
Abstract
The human microbiome has a role in the development of multiple diseases. Individual microbiome profiles are highly personalized, though many species are shared. Understanding the relationship between the human microbiome and disease may inform future individualized treatments. We hypothesize the blood microbiome signature may be a surrogate for some lung microbial characteristics. We sought associations between the blood microbiome signature and lung-relevant host factors. Based on reads not mapped to the human genome, we detected microbial nucleic acids through secondary use of peripheral blood RNA-sequencing from 2,590 current and former smokers with and without chronic obstructive pulmonary disease (COPD) from the COPDGene study. We used the Genome Analysis Toolkit (GATK) microbial pipeline PathSeq to infer microbial profiles. We tested associations between the inferred profiles and lung disease relevant phenotypes and examined links to host gene expression pathways. We replicated our analyses using a second independent set of blood RNA-seq data from 1,065 COPDGene study subjects and performed a meta-analysis across the two studies. The four phyla with highest abundance across all subjects were Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes. In our meta-analysis, we observed associations (q-value < 0.05) between Acinetobacter, Serratia, Streptococcus and Bacillus inferred abundances and Modified Medical Research Council (mMRC) dyspnea score. Current smoking status was associated (q < 0.05) with Acinetobacter, Serratia and Cutibacterium abundance. All 12 taxa investigated were associated with at least one white blood cell distribution variable. Abundance for nine of the 12 taxa was associated with sex, and seven of the 12 taxa were associated with race. Host-microbiome interaction analysis revealed clustering of genera associated with mMRC dyspnea score and smoking status, through shared links to several host pathways. This study is the first to identify a bacterial microbiome signature in the peripheral blood of current and former smokers. Understanding the relationships between systemic microbial signatures and lung-related phenotypes may inform novel interventions and aid understanding of the systemic effects of smoking.
Similar content being viewed by others

Introduction
The human microbiome has a role in human disease and overall health outcomes1,2. Individual microbiome profiles are unique, although many species are shared2,3. Knowledge of the relationship between the human microbiome and disease may serve as a component of future comprehensive individualized treatment plans4. Studies of the microbiome have typically involved 16S rRNA gene sequencing5, with metagenomic sequencing emerging more recently6.
Relevance of the lung microbiome has been demonstrated in the context of lung diseases7,8,9, including chronic obstructive pulmonary disease (COPD), asthma and idiopathic pulmonary fibrosis (IPF)10,11,12,13,14. In addition, the microbiome has been assessed in healthy lung and COPD exacerbations15,16. These studies have involved both lung tissue17,18 and the airway sampling19,20,21,22,23, with some researchers integrating the microbiome data with host gene expression13,14,17,18,21. Study of the respiratory microbiome presents many challenges24, including the low microbial biomass available in the samples25.
It has historically been believed that peripheral blood does not contain bacteria unless an acute infection was present. Through use of culture-independent sequencing methods, evidence has emerged regarding a possible healthy human blood microbiome26,27,28,29,30. Culture-independent methods in microbiome studies do not provide evidence of whether a blood microbial signature is from transient nucleic acids or from live bacteria31. A blood microbial signature has been found correlated with host disease traits in schizophrenia30, type 2 diabetes28, chronic kidney disease32 and liver fibrosis33, and it may provide a link in other tissues and diseases. Use of the microbiome for disease diagnosis and prediction has proven successful in cancer34. As with the lung microbiome, low biomass is an issue for peripheral blood microbiome studies35. Sequencing of both RNA27,30 and the 16S rRNA gene28,32,33 has been used to study the peripheral blood microbial signature.
In this study, we detected microbial signatures through secondary use of whole blood RNA-sequencing data from large subsets of the COPDGene (Genetic Epidemiology of COPD) study, repurposing sequencing reads not mapped to the human genome27,30,36. An overarching challenge in population-based microbiome studies relates to statistical power, as testing for associations between the detected microbial profiles and variables of interest places demands on sample size. Though samples were not collected as part of a traditional microbiome study, by using a large population and a meta-analysis approach, we had enhanced power to enable findings in the blood, with its typically lower microbial signals. Using statistical tools developed for microbiome analysis, we tested associations between the identified taxa and multiple COPD-related phenotypes available in COPDGene. We used network methods to integrate the microbiome signatures with the human gene expression data to highlight microbial interactions with host pathways. Our goal was to reveal microbial signatures in peripheral blood associated with lung relevant host factors and to observe lung biology relevance. A blood microbiome signature has the potential to serve as a biomarker of disease severity and progression and may inform personalized diagnostic or treatment efforts.
