
Abstract
Paralogs, arising from gene duplications, increase the functional diversity of proteins. Protein functions in paralog families have been extensively studied, but little is known about the roles that intrinsically disordered regions (IDRs) play in their paralogs. Without a folded structure to restrain them, IDRs mutate more diversely along with evolution. However, how the diversity of IDRs in a paralog family affects their functions is unexplored. Using the RNA-binding protein Musashi family as an example, we applied multiple structural techniques and phylogenetic analysis to show how members in a paralog family have evolved their IDRs to different physicochemical properties but converge to the same function. In this example, the lower prion-like tendency of Musashi-1’s IDRs, rather than Musashi-2’s, is compensated by its higher α-helical propensity to assist their assembly. Our work suggests that, no matter how diverse they become, IDRs could evolve different traits to a converged function, such as liquid-liquid phase separation.
Similar content being viewed by others

Introduction
Recurrent gene duplications over many generations create paralogs in a genome, giving rise to a myriad of new protein functions1. The redundant copy, if not silenced as a pseudogene (the non-functional copy from its ancestor), enhances survival, and many duplicated genes mutate toward new functions2. The hemoglobin, composing paralog α- and β-globin chains, is a textbook example3: The existence of two copies of the α-globin gene (HBA1 and HBA2) reduces the effect of α-thalassemia, but not the β-thalassemia, which results from the loss of the only one β-globin encoding gene (HBB); These paralogs, which have evolved from an ancestral oxygen-binding globin, collaborate in their new function of oxygen transport3. Although protein paralogs have been studied for decades, the paralogs of intrinsically disordered proteins (IDPs) or proteins with intrinsically disordered regions (IDRs), account for more than half of the eukaryotic proteome4, remain largely ignored.
Our particular interest is in RNA-binding proteins (RBPs), which are at the core of gene regulation and RNA metabolism and whose dysfunction is implicated in many diseases5. In addition to the common feature of having RNA interacting motifs6, RBPs often contain IDRs7,8. The role of these IDRs had been unclear until it was recently shown that they can organize cellular structures without a lipid membrane9. These membraneless organelles, which assemble via liquid-liquid phase separation (LLPS), have since been extensively studied and have become a working model of spatiotemporal control for many cellular functions10. Although some RBPs’ IDRs have been reported involved in forming membraneless organelles (such as RNA or stress granules), a large proportion of them are still of unknown function. The RBPs themselves are often paralogs, many of which share highly conserved RNA-binding domains but with different IDRs. In the manually curated list of RBPs (1542 RBPs in total)11, more than 22% (341/1542) are annotated in one of the paralog families in the OrthoMCL database12, and more than 48% are paralogs (749/1542) as defined in the original census study based on sequence similarity11 (see Supplementary Data 1 and Supplementary Data 2 for the lists). These paralogs have similar folded domains, hinting at similar RNA recognition mechanisms, whereas many disordered regions have greater sequence diversity.
To understand the different properties of IDRs in paralogs, we focus on those families of proteins that have two members and found that the Musashi protein family is a suitable example for this purpose. The Musashi gene was originally identified in the fruit fly (Drosophila melanogaster), responsible for sensory organ development13 and is highly conserved in animals14,15,16,17,18. In mammals, the two Musashi paralogs are translational regulators of cell fate and are involved in maintaining the stem-cell state, differentiation, and tumorigenesis19. In addition to their role in neural stem-cell development17,18,20, they also regulate several types of cancer21,22. The C-terminal domain (an IDR) is critical for forming chemoresistant stress granules in glioblastoma23 and colorectal cancer cells24. Musashi proteins cannot join stress granules without it23,24. Furthermore, the toxic oligomers formed by Musashi proteins have been implicated in Alzheimer’s disease25,26. We noticed through sequence analysis that the IDRs of Musashi-1 and −2 have different physical properties, which are conserved among vertebrate orthologs. Here, our data suggest that the decreased prion-likeness (the level of amino acid composition resembling that of prion proteins) of one paralog is compensated in assembly formation by an increase in α-helical propensity. We also compare the IDRs’ properties of other well-studied RBPs related to Musashi proteins, including TDP-43 and hnRNP A1. These results show how different properties may have evolved in IDR paralogs for the same biophysical mechanism.
Results
The IDRs of the Musashi family have different prion propensities
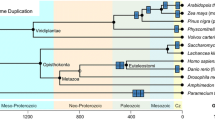
We used the primary sequence of the folded domain of fruit fly Musashi (residues 29–195, a predicted RNA recognition motif (RRM)) to identify orthologs in the UniProt database (i.e. ignoring its IDR). We selected the model organisms27 (listed in Supplementary Table 1) with similar RRM sequences and constructed a phylogenetic tree using their full-length sequences (Fig. 1a). The nematode and fruit fly have only one Musashi gene, but vertebrates have two paralogs (Fig. 1a). The C-terminal half of all these proteins are intrinsically disordered (purple bars in Fig. 1a and Supplementary Fig. 1). Note that the IDR was not used to identify orthologs but is a common feature of all Musashi proteins. Results suggest the IDR is involved in Musashi-1 joining stress granules23,24. The stress-granule-related proteins often possess a prion-like domain28,29, which is similar to prion behavior to assist assembly but does not necessarily aggregate. We, therefore, used the PLAAC algorithm30 to predict prion-likeness. Although all the IDRs of Musashi-2 in the vertebrates and lower animals are prion-like, the IDRs of Musashi-1 orthologs are less so (Fig. 1a). Figure 1b compares the human Musashi paralogs, where the IDRs cover residues 237–362 (Msi-1C) and 235–328 (Msi-2C) (red box). We separated all Musashi protein sequences into IDRs and RRMs based on their alignment to the corresponding human ortholog (Fig. 1b and Supplementary Fig. 2). The RRMs in Musashi-1 and Musashi-2 have very similar amino acid compositions (Fig. 1c, d and Supplementary Table 2). On the contrary, although the IDRs of the two paralogs have many glycines, prolines, and alanines (Fig. 1c and Supplementary Table 2), their amino acid sequences differ substantially (Fig. 1d, lower panel). Sorting the amino acids in terms of prion-forming propensity30 reveals that the Musashi-1 orthologs have fewer prion-promoting amino acids, such as glutamine and asparagine than Musashi-2 orthologs do. This amino acid composition analysis reinforces the suggestion that Musashi-2 is more prion-like (Fig. 1d).

