stable [15], contrary to phenylhydrazine derivatives (especially Glx derivatives), which deteriorate within several days [11]. We validated the suggested method and demonstrated that LOQ was 1 µM for each of five 2-oxoacids tested. The method was used for quantification of 2-oxoacids in the methanol/acetic acid extracts of rat brain, which expanded the use of HPLC for detection of 2-oxoacids not only in biological fluids, but also in animal tissue extracts. We believe that when used in combination with the previously developed methods for determination of free amino acids in brain homogenates, precise analysis of the content of 2-oxoacid as low-molecular-weight markers of the metabolic nodes, will help in predicting the functioning of mechanisms of energy production from carbohydrates and amino acids [20-22].
MATERIALS AND METHODS
All used reagents were of the highest available degree of purity. Reagents were purchased from the following manufacturers: 2,4-dinitrophenylhydrazine hydrochloride (99%, Panreac, USA), acetonitrile (HPLC, gradient grade, Panreac), phosphoric acid, concentrated for HPLC (85-90% purity, Fluke, USA), methanol (99.8% purity, Supelco, USA), chloroform (99.8% purity, Supelco), pyruvate (99%, Merck, USA), acetic acid (99.5 % purity, Panreac, Spain), KH2PO4 (99.5% purity, Biochimica, Italy), all other reagents such as 2-oxoacid standards, including 2-OM (98% purity), 2-OG (98.5% purity), MTOB (97% purity), and Glx (98% purity) were from Sigma-Aldrich (USA). Pyr and MTOB were used as sodium salts, 2-OM – as a sodium salt monohydrate, 2-OG – as an acid, and glyoxylate – as an acid monohydrate. Deionized water (Millipore Milli-Q PLUS, R ≥ 18.2 Mohm·cm) was used for solution preparation.
Preparation of solutions. To prepare derivatizing mixture, 10 µl of 85% phosphoric acid was mixed with 10 ml of acetonitrile; next, 10 mg of 2,4-dinitrophenylhydrazine hydrochloride was added. Standard stock solutions of 2-oxoacids (2 mM) were prepared in 1.5-ml tubes by placing 10-µl aliquots of the concentrated solutions (100 mM) of Glx, Pyr, 2-OM, 2-OG, and MTOB followed by adding 450 µl of water. To prepare reference solution (50 µM), 10 µl of the standard solution of 2-oxoacids (2 mM) was mixed with 390 µl of water. To prepare test solutions, 150 µl of the sample solution, blank solution (water), or reference solution were placed into a tube and 150 µl of derivatizing solution were added followed by the incubation at room temperature for 2 h.
RP-HPLC analysis of DNPH derivatives of 2-oxoacids. DNPH derivatives of 2-oxoacids were fractionated on a Synergi Max-RP column (Phenomenex, 250 mm × 4.6 mm; particle size, 5 µm) at 25°C at a flow rate of 1 ml/min using an Agilent 1200 HPLC system with a diode-array detector with detection at 360 nm. Other conditions of chromatographic fractionation were selected during the method development.
Preparation of methanol/acetic acid extracts of rat brain and removal of lipid fraction. Extracts from the brain cortex of male Wistar rats, provided by A. V. Graf (Belozersky Institute of Physicochemical Biology and Department of Biology, Moscow State University), were prepared using a methanol/acetic acid mixture as described in [20]. Briefly, tissue samples stored at –70°C were homogenized in 8 volumes of ice-cold methanol. The homogenate was diluted 2.5-fold with 0.2% acetic acid and incubated in Excella E24 shaker (New Brunswick Scientific, Germany) on ice for 30 min at a rotation speed of 180 rpm. Denatured proteins were removed by centrifugation for 20 min at 21,500g at 4°C. The supernatant was transferred into clean cooled tubes and stored at –70°C. Prior to analysis, lipid fraction was removed by extraction with chloroform [23] using a modified technique for metabolite extraction from animal cells [24]. The methanol/acetic acid extract was mixed with one volume of chloroform, thoroughly shaken for 1 min, and centrifuged for 5 min at 3500g. The upper fraction containing polar compounds was carefully separated from the bottom lipid-containing fraction, divided into 2-ml aliquots, and dried in an Eppendorf Concentrator Plus (Helicon, Russia) at room temperature. Dried aliquots of the polar fraction were stored at –70°C before analysis. Immediately before the analysis, the aliquots were dissolved in 200 µl of 50 mM potassium phosphate buffer, pH 6.0.