Methods
Study subjects
COPDGene is a longitudinal cohort study that includes non-Hispanic White and African American subjects enrolled at 21 centers across the United States37. All subjects in this study provided written consent for study procedures, including genetic analysis. COPDGene was approved by the Institutional Review Boards at all participating centers. The subjects include more than 10,000 current and former cigarette smokers with a minimum 10 pack-years smoking history, along with a small number of non-smokers. COPD cases have airflow obstruction (FEV1/FVC < 0.7), Preserved Ratio Impaired Spirometry (PRISm) cases have preserved ratio (FEV1 < 80% predicted with FEV1/FVC ≥ 0.7)38 and control subjects have normal spirometry (FEV1% predicted ≥ 80% and FEV1/FVC ≥ 0.7). The five-year follow-up visit included questionnaires, pre- and post-bronchodilator spirometry, volumetric computed tomography (CT) of the chest, and blood drawn for complete blood cell count, RNA-sequencing and biomarker studies. Subjects were at least one month removed from any exacerbation event or acute respiratory infection. Exacerbations were defined by use of antibiotics and/or systemic steroids, and severe exacerbations by emergency department visit or hospital admission39. Details of the RNA-sequencing methods are available in the online supplement40. We performed meta-analyses using a primary set of data and a second independent set of replication data from the COPDGene study.
Microbial detection
Starting from the whole blood RNA-seq data, we used reads that were not mapped to the human genome during the gene expression analysis to detect a bacterial signature. Additional filtering of the unmapped reads was performed using the PathSeq microbial detection pipeline from the Genome Analysis Toolkit (GATK4) and the host reference available from the GATK Resource Bundle41. This filtering addresses any remaining quality, host contamination or repetitive sequence issues. We subsequently used PathSeq to map these cleaned reads to bacterial genomes. The bacterial reference for mapping was created using representative genomes, chromosomes, contigs and scaffolds (277,422 total genomic entries; September 25, 2019) from the National Center for Biotechnology Information (NCBI), and the PathSeq reference creation tools. Taxonomy information for these bacterial genomic data was also obtained from NCBI (RefSeq-release95.catalog.gz). Using these mapping results and taxonomy data, the inferred bacterial abundance profiles in each sample were assembled using PathSeq. Included in these profiling data were the raw read counts, adjusted scores and normalized scores (compositional data from the adjusted scores that represent inferred relative abundance) for taxa within each taxonomic classification (genera and phyla). We used the TMM (trimmed mean of M values) method in the R/Bioconductor package edgeR42 and the RNA-seq gene expression counts from the primary analysis to normalize the PathSeq count data across samples.
Taxa associations
We tested associations between the TMM-normalized abundance for each taxon and host variables using linear models with the R/Bioconductor package MaAsLin2 (Multivariate Association with Linear Models)43. The abundance values were log-transformed prior to testing. With relatively low levels of bacterial genetic content in peripheral blood, the data is inherently sparse and MaAsLin2 is particularly well suited for analysis of such microbial data. The base statistical model included the covariates age, sex, race, pack-years of smoking, smoking status (current vs. former), RNA-seq library preparation batch and study center. Using the results from our primary and replication analyses, we performed a meta-analysis by combining the p-values from these tests using Stouffer’s method via the sumz function from R package metap44. The directions of effect in both the primary and replication analyses were required to be the same for the p-values to be combined. For each of the models, adjustment of the combined p-values for multiple testing controlled for false discovery rate (FDR < 5%). The heatmaps of taxa associations were produced using the labeledHeatmap function from the R package WGCNA45.