a Phylogenetic tree (constructed using MEGA X62) of Musashi proteins in model organisms. The red lines indicate the PLAAC prion propensity scores30. The purple bars indicate regions predicted to be disordered by the VSL2 algorithm in PONDR61. The dashed box highlights the most prion-like regions. b Disorder and prion-likeness analysis of human Musashi proteins. Residues are numbered according to the alignment with Musashi-1 and the gray shading indicates the regions that are missing in Musashi-2. The red box contains the consensus intrinsically disordered regions (residues 237–362 of Musashi-1 and 235–328 of Musashi-2) considered in this study. c Amino acid pie charts of Musashi proteins in the model organisms listed in panel (a). The RRMs and IDRs were defined as shown in panel (b). d Single amino acid population differences in Musashi-1 and -2 with the amino acids sorted by prion-likeness30. Gray bars indicate amino acids that appear fewer than ten times in the sequence.
The disordered regions of Musashi proteins have a polyalanine region that forms an α-helix
We purified the human Musashi proteins’ IDRs to investigate their differences experimentally (Supplementary Fig. 3). Their circular dichroism (CD) spectra show that they are mostly unstructured (Fig. 2a). The CD patterns of Msi-1C and Msi-2C at pH 5.5, 283 K (the conditions at which the proteins were most stable and soluble, see below), are similar overall but differ slightly around 220 nm, hinting at a potential difference in α-helicity31,32. We assigned the NMR chemical shifts of the two IDRs to obtain residue-specific structural propensities (BMRB accesses: 51207 and 51208). In the 15N-edited heteronuclear single-quantum coherence (HSQC) spectra (Fig. 2b), most of the amide proton signals are within 1 ppm, confirming the disordered nature of these domains33. Nevertheless, both regions contain a stretch of residues in which the secondary chemical shifts (the differences between the measured chemical shifts and random-coil values) are positive for Cα and C’ atoms and negative for Cβ atoms, indicating a propensity of an α-helix (Fig. 2c). The deviations from random-coil values are smaller for Msi-2C than for Msi-1C (Fig. 2c). δ2D predictions34 based only on chemical shifts (with no missing assignments in the α-helical region) indicate that the α-helical propensity of Msi-1C is higher than Msi-2C’s (residue-specific values up to ~50% vs up to ~20%; red bars in Fig. 2d). These α-helical forming regions correspond to the polyalanine stretches of the two proteins (Fig. 2d, e). Msi-1C contains an eight-alanine-repeat whereas Msi-2C’s polyalanine stretch is interrupted (and its α-helical propensity reduced) by a valine35,36. Importantly, the respectively continuous and disrupted polyalanine tracts are conserved in Musashi-1 and Musashi-2 orthologs (Fig. 2e and Supplementary Fig. 2).

a Circular dichroism (CD) spectra of the C-terminal domains of Musashi-1 (black; Msi-1C) and Musashi-2 (purple; Msi-2C). b 15N-edited NMR HSQC spectra of Msi-1C and Msi-2C with resonance assignments. c Secondary chemical shifts of Cα, Cβ, and carbonyl-carbon (C′). The most pronounced deviations are highlighted in red. d Stacked plots of the secondary structural populations derived from the chemical shifts using the δ2D algorithm34. Red: α-helix; yellow: β-sheet; gray: random coil; brown: polyproline II helix. The alanines in the polyalanine tracts are indicated by orange blocks on the x-axis. e Multiple sequence alignment of Musashi homologs. The sequences are in the same order as shown in Fig. 1a. The polyalanine stretch is highlighted in orange. The CD and HSQC spectra were repeated at least three times for both protein samples.