RESULTS
Development of HPLC method for fractionation of DNPH derivatives of 2-oxoacids. The optimal pH of the mobile phase was selected using the ACD/Phys Chem History program, ver. 2020.2.0 (ACD/Labs, Canada). This program calculates pK values and logD (hydrophobicity, octanol/water partitioning coefficient) based on the compound structure and solution pH so that at the selected pH value, all tested components are in the dominating ionic form and the differences in the logD values facilitate their separation by RP-HPLC. The pH range recommended by the manufacturer for a particular column should also be taken into consideration. The relative contents of ionic forms of 2-oxoacids at different pH values for each of the six tested compounds (5 derivatives of 2-oxoacids and derivatizing agent itself) are shown in Fig. 1.

Relative content of ionic forms of 2-OM-DNPH (a), 2-OG-DNPH (b), MTOB-DNPH (c), DNPH (d), Glx-DNPH (e), and Pyr-DNPH (f) at different pH values. Numbers next to the curves indicate ionic form charges.
The calculated dependence of the dominating ionic form relative contents on the pH value for the analyzed compounds is presented in Fig. 2a. The maximum content of the dominating ionic form for all six compounds was in the pH range from 5.9 to 6.2. The dependence of the compound hydrophobicity (logD) on the medium pH (Fig. 2b) confirmed that the best separation could be achieved at pH ~6.0. Free DNPH demonstrated the highest logD value, which suggests that the five DNPH derivatives have shorter retention times than DNPH. Since the Synergi Max-RP (Phenomenex) columns can be used with a mobile phase within the pH range from 1.5 to 10, pH 6.0 was in the acceptable pH interval.

a) pH-dependence of the relative content of the dominating ionic form for six tested compounds. b) pH-dependence of the octanol/water partition coefficient (logD) for six tested compounds: 2-OM-DNPH (1), 2-OG-DNPH (2), MTOB-DNPH (3), DNPH (4), Glx-DNPH (5), and Pyr-DNPH (6). Selected optimal pH value is shown by the vertical line. Regions outside of the operating pH range of the column are shown in red. Calculations were carried out with the ACD/Phys Chem History program (version 2020.2.0, ACD/Labs, Canada).
Based on the data presented in Fig. 2, mobile phase A was 15 mM potassium phosphate solution in water (pH 6). Acetonitrile (100%) was used as mobile phase B. The gradient of acetonitrile concentration was optimized with the ACD/AutoChrom 2020.2.0 program (ACD/Labs). In order to evaluate the dependence of peak retention times on acetonitrile concentration, three gradient regimes were tested (Table 1). The calculated and experimental retention times of analyzed compounds are presented in Table 1. The peaks were identified based on the analysis of derivatization products for each individual 2-oxoacid (Fig. 3a). Most 2-oxoacids formed two types of derivatives with DNPH, likely due to the presence of cis- and trans-isomers.
The data produced for the three used experimental gradient regimes (Table 1) could be satisfactorily describes by the linear parametric model lnk’ = a + b*X [where k’ – capacity coefficient, X – acetonitrile content in the mobile phase, (a) and (b) – regression coefficients]. The predicted optimal gradient regime (20-40% acetonitrile over 15 min) was implemented using modification of the mobile phase A. The modified phase A contained 20% acetonitrile in 12 mM potassium phosphate solution in water (pH 6.0). The optimized elution gradient is shown in Table 2.