Contamination assessment
Nucleic acids from sources other than the peripheral blood of the study subjects could impact the analyses and potentially create a false taxonomic signature. Extraction, amplification and library-preparation kits may contain nucleic acids from water and soil bacteria46. Removing taxa with inferred abundances below a specified threshold was the first step in the process of addressing contamination47. Recent studies have shown that external contaminants more consistently correlate negatively with sample nucleic acid concentration48,49. Therefore, we sought to identify additional contamination by testing the Pearson correlation between taxa abundance and RNA concentration, with a correlation coefficient < -0.4 and p-value < 0.05 demonstrating the conditions for possible contamination47. We also examined the inferred taxa abundances across the processing batches and study centers to identify patterns suggestive of contaminant introduction through laboratory kit reagents. This study did not focus on diversity measures or detection of novel organisms, as these are areas where microbial contamination may be expected to have a greater impact. In addition, our analyses involved testing associations between host binary and quantitative characteristics and microbial taxa abundance. This helps reduce the impact of batch-specific or study-wide contamination, as correlations with host variables are not expected to be consistent and significant. Our meta-analysis in two independent sets of data mitigates the effects of contamination and enhances the ability to detect biologically relevant signatures.
Host microbe interactions
We projected the human gene expression data onto the pathways in the Hallmark gene set collection using gene set variation analysis via the R/Bioconductor package GSVA50. The genes represented in both the gene expression data and the Hallmark gene sets were included in the GSVA procedure (Methods in the online supplement). The Hallmark canonical pathway set reduces redundancy found in public gene sets to enhance enrichment analyses. GSVA output is a pathway-by-subject matrix of expression data for observation of host-microbiome interactions. We used the pathways in this matrix as variables in MaAsLin2 models. Similar to the taxa-association analysis, we performed a meta-analysis by combining the p-values from these tests using Stouffer’s method44. The directions of effect in both the primary and replication analyses were required to be the same. We constructed a bipartite network (edges connecting taxa and pathways) using the results from these models. Communities within this network were identified using the R package CONDOR51. Networks and communities were visualized using the R package igraph52, with the GEM (graph embedder) force-directed layout algorithm.
Ethics statement
All subjects in this study provided written consent for study procedures, including genetic analysis. The study was approved at all clinical centers by the following Institutional Review Boards: National Jewish IRB, Partners Human Research Committee, Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals, Columbia University Medical Center IRB, The Duke University Health System Institutional Review Board for Clinical Investigations (DUHS IRB), Johns Hopkins Medicine Institutional Review Boards (JHM IRB), The John F. Wolf MD Human Subjects Committee of Harbor-UCLA Medical Center, Morehouse School of Medicine Institutional Review Board, Temple University Office for Human Subjects Protections Institutional Review Board, The University of Alabama at Birmingham Institutional Review Board for Human Use, University of California San Diego Human Research Protections Program, The University of Iowa Human Subjects Office, VA Ann Arbor Healthcare System IRB, University of Minnesota Research Subjects’ Protection Programs (RSPP), University of Pittsburgh Institutional Review Board, UT Health Science Center San Antonio Institutional Review Board, Health Partners Research Foundation Institutional Review Board, Medical School Institutional Review Board (IRBMED), Minneapolis VAMC IRB, and Institutional Review Board/Research Review Committee Saint Vincent Hospital – Fallon Clinic – Fallon Community Health Plan. The research methods were carried out in accordance with the relevant guidelines.
Ethics approval and consent to participate
All subjects in this study provided written informed consent. COPDGene was approved by the Institutional Review Boards at all participating centers.
Clinical center | Institution title | Protocol number |
|---|---|---|
National Jewish Health | National Jewish IRB | HS-1883a |
Brigham and Women’s Hospital | Partners Human Research Committee | 2007-P-000554/2; BWH |
Baylor College of Medicine | Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals | H-22209 |
Michael E. DeBakey VAMC | Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals | H-22202 |
Columbia University Medical Center | Columbia University Medical Center IRB | IRB-AAAC9324 |
Duke University Medical Center | The Duke University Health System Institutional Review Board for Clinical Investigations (DUHS IRB) | Pro00004464 |
Johns Hopkins University | Johns Hopkins Medicine Institutional Review Boards (JHM IRB) | NA_00011524 |
Los Angeles Biomedical Research Institute | The John F. Wolf, MD Human Subjects Committee of Harbor-UCLA Medical Center | 12756–01 |
Morehouse School of Medicine | Morehouse School of Medicine Institutional Review Board | 07–1029 |
Temple University | Temple University Office for Human Subjects Protections Institutional Review Board | 11369 |
University of Alabama at Birmingham | The University of Alabama at Birmingham Institutional Review Board for Human Use | FO70712014 |
University of California, San Diego | University of California, San Diego Human Research Protections Program | 070876 |
University of Iowa | The University of Iowa Human Subjects Office | 200710717 |
Ann Arbor VA | VA Ann Arbor Healthcare System IRB | PCC 2008–110732 |
University of Minnesota | University of Minnesota Research Subjects’ Protection Programs (RSPP) | 0801M24949 |
University of Pittsburgh | University of Pittsburgh Institutional Review Board | PRO07120059 |
University of Texas Health Sciences Center at San Antonio | UT Health Science Center San Antonio Institutional Review Board | HSC20070644H |
Health Partners Research Foundation | Health Partners Research Foundation Institutional Review Board | 07–127 |
University of Michigan | Medical School Institutional Review Board (IRBMED) | HUM00014973 |
Minneapolis VA Medical Center | Minneapolis VAMC IRB | 4128-A |
Fallon Clinic | Institutional Review Board/Research Review Committee Saint Vincent Hospital – Fallon Clinic – Fallon Community Health Plan | 1143 |
Consent for publication
Not applicable.