The non-conserved regions flanking the polyalanine tracts tune their α-helical propensity
Musashi-1 has two additional regions not found in Musashi-2 around the polyalanine tract (denoted Seq1 and Seq2; Fig. 3a). We created three constructs without Seq1, Seq2, or both, to investigate their effect (Fig. 3b; ΔSeq1, ΔSeq2, and ΔSeq1ΔSeq2). The CD patterns of these variants are similar to those of Msi-1C (Fig. 3c) and in the 15N-1H HSQC spectra, most peaks overlap with those of Msi-1C. However, there are pronounced changes between Seq1 and Seq2 (Fig. 3d). The adjacency of these regions to the mutation sites is not the only cause because residues further away from the polyalanine region show smaller chemical shift perturbations than those within the polyalanine region (as highlighted in Fig. 3d as an example and quantified in Fig. 3e). These changes show systematic patterns with downfield chemical shifts in the proton and nitrogen dimensions for ΔSeq1 and the opposite trend for ΔSeq2 (Fig. 3d, e), indicating a shifting equilibrium between different conformations in the fast exchange regime. To confirm this, we assigned the 13C chemical shifts of the three variants (BMRB accesses: 51204, 51205, and 51206; Supplementary Fig. 4a), which are mostly similar, except those close to the polyalanine region (Supplementary Fig. 4b). We calculated the secondary chemical shift difference of Cα and Cβ atoms (ΔδCα-ΔδCβ; which minimizes the error from chemical shift referencing) and compared these values in the variants to those in the wildtype (Δ(ΔδCα-ΔδCβ); Fig. 3f). Since larger (ΔδCα-ΔδCβ) values indicate a stronger α-helical tendency, these results indicate that the wild-type’s α-helical propensity is lower than ΔSeq1’s but higher than ΔSeq2’s. The estimated α-helical propensities (using the δ2D program) are up to 6% higher than in the wildtype for ΔSeq1 but up to 5% lower than in the wildtype for ΔSeq2 (Fig. 3g). These results are in keeping with the indistinguishable CD data because, relative to the entire length of the sequences, the difference in α-helical propensity is negligible (<10% difference over just 10% of the sequence). The ΔSeq1ΔSeq2 variant has a similar predicted secondary structural population as the wildtype (Fig. 3g). These results indicate that α-helical propensity around the polyalanine stretch is altered by these non-conserved variations in primary sequence with, in order of α-helical propensity, ΔSeq1 > wildtype ≈ ΔSeq1ΔSeq2 > ΔSeq2. Both these regions modify the α-helical propensity probably by altering the equilibrium between the monomeric and condensed states because of their physical properties, either many charged residues (Seq1) or a majority of hydrophobic residues (Seq2), would change the tendency to assemble. Although the helicity correlates with self-assembly in some cases37, the correlation is difficult to identify in our studies. Nevertheless, it is notable that there are no species in which Seq1 or Seq2 are missing alone, hinting that this α-helical propensity may have been fine-tuned.

a Sequence alignment of the human Musashi proteins. The main difference is the absence of the Seq1 and Seq2 regions in Musashi-2. b The designed constructs, with only the preserved residues indicated. c Circular dichroism spectra of the different constructs (in black, red, green, and yellow) overlaid on the wild types (in gray). d Overlaid NMR HSQC spectra (color-coded as in panel (b)), with an expanded view of overlapping (Val-329) and shifted (Val-282) cross-peaks. e–g Difference between the deletion constructs and the wildtype for e proton (ΔδH) and nitrogen (ΔδN) chemical shifts, f secondary chemical shift differences between Cα and Cβ; g δ2D34 α-helical propensity scores. The Seq1 and Seq2 regions are indicated in light gray if present, dark gray if missing; the polyalanine region is highlighted in orange. The CD and HSQC spectra were repeated at least three times for all protein samples.
The polyalanine region promotes Musashi-1 assembly
Msi-1C and Msi-2C differ in α-helical propensity and the regions in which they differ most change this propensity, and thus we investigated the effects of removing the polyalanine stretch between Seq1 and Seq2. The CD curve of this variant, denoted ΔSeqA, differs from that of Msi-1C around 220 nm (Fig. 4a), in a similar manner as Msi-2C’s does (Fig. 2a), indicating a loss of α-helicity. Except for the truncation sites, the HSQC spectrum overlaps well with Msi-1C’s, indicating no further structural change (Fig. 4b, c).

Overlaid a circular dichroism and b NMR HSQC spectra of the ΔSeqA construct (orange) and Msi-1C (gray). c Chemical shift differences in the proton (ΔδH) and nitrogen (ΔδN) dimensions between ΔSeqA and Msi-1C. The polyalanine region is highlighted in orange. d Light micrographs of Msi-1C, Msi-2C, and ΔSeqA at pH 5.5 or 6.5. Scale bar: 10 µm. Experiments were performed at least three times for each protein sample. e Fluorescence recovery after photobleaching (FRAP) results in pH 5.5 and 6.5. The colored lines represent the mean ± SD and the gray lines are individual recovery profiles. At least 20 condensates were recorded for each sample. f Precipitation assays of Msi-1C, Msi-2C, and ΔSeqA at pH 5.5 and 6.5 after incubation at room temperature for different time periods. Supernatant concentrations were determined after centrifugation (triplicate; mean ± SD) and confirmed by SDS-PAGE. g Energy landscape representation of the results.