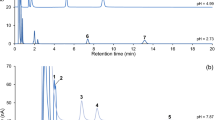
Fractionation of the standard mixture (Fig. 3b) using the optimized elution regime provided satisfactory resolution (>1.0) of 2-OM (3.5), 2-OG (9.8), MTOB (1.7), Glx-1 (13.1), Glx-2 (10.1), and Pyr-1 (16.0).

a) RP-HPLC fractionation of standard mixture of 2-oxoacids using three elution regimes: 5-50% B over 20 min (1), 5-50% B over 40 min (2), and 20-60% B over 30 min (3) in mobile phase A. b) Chromatogram of standard mixture of 2-oxoacids using modified mobile phase A and optimized elution regime (Table 2). Peak Y is a by-product of derivatization of 2-oxoacids (not identified)
The validation parameters of the developed fractionation method are presented in Table 3.
It was found that for the compounds producing two peaks of derivatization products, the areas under single peaks displayed higher variability than the sum of the areas under these peaks. Based on the obtained validation parameters, it can be concluded that the developed method allows determination of all tested 2-oxoacids with a sufficiently high accuracy, except the sulfur-containing MTOB, which is unstable and whose peak does not resolve well from DNPH peak.
Analysis of 2-oxoacids in the samples of rat brain homogenates. Despite the reliability of the developed method, HPLC-based quantification of 2-oxoacids in biological fluids and tissues is not always a simple task. The first problem is associated with the sample preparation. First of all, this includes the efficiency of the total protein precipitation using specialized reagents, as well as necessity for the removal of admixtures, primarily of lipid nature, that interfere with the subsequent analysis. In view of this, pre-processing of animal brain samples is especially challenging due to the high content of lipids of different nature in the brain. Despite the selection of conditions that facilitate the maximal removal of brain lipids, even trace amounts of lipids in the samples could cause fast deterioration of chromatographic column (as based on our experience).
The selected chromatographic conditions were used for separating 2-oxoacids and measuring their content in the methanol/acetic acid extracts of the rat brain (representative chromatogram is shown in Fig. 4).

Averaging the results of analysis of the brain extracts from different animals (Table 4) demonstrated that the determined content of 2-oxoacids was reproducible; the standard error of mean for the investigated samples of brain extracts from different animals was: 8, 13, 17, 20, and 22% of mean for 2-OM, MTOB, Pyr, 2-OG, and Glx, respectively. The observed high dispersion for some of the compounds (Glx, 2-OG, and Pyr) was likely associated with the individual features of brain metabolism.
DISCUSSION
Metabolomics (systemic investigation of metabolomes) is a promising research area, as important as genomics and proteomics. However, the metabolomics database is still relatively small, and quantitative analysis of particular metabolite requires special techniques that still have to be developed. In many cases, the use of targeted approach, such as determination of a relatively small number of 2-oxoacids, seems to be more relevant.
The earlier methods used for quantitative assay of 2-oxoacids in clinical and research laboratories involved spectrophotometric and fluorometric monitoring of enzymatic NADH-dependent reactions. These reactions are easy to conduct and sufficiently reliable, at least for one-step techniques. The multistep techniques are used less often, as they require more reagents and enzymes and, hence, are more expensive and less reliable. Also, most of these enzymatic assays are applicable for the quantification of a single individual compound. In the case of 2-oxoacids, the above issues were partially resolved by using analytical HPLC combined with the derivatization of analyzed compounds [10, 11]. In particular, the use of o-phenylenediamine allowed to identify 2-oxoacid derivatives by spectrophotometry or fluorimetry [16]. The results of chromatographic analysis of 2-oxoacids with branched chain (2-oxoisovalerate, 2-oxoisocaproate, and 2-oxo-3-methylvalerate) in the blood plasma were comparable with the results of traditional enzymatic assays [35], which suggested the applicability of chromatography for the quantitative assay of other 2-oxoacids [17, 36]. Techniques based on derivatization with o-phenylenediamine and 4,5-methylenedioxy-1,2-diaminobenzene have demonstrated high sensitivity [6]. However, although phenylhydrazine has been used for determination of 2-oxoacids in biological samples by RP-HPLC [10, 11], no studies have employed its more stable derivative DNPH for the same purpose.