Results
After quality control procedures, RNA-seq data were available for 2,647 samples from current and former smokers from the COPDGene five-year follow-up visit. Approximately two-thirds of subjects were former smokers and twenty-five percent were African American (Table 1). There were slightly more males than females and the average age of these subjects was 65.5 years. The overall disease burden in the population was summarized in Table 1 by a comorbidity index (range 0 to 14, mean = 2.97 and standard deviation = 1.98)53. We performed microbial signature profiling using PathSeq and excluded 57 samples with outlying unmapped read counts (Methods in the online supplement). We then visualized the inferred relative abundance profiles and tested host associations for these 2,590 subjects (Fig. 1). Ordered by mean normalized score from PathSeq, the four taxa observed at the phylum level above an abundance-filtering 1% threshold across all subjects were Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes. In the abundance plot of the normalized scores for these four phyla, ordered by RNA-seq library batch and study center, we observed consistent taxon distributions across the batches and study centers (Figures S1-S2 in the online supplement). Twenty genera had mean normalized scores that eclipsed the 1% threshold chosen to remove low-level contamination. We observed batch specific contamination profiles (Figures S3-S10) for eight genera (Flavobacterium, Pseudomonas, Methylobacterium, Methyloversatilis, Streptomyces, Methylorubrum, Ralstonia and Nevskia). All of these genera are known possible contaminants24,46 and were excluded from the analyses. We also sought to identify remaining contamination by observing the relationship between inferred abundance and nucleic acid concentration using the computation approach outlined in Methods. We again identified the aforementioned genus Methyloversatilis (correlation coefficient = -0.44 and p < 0.0001) as a possible contaminant.

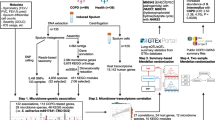
Overview of the study design illustrating the sequencing, statistical and gene enrichment framework. This illustrates the integration with host characteristics and gene expression for observations of host microbiome interaction (GATK = Genome Analysis Toolkit; MaAsLin2 = Multivariate Association with Linear Models).
Genera abundance and host phenotype
We normalized the taxa counts at the genus level from PathSeq using the TMM method. We created a summary of the reads from the gene expression and PathSeq analyses for each of the 12 taxa (Table S1 in the online supplement). Using the TMM-normalized taxa abundances, we created a heatmap with clustering of samples in the columns by Bray–Curtis dissimilarity (Figure S11 in the online supplement). In the color coded tracks for BMI, race, sex, library preparation batch, study center, COPD status and smoking status, we observed visual clustering only by batch (grouping of samples from the same batch). A variable for library batch was included as a covariate in the statistical models to mitigate batch effects and reduce spurious findings. We tested associations between the TMM-normalized abundances for each taxon at the genus level and host phenotype, exposure, treatment and trait variables using linear models with MaAsLin2 (Table S2 in the online supplement). We summarized the findings in a heatmap of the p-values and effect sizes (Figure S12 in the online supplement).
Using an independent replication set of 1,065 samples from the COPDGene five-year follow-up visit (Table S3 in the online supplement), we detected microbial signatures using PathSeq and normalized the taxa counts at the genus level using the TMM method (Table S1 in the online supplement). Contamination was not observed in these data for the 12 taxa using the same methods as in the initial dataset. We performed association tests using the models and methods from the primary analysis and the TMM-normalized taxa abundances for the 12 taxa in the replication set. We summarized the findings in a heatmap of the p-values and effect sizes (Figure S13 in the online supplement).