In all the NMR and CD studies, we noticed that the signal intensities did not correlate with protein concentrations. For example, the intensity ratio of HSQC spectra did not match their molar ratio (Supplementary Fig. 5). We thus speculate that higher-order oligomers (NMR-undetectable) are present. We used optical microscopy and indeed observed condensates in each case (Fig. 4d and Supplementary Fig. 6). At pH 5.5, the condensates were all spherical, and fluorescence recovery after photobleaching (FRAP) measurements on proteins labeled with a fluorescent probe (Cy3-NHS) showed that the condensates are dynamic (Fig. 4e). At pH 6.5, on the contrary, the condensates observed for Msi-1C were irregular and showed no fluorescence recovery (Fig. 4d, e), whereas, for Msi-2C and ΔSeq, the condensates remained spherical and dynamic (Fig. 4d and Supplementary Fig. 6). In order to avoid ambiguity in the following discussion, the term “condensates” is used for the dynamic states and “aggregates” for the less dynamic and irreversible state (irregular shapes under the microscope), as suggested38,39. We also noticed that the level of fluorescence recovery varied between samples (gray lines in Fig. 4e), indicating that the condensates undergo very rapid sol-gel transitions (Fig. 4e), especially for Msi-1C at pH 5.5 (i.e., quantifying the level of recovery would not be informative). Rapid aggregation has also been reported for other proteins40,41,42. Therefore, to compare the aggregation tendency, we prepared 20 µM samples at pH 5.5 and 6.5 and incubated them at room temperature for different periods. We then centrifuged the samples (at ~12,000 × g, to remove large aggregates but leave small condensates in the supernatant, as confirmed by microscopy) and measured the concentration of the soluble fraction. At pH 5.5, the proteins remained mostly in the supernatant for all incubation times (Fig. 4f, as determined by the absorbance at 280 nm). Condensates in the soluble fraction may affect the absorbance accuracy, thus we have also confirmed the amount of supernatant protein using SDS-PAGE gel (Fig. 4f; uncropped images in Supplementary Fig. 6). At pH 6.5, nearly all Msi-1C molecules aggregated within 1 h of incubation but Msi-2C, and to a lesser extent, the ΔSeqA construct remained partially in the supernatant for longer (at least 24 h; Fig. 4f). These results suggest that the polyalanine region in Msi-1C promotes aggregation. Figure 4g shows an energy landscape representation of the metastable nature of LLPS in these proteins40. Their three main states are the monomeric form, the LLPS state (dynamic condensate), and the aggregate form. At pH 5.5, the energy barrier between LLPS and aggregation is high, leaving Msi-1C trapped in the dynamic condensate. At pH 6.5, however, the energy barrier is lower, and Msi-1C aggregates quickly. However, at the same pH, ΔSeqA remains in the dynamic soluble state for longer, indicating that removing the α-helical region restores the energy barrier between the LLPS and aggregate states. These results suggest that although Msi-1C is less prion-like, its stronger α-helical propensity promotes assembly, regardless of the “price paid” in terms of aggregation42,43.
Discussion
Various properties contribute to functional assembly in IDRs
Without the constraints of a fixed shape, the IDRs in a paralog family can evolve more freely than structured regions, either gaining new functions or compensating for lost ones. The results obtained here for the Musashi protein family suggest IDRs may have evolved different means of functional assembly. Musashi-1 is found in stress granules, but its IDR has fewer prion-promoting amino acids than many others with this property (e.g., FUS, TDP-43, and hnRNP A1). However, our results suggest that the stronger α-helical propensity of Musashi-1’s polyalanine stretch may assist its assembly, as polyalanine tracts are known to contribute to protein self-assembly44. On the other hand, Musashi-2’s IDR is sufficiently prion-like for assembly despite a lower α-helical propensity (Fig. 4).
Our results also explain a number of biological observations. Musashi proteins are overexpressed for cell renewal and stemness maintenance22. Although certain cell types express one or other Musashi paralogs (e.g., Musashi-2 in hematopoietic stem cells45), they are functionally redundant when they appear together. For example, Musashi-2 compensates for the proliferation of neural progenitor cells in Musashi-1 double-knockout mice20; both Musashi proteins promote colorectal cancer cell growth through the same signaling pathway46; complete loss of visual function in photoreceptor cells is only observed in Musashi-1,2 double-knockout mice47. The IDRs’ different physical properties compensate for the loss of the other, but these differences (e.g., α-helix dominant vs more prion-like) may nevertheless lead to specific interaction mechanisms, for instance, tau protein only interacts with Musashi-1 for transportation into nuclei, whereas tau’s interaction with Musashi-1 or −2 leads to different pathological stages in tauopathies26.
Polyalanine is commonly involved in promoting self-assembly
Polyalanine sequences may be a common evolved trait through which RBPs assemble or join membraneless organelles. For example, a sufficiently long polyalanine tract in its IDR enhances the subnuclear targeting properties of RBM4 (RNA-binding motif 4)48. Indeed, with glutamine and asparagine, alanine is one of the most frequently repeated amino acids in proteins, and polyalanine stretches promote self-assembly44. Several other amino acids are also helix-promoting, such as methionine, leucine, and glutamine35. In the IDR of the extensively studied TDP-43, for example, the “AMMAAAQAALQ” amino acid motif has a strong tendency to form an α-helix, promoting condensation49,50,51 regardless of its short polyalanine tract. In order to estimate the frequency of polyalanine appearance, we rapidly searched for this trait in the proteome using a simple scoring function based on amino acid α-helical propensity values derived from our experimental observations and consensus studies (Fig. 5a)35,36. Using these tentative scores and aggregating for repeats (examples shown in Fig. 5b), we calculated the portion of all IDRs in the human proteome and in RBPs (1542 in total) in which the aggregated polyalanine score reaches 1.5, 2.0, or 2.5 (Fig. 5c). We also analyzed a group of 692 mRNA-binding proteins (mRBPs) because many reported RBPs42,52,53 with LLPS-related functions reside in this category. Our analysis shows that polyalanine stretches are more common in RBPs, especially mRBPs, than in the general human proteome (Fig. 5c). We also used RaptorX54 to predict the residues with α-helical propensity among the IDRs of the human proteome, RBPs, and mRBPs. We set arbitrary criteria (all other criteria show the same trend) to count those IDRs with at least five consecutive residues predicted higher than 0.8 (1.0 is the full scale) α-helical propensity in the program. Figure 5d shows the same tendency as the polyalanine score: mRBPs’ or RBPs’ IDRs are more likely to have α-helical elements within them. In these analyses, the difference is higher than the deviation of 100 random selections of 1542 or 689 proteins from the human proteome (the sample sizes considered for the RBPs and mRBPs, respectively; Fig. 5c, d). We attribute the results to that RBPs often join biomolecular condensates for their functions55, and polyalanine/α-helix is one feature of IDRs that contributes to self-assembly.