An average content of Pyr in the rat brain determined by RP-HPLC with DNPH derivatization in this study was close to that determined using traditional enzymatic assay (reported in the literature) (Table 4). Similarly to other tissues (except kidneys), Glx was not detected in the brain under normal conditions (Table 4); however, it was found in animals experiencing thiamine deficit [34]. No assays of the 2-OM and MTOB content in the animal brain have been described in the literature, although it was found that the concentration of the latter in the blood plasma of healthy humans is ~0.15 µM [37]. However, analysis of rat brain extracts revealed that the concentration of 2-OG was much lower than the concentration of Pyr, while according to the earlier published data, the content of these two 2-oxoacids should be similar (Table 4). This observation could be explained by the fact that 2-OG was not identified at some stages of analysis. The reasons for the low levels of this metabolite in the analyzed brain extracts could be the low content of this compound in the brain in comparison with other tissues, its loss during storage and drying of the extracts, and rapid degradation during the period between decapitation and full freezing of the cerebral cortex (less than several minutes). In particular, low content of 2-OG in the brain extracts could be explained by its reductive amination to glutamate, which could proceed non-enzymatically in the methanol-containing tissue extracts in the presence of pyridoxamine and especially urea as amino group donors [38]. In some studies, the concentration of urea in mammalian brain was found to be in a millimolar range [39], which, according to the published data, is one order of magnitude higher than the concentration of 2-OG (Table 4). Hence, non-enzymatic transformation of 2-OG into glutamate during sample processing and storage of the methanol/acetic acid extracts cannot be ruled out.
CONCLUSIONS
Here, we developed a new RP-HPLC method for quantification of 2-oxoacids (Glx, 2-OG, 2-OM, Pyr, and MTOB) that involves spectrophotometric detection of the products of their chemical derivatization with DNPH and determined its validation parameters. The developed method was used to evaluate the content of 2-oxoacids in the methanol/acetic acid extracts from the rat brains. In addition to the previously employed techniques for amino acid determination [20, 21], this method could be a useful tool for evaluating the role of 2-oxoacids as markers of interaction between metabolic pathways of carbohydrates and amino acids. It is known that the nodes common for such pathways involve the multienzyme complexes of 2-oxoacid dehydrogenases, which control the content of 2-oxoacids in the cells by irreversibly degrading these compounds. Moreover, suppression of 2-oxoglutarate dehydrogenase and pyruvate dehydrogenase complexes has been demonstrated in neurodegenerative diseases, which is associated with the metabolic stress characteristic for these pathologies [40]. The information on the levels of 2-oxoacids in combination with the knowledge on the content of amino acids and enzymatic activities in the samples could facilitate understanding the metabolic process in the animal brain under normal and pathological conditions.
Abbreviations
- 2-OG:
-
2-oxoglutarate
- 2-OM:
-
2-oxomalonate
- DNPH:
-
2,4-dinitrophenylhydrazine
- Glx:
-
glyoxylate
- MTOB:
-
4-methylthio-2-oxobutyrate
- Pyr:
-
pyruvate
- RP-HPLC:
-
reversed-phase high-performance liquid chromatography
References
Graham, D. E. (2011) 2-oxoacid metabolism in methanogenic CoM and CoB biosynthesis, Methods Enzymol., 494, 301-326, https://doi.org/10.1016/B978-0-12-385112-3.00015-9.
Muhling, J., Paddenberg, R., Hempelmann, G., and Kummer, W. (2006) Hypobaric hypoxia affects endogenous levels of alpha-keto acids in murine heart ventricles, Biochem. Biophys. Res. Commun., 342, 935-939, https://doi.org/10.1016/j.bbrc.2006.02.054.