Meta-analysis
The p-values from the primary and replication analyses were combined for each of the association tests using Stouffer’s method requiring the directions of effect be the same. A heatmap was created to summarize the meta-analysis results (Fig. 2, Figure S14 in the online supplement) with the color intensity indicating significance (negative log transformed q-values) and gray or blue shading indicating the effect direction. Scatter or box plots of the model residuals of the inferred TMM abundance for the significant (FDR < 5%) meta-analysis findings were created in the primary (Figure S15 in the online supplement) and replication (Figure S16 in the online supplement) sets of data to illustrate the relationships between taxa abundance and the variables of interest.

Heatmap of the associations between genera inferred abundance and host-related variables for the meta-analysis. Variables with at least one finding with FDR < 10% were included. The value in each cell is the adjusted q-value. The color scale for the cells represents the sign of the effect multiplied by negative log10 of the q-values, with intensity proportional to significance and gray shading representing positively correlated associations and blue shading representing negatively correlated associations. Results with discordant directions of effect in the meta-analysis are set to q = 1 (white) (heatmap produced using the labeledHeatmap function from the R package WGCNA45). Variables with at least one significant association are included (WBC = white blood cell count, Lymphocytes = lymphocyte count, NeutroLymph_Ratio = ratio of neutrophil counts to lymphocyte counts, Lymphocyte_pct = percentage of lymphocytes, Neutrophil_pct = percentage of neutrophils, 6 MW = six-minute walk distance, mMRC = Modified Medical Research Council dyspnea score, COPD (case–control) = COPD cases vs. controls, PackYears = pack-years history of smoking, Smoking (current-former) = current vs. former smoking status).
From the meta-analysis (Fig. 2), we observed associations between smoking status (current vs. former) and Acinetobacter (q = 0.017), Serratia (q = 0.0057) and Cutibacterium (q = 0.017) abundance. Two measures of functional capacity (6-min walk distance and mMRC dyspnea scale) were associated with at least one taxon. Acinetobacter (q = 0.042), Serratia (q = 0.0093), Streptococcus (q = 0.042) and Bacillus (q = 0.048) abundances were associated with mMRC, with a higher dyspnea score corresponding to higher bacterial abundance. Serratia (q = 0.042) abundance was associated with 6-min walk distance (6 MW), with higher bacterial abundance corresponding to lower 6-min walk distances. All 12 taxa were associated (q < 0.05) with at least one white blood cell distribution variable. Neutrophil levels and bacterial abundance were positively correlated. Conversely, lymphocyte levels were negatively correlated with abundance. Abundance for nine of the 12 taxa was associated (q < 0.05) with sex, with lower bacterial abundance in males. Seven of the 12 taxa were associated with race, with bacterial abundance lower in non-Hispanic white participants.
Host-microbiome interactions
We sought to highlight host-microbiome interactions using microbial abundance profiles and host gene expression pathways. We created a matrix of pathway expression for the Hallmark sets from MSigDB using the R/Bioconductor package GSVA and the human blood RNA-seq data in both the primary and replication data. We tested the association between TMM-normalized taxa abundance and host pathways in both sets of data for each of the 12 genera using models, adjusting for age, sex, race, pack-years of smoking, current smoking status (vs, former), library prep batch and study center. The associations across all taxa and pathways were summarized for both sets of data in a heatmaps (Figures S17 and S18 in the online supplement). The p-values from the primary and replication analyses were combined for each of the association tests using Stouffer’s method requiring the directions of effect be the same and a heatmap was created to summarize the results (Figure S19 in the online supplement). We used network methods to visualize the large set of significant findings. We constructed a bipartite network using the significant (FDR < 5%) associations as edges (edge weights = -log10(p-value)) between taxa and pathways (Figure S20 in the online supplement). Using CONDOR (see Methods), we identified three communities within this network (Figures S21 and S22 in the online supplement) with one of particular relevance to our taxa-association findings (Fig. 3). This community has six genera (Streptococcus, Cutibacterium, Corynebacterium, Lactobacillus, Staphylococcus, and Bacillus) and 15 host pathways, including WNT BETA CATENIN SIGNALING, MTORC1 SIGNALING, and OXIDATIVE PHOSPHORYLATION . Within these communities we observe clustering of genera with shared pathway associations, suggesting joint influence on the host processes.