a Tentative scores are assigned to the amino acids according to the experimental observation to estimate the frequency of polyalanine appearance. b Examples of how polyalanine propensity scores were calculated for continuous residue stretches of increasing length in proteins with α-helix-forming regions (Musashi-1, Musashi-2, and TDP-43) or without (hnRNP A1). c Prevalence of IDRs with polyalanine scores greater than 2.5, 2.0, and 1.5, in the human proteome, RNA-binding proteins (RBPs), and mRNA-binding proteins (mRBPs). d Population of IDRs with α-helical elements longer than five consecutive residues in the human proteome, RBPs, and mRBPs. The point clouds, square points, and error bars in the RBP and mRBP panels are the distribution, mean, and standard deviation of the polyalanine (panel c) and α-helix scores (panel d) of 100 random selections of sets of 1542 or 689 proteins (the RBP and mRBP sample sizes) from the human proteome (as negative controls).
When did Musashi IDRs diverge?
The IDRs of Musashi proteins may have respectively gained α-helicity or become less prion-like. Which came first? The answer is perhaps hidden in the primary sequence. Using the RRM from D. melanogaster Musashi once again, we searched for orthologs in the phylum Chordata, a higher taxonomic rank than the subphylum of vertebrates in which the Musashi paralogs arise (Fig. 1a). Amphioxus (the lancelet), a model organism for primitive chordates, has a Musashi homolog which does not have a long polyalanine tract but is prion-like, similar to that of the nematode and fruit fly (Fig. 6a, b and Supplementary Fig. 7). This suggests that the Musashi paralogs may have arisen after the appearance of chordates. In ghost sharks indeed, primitive vertebrates with the two Musashi paralogs, Musashi-1 has a polyalanine tract that is not prion-like (Fig. 6c and Supplementary Fig. 7), whereas interestingly, Musashi-2 has a polyalanine tract and is prion-like. Collectively, these results suggest that the polyalanine stretch may have appeared in the paralogs alongside primitive prion propensity before Musashi-l lost its prion-promoting amino acids, whereas a disruption in the α-helical region reduced Musashi-2’s tendency to self-assemble.

a Phylogenetic tree of the Musashi family as shown in Fig. 1a with the additional lineages analyzed: a primitive chordate (the lancelet, blue box, panel (b)) and a primitive vertebrate (the ghost shark, green circle, panel (c)). b, c PONDR sequence disorder and PLAAC prion-likeness scores of b the lancelet (Buccinum belcheri) and c the ghost shark (Callorhinchus milii). The RNA recognition motifs (based on PROSITE analysis73) are indicated by gray squares. The alignment of the polyalanine regions (highlighted in orange) of human and ghost shark Musashi proteins are also shown. d Phylogenetic tree of Musashi paralogs with high sequence similarity found in the human proteome. The percentages shown are the levels of sequence similarity. The importance of different drivers of assembly in their IDRs is indicated by colored bars representing their α-helical propensity (orange), prion-likeness (red), and the number of aromatic residues (purple). e α-helical propensity predicted using RaptorX54. f Sequence disorder and prion-likeness for DAZAP-1, hnRNP A1, and TDP-43 (c.f. Fig. 1b for Musashi-1 and −2).
“Distant relatives” in the Musashi family
According to the RBP census study11, many other RBPs were grouped in the paralog family of Musashi proteins based on sequence similarity. We confirmed this by searching for sequences similar to Musashi-1’s in the human proteome. Other than Musashi-2 (71.3% identity), the top hits are DAZ-associated protein 1 (DAZAP-1, 42.6%), several heterogeneous nuclear ribonucleoproteins (hnRNP A0, A1, A2…; 39.0–42.5%) and TDP-43 (34.9%) (Fig. 6d), agreeing with the previous study11. Accordingly, we compared the IDRs’ properties of these Musashi protein’s “distant relatives”: Studies of the low complexity IDRs of hnRNP A1 and A2 are pioneering examples in the emerging field of protein LLPS41,52,56. They are also a good model for studying LLPS theory, such as the effects of the number and distribution of aromatic residues57. TDP-43’s IDR is another extensively studied example. TDP-43’s IDR differs from hnRNP A1 or A2’s in that, as well as being prion-like, it encompasses a short α-helical region that contributes to self-association and LLPS49,51. Furthermore, TDP-43’s LLPS is mediated by just a few aromatic residues50, a feature we have attributed to the presence of the α-helix, which promotes intermolecular contacts50 (Fig. 6d). The present study adds the biophysical properties of the two Musashi proteins to existing information on this distantly related protein family. Although DAZPA1 has been far less studied than other members, its C-terminal domain is critical to its function in interacting with eIF4G58 and potentially interacts with many RBPs, including hnRNP A159. Moreover, bioinformatic analysis shows that this protein’s C-terminal domain is disordered and prion-like, with many aromatic residues but no predicted α-helix, similar to that of hnRNP A1 (Fig. 6e, f and Supplementary Fig. 8). Although the IDRs of these RBPs have different properties, they undergo LLPS. As a conclusion, we suggest that IDRs evolve whatever traits are beneficial to function, which accords with François Jacob’s statement that “evolution does not produce novelties from scratch”60. He was referring to the diversity of all lifeforms but his words also apply to IDRs, which acquire new functions through evolutionary “tinkering”60 between prion-likeness, α-helicity, aromatic residues, etc. Our work could be used as a template to investigate IDRs in other paralogs and how their functions have diversified or been preserved during evolution.