Hutson, S. M., and Harper, A. E. (1981) Blood and tissue branched-chain amino and alpha-keto acid concentrations: Effect of diet, starvation, and disease, Am. J. Clin. Nutr., 34, 173-183, https://doi.org/10.1093/ajcn/34.2.173.
Hoang, M., and Joseph, J. W. (2020) The role of alpha-ketoglutarate and the hypoxia sensing pathway in the regulation of pancreatic beta-cell function, Islets, 12, 108-119, https://doi.org/10.1080/19382014.2020.1802183.
Kronberger, L., Semmelrock, H. J., Schaur, R. J., Schauenstein, E., Schreibmayer, W., et al. (1980) Tumor host relations. VI. Is alpha-ketoglutarate a tumor marker? Association with tumor extent in humans – correlation with tumor size in rats, J. Cancer Res. Clin. Oncol., 97, 295-299, https://doi.org/10.1007/BF00405781.
Shibata, K., Nakata, C., and Fukuwatari, T. (2016) High-performance liquid chromatographic method for profiling 2-oxo acids in urine and its application in evaluating vitamin status in rats, Biosci. Biotechnol. Biochem., 80, 304-312, https://doi.org/10.1080/09168451.2015.1083395.
Bertrand, M., Decoville, M., Meudal, H., Birman, S., and Landon, C. (2020) Metabolomic nuclear magnetic resonance studies at presymptomatic and symptomatic stages of Huntington’s disease on a Drosophila model, J. Proteome Res., 19, 4034-4045, https://doi.org/10.1021/acs.jproteome.0c00335.
Kondoh, H., Teruya, T., and Yanagida, M. (2020) Metabolomics of human fasting: new insights about old questions, Open Biol., 10, 200176, https://doi.org/10.1098/rsob.200176.
Losman, J. A., Koivunen, P., and Kaelin, W. G., Jr. (2020) 2-Oxoglutarate-dependent dioxygenases in cancer, Nat. Rev. Cancer, 20, 710-726, https://doi.org/10.1038/s41568-020-00303-3.
Petrarulo, M., Pellegrino, S., Bianco, O., Marangella, M., Linari, F., et al. (1988) High-performance liquid chromatographic determination of glyoxylic acid in urine, J. Chromatography, 432, 37-46, https://doi.org/10.1016/s0378-4347(00)80631-3.
Lange, M., and Malyusz, M. (1994) Fast method for the simultaneous determination of 2-oxo acids in biological fluids by high-performance liquid chromatography, J. Chromatography. B Biomed. Appl., 662, 97-102, https://doi.org/10.1016/0378-4347(94)00383-1.
Tonhazy, N. E., White, N. G., and Umbreit, W. W. (1950) A rapid method for the estimation of the glutamic-aspartic transaminase in tissues and its application to radiation sickness, Arch. Biochem., 28, 36-42.
Reitman, S., and Frankel, S. (1957) A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases, Am. J. Clin. Pathol., 28, 56-63, https://doi.org/10.1093/ajcp/28.1.56.
De Ochs, S. M., Fasciotti, M., Barreto, R. P., de Figueiredo, N. G., Albuquerque, F. C., et al. (2010) Optimization and comparison of HPLC and RRLC conditions for the analysis of carbonyl-DNPH derivatives, Talanta, 81, 521-529, https://doi.org/10.1016/j.talanta.2009.12.036.
Wahed, P., Razzaq, M. A., Dharmapuri, S., and Corrales, M. (2016) Determination of formaldehyde in food and feed by an in-house validated HPLC method, Food Chem., 202, 476-483, https://doi.org/10.1016/j.foodchem.2016.01.136.
Singh, B. K., Szamosi, I., and Shaner, D. (1993) A high-performance liquid chromatography assay for threonine/serine dehydratase, Anal. Biochem., 208, 260-263, https://doi.org/10.1006/abio.1993.1043.