Community from the bipartite network from the host-microbiome interaction analysis with relevance to COPD, dyspnea and smoking associations. Edges represent a significant (FDR < 5%) association between genus abundance (blue circles) and the expression of the human Hallmark pathway (red squares) in the meta-analysis (figure produced using the R package igraph52).
Discussion
We re-purposed peripheral blood RNA-sequencing data in a large sample set from the COPDGene Study. Using RNA-sequencing reads that did not map to the human genome, we identified microbial signatures at both the phylum and genus levels. We tested associations between inferred abundance and host-related variables. At the phylum level, we identified Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes. Recent studies using both 16S rRNA gene sequencing and unmapped human RNA-seq data have shown that peripheral blood typically includes a nucleic acid signature of these phyla26,27,35.
Taxa associations
Detection at the genus level produced a larger set of taxa, with all 12 taxa significantly associated with at least one host-related variable. Eight of the genera had at least six significant findings. For the associations between taxa abundance and white blood cell composition, we observed positive correlation for neutrophil percentage and neutrophil-to-lymphocyte ratio. Although the role of neutrophils in the establishment of the microbiome can be complex54 the positive correlation is plausible given the role of neutrophils in the defense against bacterial infections. We observed a positive association between the genera Acinetobacter and Streptococcus and mMRC dyspnea score. Acinetobacter is a known cause of acute exacerbations and lung infections55,56,57. Acinetobacter airway abundance may also be a marker of outcome for critically ill COPD patients58. Streptococcus pneumoniae is a common cause of respiratory infections and has been observed in the airway of patients with exacerbations57 and has been isolated from sputum samples in COPD patients in both a stable and an exacerbation state59. The abundance of Serratia and Bacillus was also associated with mMRC dyspnea score. Although Serratia and Bacillus species are less frequently associated with lung infections, Serratia has been identified in patients with exacerbations of COPD60,61. Bacillus was isolated from the lung of stable COPD subjects62 and subjects with more variable microbiomes during a longitudinal study of sputum in COPD63. In the study by Bouquet et al.63, microbiota variability corresponded to higher exacerbation frequency and frequent viral infections in stable COPD. The association between Serratia abundance and six minute walk distance highlights another association with relevance to pulmonary functional capacity and outcomes in COPD64.
Acinetobacter, Serratia and Cutibacterium abundance was associated with current smoking status, compared to former smokers. Species in the Acinetobacter and Serratia have been identified in cigarettes65 providing a possible mechanism for introduction of these taxa, though an explanation for higher abundance in the peripheral blood of former smokers is not apparent at this time. Community acquired Acinetobacter infections, including bacteremia, were also found more in patients with a history of heavy smoking66. Cutibacterium species are members of the upper respiratory tract microbiome67,68 and although smoking has an impact on the microbiome of the upper respiratory tract69,70 evidence regarding the influence of smoking on Cutibacterium is lacking. Irrespective of individual taxa, the impact of smoking on bacterial infections and the microbiome are complex71,72, particularly in the context of COPD73,74. Together, this information suggests relevance for the identified taxa in the lung microbiome and respiratory infections with possible implications in chronic or persistent dyspnea and inflammation.
Further efforts will be required to determine whether these associations in peripheral blood highlight cross-tissue mechanisms similar to the immunomodulatory effects observed in the gut-lung axis75,76, or perhaps similar to interactions or microbial translocations observed between liver and gut in liver disease77. Despite any direction of effect ambiguity, together these findings suggest we may be capturing lung disease relevant microbial signatures in peripheral blood.
The associations between nine taxa and sex are supported by previous findings regarding sex-specific microbiome characteristics in the gut78,79. Previous studies highlighted sex differences with respect to bacterial infections, including respiratory infections80, and relevance in the relationship between airway microbiome and asthma81. Likewise, gut microbiota diversity may vary across ethnicity82,83, supporting our taxa abundance associations with race. The associations between blood taxa and both sex and race may provide insight into systemic host bacterial responses and inform development of personalized therapeutics.