Methods
Bioinformatics analysis
The primary sequences were obtained from UniProt with the associated entries listed in Supplementary Table 1. Levels of structural disorder and prion-likeness were respectively analyzed with the PONDR61 and PLAAC30 webservers. The phylogenic trees were construed using the neighbor-joining method in MEGA X62.
In analyzing the polyalanine and α-helical propensities, all human protein sequences were retrieved from UniProt (UniProtKB_2021_01, download date: 2021/02/06, 20396 sequences in total) and separated into disordered and ordered regions based on PONDR predictions (VSL2)61. The predicted disordered regions longer than 40 consecutive residues were analyzed. The tentative polyalanine scoring (Fig. 5a) of the sequences was done using in-house scripts to estimate the frequency of polyalanine appearance. The α-helical propensities were predicted using RaptorX54. An IDR in a protein having at least five residues in a raw predicted as α-helix (with a value higher than 0.8 out of 1.0) is counted in our analysis.
DNA constructs
The Msi-1C construct in this study (residues number 237 to 362) was cloned from a previous longer construct (194–362)63. We removed a strongly hydrophobic region reported to contain binding sites for PABP and GLD222, which is irrelevant to the present study and whose removal improves the protein’s solubility. The ΔSeq1, ΔSeq2, ΔSeq1ΔSeq2, and ΔSeqA constructs were prepared with designed primers (Supplementary Table 3). Msi-2 cDNA (residues 234–328, according to the alignment with Msi-1C) was purchased from OriGene. All these variants were constructed in a pET21 vector backbone with a hexahistidine tag on the C-terminus of the expressed protein. All constructs were fully sequenced.
Protein expression and purification
All constructs were purified using the same protocol, which has been described in detail elsewhere63. In short, transformed E. coli BL21(DE3) cells were grown at 37 °C until the OD reached 0.6 and were induced with a final concentration of 1 mM isopropyl-β-d-1-thiogalactopyranoside at 25 °C overnight. The cells were harvested and lysed by sonication. After centrifugation, the inclusion bodies were dissolved using 20 mM Tris buffer at pH 8 with 8 M urea (buffer A). After a second centrifugation, the supernatant was filtered with a 0.45 µm filter and loaded onto a nickel-charged immobilized metal affinity chromatography column (Qiagen). After washing with ten column volumes of buffer A, the samples were eluted with 5 column volumes of buffer B (buffer A with 500 mM imidazole). The eluted samples were acidified with trifluoroacetic acid (down to pH ~3), loaded onto a C4 reverse-phase column (Thermo Scientific Inc.), and eluted with a gradient of acetonitrile (from 0 to 100%, mixed with triple-distilled water) by HPLC and then lyophilized. The lyophilized samples were stored in a dry cabinet until use. For all experiments, powder samples were dissolved in 20 mM MES-NaOH at pH 5.5 or 6.5. The protein concentration was determined from the absorbance at 280 nm measured with a Nanodrop spectrometer (Thermo Scientific Inc.).
Circular dichroism spectroscopy
Circular dichroism spectra were recorded using an AVIV model 410 spectropolarimeter with a 0.1 mm cuvette. Data were collected between 190 and 260 nm with an interval of 1 nm. Ten measurements were co-added for each data point. All spectra were recorded at 283 K and the samples were kept in a water bath at 283 K between measurements. All experiments were performed in triplicate.
NMR spectroscopy, chemical shift assignment, and data analysis
15N-edited HSQC spectra were recorded using the standard pulse sequence with WATERGATE solvent suppression64,65. Chemical shifts were assigned using standard HNCA, HN(CO)CA, HNCO, HN(CA)CO, CBCA(CO)NH, and HNCACB experiments acquired with non-uniform sampling (25%)66,67. All data were recorded using a Bruker AVIII 600 MHz spectrometer with a cryogenic probe.
The data were processed using NMRPipe68. Chemical shifts were assigned using the automated assignment scheme69 implemented in NMRFAM-Sparky70, and then confirmed manually. Secondary chemical shift analysis was performed using Kjaergaard et al.’s database of random-coil shifts71. Secondary structure populations were estimated using δ2D34. No chemical shifts were missing around the critical α-helical region, such that the secondary structure estimates for the constructs were made using the same number of chemical shifts.
Microscopy
Protein samples were loaded onto mPEG-passivated slides72. Micrographs were collected using an Olympus BX51 microscope with a 40× long-working-distance objective lens.
Microscopy and fluorescence recovery after photobleaching (FRAP) experiments
For the fluorescent dye labeling, 2.5 mg lyophilized protein was dissolved in 0.1 M sodium phosphate with 6 M guanidine hydrochloride at pH 8.3 and mixed with 15.6 mM (~1 mg in total) of Cy3-NHS (Lumiprobe) overnight at room temperature. The excess Cy3-NHS was removed and the buffer was exchanged with a 20 mM MES buffer at pH 5.5 using a PD-10 column (GE Healthcare). The typical labeling efficiency, determined from the ratio of extinction coefficients measured for the protein and the fluorescence dye (Cy3: 150,000 M−1 cm−1 at 550 nm) was ~20%. The Cy3-labeled sample was aliquoted, flash-frozen, and stored at −80 °C before usage.