Hayashi, T., Tsuchiya, H., Todoriki, H., and Naruse, H. (1982) High-performance liquid chromatographic determination of alpha-keto acids in human urine and plasma, Anal. Biochem., 122, 173-179, https://doi.org/10.1016/0003-2697(82)90267-6.
Martis, S., Droux, B. M., Deboudard, F., Nasser, W., Meyer, S., et al. (2020) Separation and quantification of 2-keto-3-deoxy-gluconate (KDG) a major metabolite in pectin and alginate degradation pathways, Anal. Biochem., 619, 114061, https://doi.org/10.1016/j.ab.2020.114061.
Marangella, M., Petrarulo, M., Bianco, O., Vitale, C., Finocchiaro, P., et al. (1991) Glycolate determination detects type I primary hyperoxaluria in dialysis patients, Kidney Int., 39, 149-154, https://doi.org/10.1038/ki.1991.19.
Ksenofontov, A. L., Boyko, A. I., Mkrtchyan, G. V., Tashlitsky, V. N., Timofeeva, A. V., et al. (2017) Analysis of free amino acids in mammalian brain extracts, Biochemistry (Moscow), 82, 1183-1192, https://doi.org/10.1134/S000629791710011X.
Trofimova, L., Ksenofontov, A., Mkrtchyan, G., Graf, A., Baratova, L., et al. (2016) Quantification of rat brain amino acids, Anal. Data Consistency, 12, 349-356, https://doi.org/10.2174/1573411011666151006220356.
Artiukhov, A. V., Kazantsev, A. V., Lukashev, N. V., Bellinzoni, M., and Bunik, V. I. (2020) Selective inhibition of 2-oxoglutarate and 2-oxoadipate dehydrogenases by the phosphonate analogs of their 2-oxo acid substrates, Front. Chem., 8, 596187, https://doi.org/10.3389/fchem.2020.596187.
Bligh, E. G., and Dyer, W. J. (1959) A rapid method of total lipid extraction and purification, Can. J. Biochem. Physiol., 37, 911-917, https://doi.org/10.1139/o59-099.
Trofimova, L. K., Araujo, W. L., Strokina, A. A., Fernie, A. R., Bettendorff, L., et al. (2012) Consequences of the alpha-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases, Curr. Med. Chem., 19, 5895-5906, https://doi.org/10.2174/092986712804143367.
Schenker, S., McCandless, D. W., Brophy, E., and Lewis, M. S. (1967) Studies on the intracerebral toxicity of ammonia, J. Clin. Invest., 46, 838-848, https://doi.org/10.1172/JCI105583.
Dale, R. A. (1965) Effects of sampling procedures on the contents of some intermediate metabolities of glycolysis in rat tissues, J. Physiol., 181, 701-711, https://doi.org/10.1113/jphysiol.1965.sp007792.
Miller, A. L., Hawkins, R. A., Harris, R. L., and Veech, R. L. (1972) The effects of acute and chronic morphine treatment and of morphine withdrawal on rat brain in vivo, Biochem. J., 129, 463-469, https://doi.org/10.1042/bj1290463.
Goldberg, N. D., Passonneau, J. V., and Lowry, O. H. (1966) Effects of changes in brain metabolism on the levels of citric acid cycle intermediates, J. Biol. Chem., 241, 3997-4003.
Holowach, J., Kauffman, F., Ikossi, M. G., Thomas, C., and McDougal, D. B., Jr. (1968) The effects of a thiamine antagonist, pyrithiamine, on levels of selected metabolic intermediates and on activities of thiamine-dependent enzymes in brain and liver, J. Neurochem., 15, 621-631, https://doi.org/10.1111/j.1471-4159.1968.tb08961.x.
Gardiner, M., Smith, M. L., Kagstrom, E., Shohami, E., and Siesjo, B. K. (1982) Influence of blood glucose concentration on brain lactate accumulation during severe hypoxia and subsequent recovery of brain energy metabolism, J. Cereb. Blood Flow Metab., 2, 429-438, https://doi.org/10.1038/jcbfm.1982.49.