Host-microbiome networks
We leveraged the human RNA-seq data from the same samples to explore host-microbiome interactions using network methods for significant taxa and host pathway associations. Within the communities of the bipartite network, genera with common pathway associations were clustered, providing insight into shared influence on the host processes. For one particular community within the bipartite network (Fig. 3), we observed clustering of Streptococcus (associated with mMRC dyspnea score) with Cutibacterium (associated with current smoking status) through several host pathways, including OXIDATIVE PHOSPHORYLATION, WNT BETA CATENIN SIGNALING, and MTORC1 SIGNALING. Pathways in Fig. 3 are involved in aspects of COPD. In regards to oxidative phosphorylation, mitochondrial reactive oxygen species production and mitochondrial dysfunction are believed to have a role in the development of lung diseases including COPD84, with implication in exercise capacity85. It has been suggested that cross-talk between the bacterial microbiome and mitochondria is a component of overall microbiome interactions with the host86.
The mTORC1 signaling pathway has been implicated in lung cell senescence and emphysema87 and is involved in airway inflammation88 and development of corticosteroid resistance driven by cigarette smoke89. Having a prominent role in regulation of immune responses90,91, the mTOR pathway, in particular, responds to environmental changes and regulates intracellular processes92. The mTOR pathway may have a role in determining the composition of the gut microbiome93,94.
Airway down-regulation of the Wnt/beta-catenin pathway has been observed in smokers95, suggesting a role in the development of smoking-related airway disease and airway inflammation in COPD96. With a role in cell proliferation and cellular morphology97, Wnt/beta-catenin signaling is a process bacterial pathogens may exploit to better establish infection98, providing a possible target for future antimicrobial therapeutics99. Although the microbial signature observed in our study does not appear to be pathogen-specific, both the establishment and maintenance of the bacterial microbiome and the regulation of a host pathogen defense involve a shared complex relationship with host immune responses100.
Together, these findings suggest we have detected a systemic blood signature of host-microbe interactions with pathogenic relevance and perhaps linked to the COPD-relevant associations we identified. This bipartite network approach demonstrates a versatile method for observation of these host-microbiome interactions. The approach is similar to previous airway host-microbiome interaction studies, though focused on a knowledge-based pathway approach instead of unsupervised dimensionality reduction of gene expression data using principal component analysis (PCA)21. The edges in this network may highlight taxa with shared interactions or influence on host biological processes. Both the blood microbial signatures and the structure of these host interactions may inform patient stratification or personalized medicine efforts related to COPD and exacerbations. These efforts could involve particular host pathway or gene targets, identified by their relationship to COPD-relevant microbial taxa using these methods.
Limitations
There are several limitations to the current study. In this secondary analysis of blood RNA-sequencing data, we are capturing RNA from bacterial genes. These mapped reads are serving as a proxy for abundance. Future studies involving 16S rRNA gene or whole-genome shotgun sequencing in parallel with the host transcriptome analysis will provide further insight into the blood microbial signatures. Although the existence of a healthy blood microbiome remains a subject of debate35, we have replicated taxa from previous blood microbiome 16S rRNA gene and RNA sequencing studies26,27,30, demonstrating the generalizability of this approach. Future metagenomic studies with concurrent blood and lung or airway samples, perhaps in a longitudinal context, will be required to determine to what extent peripheral blood recapitulates the lung microbiome. This may also reveal mechanisms responsible for overlapping microbial signatures, such as bacterial translocation, and further identify any transient behavior of these signatures. Given the relatively small effect sizes, the applicability of these findings in a clinical context will be considered in future studies. The sequencing data from this study was not obtained for use in a microbiome study. Therefore, specific bacterial contamination mitigation procedures were not included in the COPDGene protocol, beyond sterile blood acquisition. We assessed for contamination using visual inspection of our data and statistical testing, and we excluded taxa with any potential evidence of contamination. A replication dataset was included to ensure validity of our results. In future studies, protocols involving the inclusion of negative controls and treatment of kit reagents to reduce contaminating nucleic acid content and other measures will help to address the issue of sample contamination24,46.
Conclusions
In this study of the blood microbiome, we were able to identify COPD-relevant bacterial signatures in a secondary analysis of peripheral blood RNA-seq data from a large cohort of smokers. Analyses at the genus level found associations between blood microbial signals and multiple COPD-relevant traits. Using a network approach on the paired human RNA-seq and microbial datasets, we identified host transcriptomic pathways linking multiple taxa, highlighting a useful method for future studies of the human microbiome and transcriptome. Together these findings demonstrate that the peripheral blood microbial signature and host-microbiome interactions may have the potential to capture relevant lung microbiome features and biology. This study provides an initial step toward discovery of composite blood biomarkers for use in predictive disease models to inform personalized treatments of chronic smoking-related diseases.