The lyophilized samples were dissolved in 20 mM MES buffer at pH 5.5 or pH 6.5 and mixed with a Cy3-labeled sample with a final concentration of 20 µM and ~1% Cy3-labeled protein. The samples were then loaded onto ultraclean coverslips and observed with an iLas multi-modal of total internal reflection fluorescence (Roper Scientific, Inc.)/spinning disk (CSUX1, Yokogawa) confocal microscope (Ti-E, Nikon) equipped with 100 × 1.49NA plan objective lens (Nikon). The condensates were bleached with a 561 nm laser. Images were acquired at one-second intervals using an Evolve EMCCD camera (Photometrics) and were analyzed using the Metamorph software (Molecular Devices, LLC).
Aggregation assays
Lyophilized samples were dissolved in 20 mM MES buffer at pH 5.5 or pH 6.5 and stored at room temperature for different times. The samples were then centrifuged at 12,000 × g for 5 min. Supernatant concentrations were determined using a NanoDrop spectrometer. Supernatant samples were also analyzed using SDS-PAGE gels to confirm the amount of protein present. All experiments were performed in triplicate.
Statistics and reproducibility
All NMR HSQC spectra, CD experiments, and microscope observations were repeated at least three times. The reproducibility of these types of biophysical experiments is high. The FRAP experiments were repeated from three independently prepared samples. At least ten condensates were recorded to obtain the mean ± standard deviation recovery curves. Precipitation assays were performed at least three times from independently prepared samples. The data were given as the mean ± standard deviation. The individual data points were also shown to present data distribution.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Ohno, S. Evolution by Gene Duplication (Springer, 1970).
Wilson, E. B. The structure of protoplasm. Science 10, 33–45 (1899).
Lodish, H. et al. Molecular Cell Biology (W. H. Freeman and Company, 2016).
Uversky, V. N. & Dunker, A. K. Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264 (2010).
Gebauer, F., Schwarzl, T., Valcarcel, J. & Hentze, M. W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 22, 185–198 (2020).
Dominguez, D. et al. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell 70, 854–867.e9 (2018).
Varadi, M., Zsolyomi, F., Guharoy, M. & Tompa, P. Functional advantages of conserved intrinsic disorder in RNA-binding proteins. PLoS ONE 10, e0139731 (2015).
Zagrovic, B., Bartonek, L. & Polyansky, A. A. RNA-protein interactions in an unstructured context. FEBS Lett. 592, 2901–2916 (2018).
Kwon, I. et al. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 (2013).
Alberti, S. & Hyman, A. A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213 (2021).
Gerstberger, S., Hafner, M. & Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 15, 829–845 (2014).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Nakamura, M., Okano, H., Blendy, J. A. & Montell, C. Musashi, a neural RNA-binding protein required for Drosophila adult external sensory organ development. Neuron 13, 67–81 (1994).
Yoda, A., Sawa, H. & Okano, H. MSI-1, a neural RNA-binding protein, is involved in male mating behaviour in Caenorhabditis elegans. Genes Cells 5, 885–895 (2000).
Hirota, Y. et al. Musashi and seven in absentia downregulate Tramtrack through distinct mechanisms in Drosophila eye development. Mech. Dev. 87, 93–101 (1999).
Kawashima, T. et al. Expression patterns of musashi homologs of the ascidians, Halocynthia roretzi and Ciona intestinalis. Dev. Genes Evol. 210, 162–165 (2000).
Shibata, S. et al. Characterization of the RNA-binding protein Musashi1 in zebrafish. Brain Res. 1462, 162–173 (2012).
Sakakibara, S. et al. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev. Biol. 176, 230–242 (1996).
Okano, H., Imai, T. & Okabe, M. Musashi: a translational regulator of cell fate. J. Cell Sci. 115, 1355–1359 (2002).
Sakakibara, S. et al. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc. Natl Acad. Sci. USA 99, 15194–15199 (2002).
Lagadec, C. et al. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells 32, 135–144 (2014).
Fox, R. G., Park, F. D., Koechlein, C. S., Kritzik, M. & Reya, T. Musashi signaling in stem cells and cancer. Annu. Rev. Cell Dev. Biol. 31, 249–267 (2015).
Chiou, G. Y. et al. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 7, 2172 (2017).
Chen, H. Y. et al. Musashi-1 promotes chemoresistant granule formation by PKR/eIF2alpha signalling cascade in refractory glioblastoma. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 1850–1861 (2018).
Sengupta, U. et al. Formation of toxic oligomeric assemblies of RNA-binding protein: Musashi in Alzheimer’s disease. Acta Neuropathol. Commun. 6, 113 (2018).
Montalbano, M. et al. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat. Commun. 11, 4305 (2020).
Hedges, S. B. The origin and evolution of model organisms. Nat. Rev. Genet. 3, 838–849 (2002).
King, O. D., Gitler, A. D. & Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 (2012).
Harrison, A. F. & Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474, 1417–1438 (2017).
Lancaster, A. K., Nutter-Upham, A., Lindquist, S. & King, O. D. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502 (2014).
Greenland, K. N. et al. Order, disorder, and temperature-driven compaction in a designed elastin protein. J. Phys. Chem. B 122, 2725–2736 (2018).
Uversky, V. N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756 (2002).
Prestel, A., Bugge, K., Staby, L., Hendus-Altenburger, R. & Kragelund, B. B. Characterization of dynamic IDP complexes by NMR spectroscopy. Methods Enzymol. 611, 193–226 (2018).