Folbergrova, J., MacMillan, V., and Siesjo, B. K. (1972) The effect of hypercapnic acidosis upon some glycolytic and Krebs cycle-associated intermediates in the rat brain, J. Neurochem., 19, 2507-2517, https://doi.org/10.1111/j.1471-4159.1972.tb01310.x.
Thurston, J. H., Hauhart, R. E., and Schiro, J. A. (1983) Lactate reverses insulin-induced hypoglycemic stupor in suckling-weanling mice: Biochemical correlates in blood, liver, and brain, J. Cereb. Blood Flow Metab., 3, 498-506, https://doi.org/10.1038/jcbfm.1983.77.
Soto, M., Orliaguet, L., Reyzer, M. L., Manier, M. L., Caprioli, R. M., et al. (2018) Pyruvate induces torpor in obese mice, Proc. Natl. Acad. Sci. USA, 115, 810-815, https://doi.org/10.1073/pnas.1717507115.
Liang, C. C. (1962) Studies on experimental thiamine deficiency. Trends of keto acid formtion and detection of glyoxylic acid, Biochem. J., 82, 429-434, https://doi.org/10.1042/bj0820429.
Schadewaldt, P., Hummel, W., Trautvetter, U., and Wendel, U. (1989) A convenient enzymatic method for the determination of 4-methyl-2-oxopentanoate in plasma: Comparison with high performance liquid chromatographic analysis, Clin. Chim. Acta, 183, 171-182, https://doi.org/10.1016/0009-8981(89)90333-1.
Wendel, U., Even, G., Langenbeck, U., Schadewaldt, P., and Hummel, W. (1992) Determination of (S)- and (R)-2-oxo-3-methylvaleric acid in plasma of patients with maple syrup urine disease, Clin. Chim. Acta, 208, 85-91, https://doi.org/10.1016/0009-8981(92)90024-k.
Blom, H. J., Ferenci, P., Grimm, G., Yap, S. H., and Tangerman, A. (1991) The role of methanethiol in the pathogenesis of hepatic encephalopathy, Hepatology, 13, 445-454.
Wiese, E. K., Hitosugi, S., Buhrow, S. A., Loa, S. T., Sreedhar, A., et al. (2020) Reductive amination of alpha-Ketoglutarate in metabolite extracts results in glutamate overestimation, J. Chromatogr. A, 1623, 461169, https://doi.org/10.1016/j.chroma.2020.461169.
Arieff, A. I., Massry, S. G., Barrientos, A., and Kleeman, C. R. (1973) Brain water and electrolyte metabolism in uremia: Effects of slow and rapid hemodialysis, Kidney Int., 4, 177-187, https://doi.org/10.1038/ki.1973.100.
Bunik, V. (2017) Vitamin-Dependent Multienzyme Complexes of 2-Oxoacid Dehydrogenases: Structure, Function, Regulation and Medical Implications, Happauge: Nova Science Publishers.
Acknowledgments
The authors are grateful to A. V. Graf (Belozersky Institute of Physico-Chemical Biology and Faculty of Biology, Moscow State University) for providing rat brain samples.
Funding
This work was supported by the Russian Science Foundation (project no. 18-14-00116, V. I. Bunik, leading investigator).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare no conflicts of interest in financial or any other sphere. Animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the European Union Directives 86/609/EEC and 2010/63/EU and were approved by the Bioethics Committee of Lomonosov Moscow State University (protocol no. 69-o from 09.06.2016).
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tashlitsky, V.N., Artiukhov, A.V., Fedorova, N.V. et al. Analysis of Content of 2-Oxoacids in Rat Brain Extracts Using High-Performance Liquid Chromatography. Biochemistry Moscow 87, 356–365 (2022). https://doi.org/10.1134/S0006297922040058
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922040058