Camilloni, C., De Simone, A., Vranken, W. F. & Vendruscolo, M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 51, 2224–2231 (2012).
Pace, C. N. & Scholtz, J. M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 (1998).
Levitt, M. Conformational preferences of amino acids in globular proteins. Biochemistry 17, 4277–4285 (1978).
Conicella, A. E. et al. TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc. Natl Acad. Sci. USA 117, 5883–5894 (2020).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Hardenberg, M., Horvath, A., Ambrus, V., Fuxreiter, M. & Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl Acad. Sci. USA 117, 33254–33262 (2020).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Patel, A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015).
Itakura, A. K., Futia, R. A. & Jarosz, D. F. It pays to be in phase. Biochemistry 57, 2520–2529 (2018).
Polling, S. et al. Polyalanine expansions drive a shift into alpha-helical clusters without amyloid-fibril formation. Nat. Struct. Mol. Biol. 22, 1008–1015 (2015).
Kharas, M. G. et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 16, 903–908 (2010).
Li, N. et al. The Msi family of RNA-binding proteins function redundantly as intestinal oncoproteins. Cell Rep. 13, 2440–2455 (2015).
Sundar, J., Matalkah, F., Jeong, B., Stoilov, P. & Ramamurthy, V. The Musashi proteins MSI1 and MSI2 are required for photoreceptor morphogenesis and vision in mice. J. Biol. Chem. 296, 100048 (2020).
Chang, S. H., Chang, W. L., Lu, C. C. & Tarn, W. Y. Alanine repeats influence protein localization in splicing speckles and paraspeckles. Nucleic Acids Res. 42, 13788–13798 (2014).
Li, H. R. et al. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta 1866, 214–223 (2018).
Li, H. R., Chiang, W. C., Chou, P. C., Wang, W. J. & Huang, J. R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293, 6090–6098 (2018).
Conicella, A. E., Zerze, G. H., Mittal, J. & Fawzi, N. L. ALS mutations disrupt phase separation mediated by alpha-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549 (2016).
Xiang, S. et al. The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell 163, 829–839 (2015).
Sun, Y. & Chakrabartty, A. Phase to phase with TDP-43. Biochemistry 56, 809–823 (2017).
Yang, Y. et al. Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Brief. Bioinform. 19, 482–494 (2018).
Wiedner, H. J. & Giudice, J. It’s not just a phase: function and characteristics of RNA-binding proteins in phase separation. Nat. Struct. Mol. Biol. 28, 465–473 (2021).
Kato, M. et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012).
Martin, E. W. et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699 (2020).
Smith, R. W. et al. DAZAP1, an RNA-binding protein required for development and spermatogenesis, can regulate mRNA translation. RNA 17, 1282–1295 (2011).
Yang, H. T., Peggie, M., Cohen, P. & Rousseau, S. DAZAP1 interacts via its RNA-recognition motifs with the C-termini of other RNA-binding proteins. Biochem. Biophys. Res. Commun. 380, 705–709 (2009).
Jacob, F. Evolution and tinkering. Science 196, 1161–1166 (1977).
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P. & Dunker, A. K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 61, 176–182 (2005).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Chen, T. C. & Huang, J. R. Musashi-1: an example of how polyalanine tracts contribute to self-association in the intrinsically disordered regions of RNA-binding proteins. Int. J. Mol. Sci. 21, 2289 (2020).
Piotto, M., Saudek, V. & Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 (1992).
Bodenhausen, G. & Ruben, D. J. Natural abundance N-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189 (1980).
Hyberts, S. G., Milbradt, A. G., Wagner, A. B., Arthanari, H. & Wagner, G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 (2012).
Hyberts, S. G., Frueh, D. P., Arthanari, H. & Wagner, G. FM reconstruction of non-uniformly sampled protein NMR data at higher dimensions and optimization by distillation. J. Biomol. NMR 45, 283–294 (2009).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Lee, W., Westler, W. M., Bahrami, A., Eghbalnia, H. R. & Markley, J. L. PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics 25, 2085–2087 (2009).
Lee, W., Tonelli, M. & Markley, J. L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015).
Kjaergaard, M., Brander, S. & Poulsen, F. M. Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J. Biomol. NMR 49, 139–149 (2011).
Alberti, S. et al. A user’s guide for phase separation assays with purified proteins. J. Mol. Biol. 430, 4806–4820 (2018).
Sigrist, C. J. et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 38, D161–D166 (2010).
Acknowledgements
The authors thank Prof. Won-Jing Wang (NYCU) for access to the microscope, Academia Sinica High-Field NMR Center for technical support (HFNMRC is funded by Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-112)), Chen-Yu Hung for microscope setting, and Dr. Tsai-Chen Chen for initial work on this project. This work was supported by the Ministry of Science and Technology of Taiwan (109-2113-M-010-003 and 110-2113-M-A49A-504-MY3 to J.-r.H. and 109-2326-B-010-002 -MY3 to J.-C.K.).
Author information
Authors and Affiliations
Contributions
J.-r.H. conceived the project and wrote the manuscript. S.-H.C., W.-L.H., Y.-C.S., J.-C.K., and J.-r.H. collected and analyzed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Sourav Chowdhury and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Krishnananda Chattopadhyay and Manuel Breuer. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiu, SH., Ho, WL., Sun, YC. et al. Phase separation driven by interchangeable properties in the intrinsically disordered regions of protein paralogs. Commun Biol 5, 400 (2022). https://doi.org/10.1038/s42003-022-03354-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03354-4
- Springer Nature Limited