Data availability
Phenotype and the primary RNA sequencing data are available in dbGaP, accessions phs000179 and phs000765. The replication data will be available in dbGaP when processing is completed.
Abbreviations
- AWT:
-
Airway Wall Thickness
- COPD:
-
Chronic Obstructive Pulmonary Disease
- CONDOR:
-
COmplex Network Description Of Regulators
- COPDGene:
-
Genetic Epidemiology of COPD
- CT:
-
Computed Tomography
- FEV1:
-
Forced Expiratory Volume in 1 s
- FVC:
-
Forced Vital Capacity
- FDR:
-
False Discovery Rate
- GATK:
-
Genome Analysis Toolkit
- GSVA:
-
Gene Set Variation Analysis
- IPF:
-
Idiopathic Pulmonary Fibrosis
- MaAsLin2:
-
Multivariate Association with Linear Models
- mMRC:
-
Modified Medical Research Council dyspnea score
- NCBI:
-
National Center for Biotechnology Information
- Pi10:
-
SRWA-Pi10; square root wall area of a hypothetical airway with 10 mm internal perimeter
- PRISm:
-
Preserved Ratio Impaired Spirometry
- 6 MW:
-
6-Minute Walk distance
- TMM:
-
Trimmed Mean of M values
- WBC:
-
White Blood Cell
References
Gevers, D. et al. The human microbiome project: A community resource for the healthy human microbiome. PLOS Biol. 10, e1001377 (2012).
Proctor, L. M. et al. The integrative human microbiome project. Nature 569, 641–648 (2019).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649-662.e20 (2019).
Rose, S.M.S.-F. et al. A longitudinal big data approach for precision health. Nat. Med. 25, 792–804 (2019).
Morgan, X. C. & Huttenhower, C. Chapter 12: Human microbiome analysis. PLOS Comput Biol. 8, e1002808 (2012).
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844 (2017).
Budden, K. F. et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 7, 907–920 (2019).
Dickson, R. P., Erb-Downward, J. R. & Huffnagle, G. B. The role of the bacterial microbiome in lung disease. Expert Rev. Respir. Med. 7, 245–257 (2013).
O’Dwyer, D. N., Dickson, R. P. & Moore, B. B. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J. Immunol. 196, 4839–4847 (2016).
Jacobs, D. M., Ochs-Balcom, H. M., Zhao, J., Murphy, T. F. & Sethi, S. Lower airway bacterial colonization patterns and species-specific interactions in chronic obstructive pulmonary disease. J. Clin. Microbiol. 56, e00330-e418 (2018).
Pragman, A. A. et al. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome 6, 7 (2018).
Lee, J. et al. Different upper airway microbiome and their functional genes associated with asthma in young adults and elderly individuals. Allergy 74, 709–719 (2019).
Pérez-Losada, M., Castro-Nallar, E., Bendall, M. L., Freishtat, R. J. & Crandall, K. A. Dual transcriptomic profiling of host and microbiota during health and disease in pediatric asthma. PLoS ONE 10, e0131819 (2015).
Molyneaux, P. L. et al. Host-microbial interactions in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 195, 1640–1650 (2017).
Wang, Z. et al. Lung microbiome dynamics in COPD exacerbations. Eur. Respir. J. 47, 1082–1092 (2016).
Dickson, R. P. et al. Bacterial topography of the healthy human lower respiratory tract. MBio 8, e02287-16 (2017).
Ren, L. et al. Transcriptionally active lung microbiome and its association with bacterial biomass and host inflammatory status. mSystems 3, e00199-18 (2018).
Sze, M. A. et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 192, 438–445 (2015).
Cabrera-Rubio, R. et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J. Clin. Microbiol. 50, 3562–3568 (2012).
Pragman, A. A. et al. Chronic obstructive pulmonary disease upper airway microbiome is associated with select clinical characteristics. PLoS ONE 14, e0219962 (2019).
Wang, Z. et al. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir. Res. 20, 113 (2019).
Haldar, K. et al. The sputum microbiome is distinct between COPD and health, independent of smoking history. Respir. Res. 21, 183 (2020).
Einarsson, G. G. et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax 71, 795–803 (2016).
Carney, S. M. et al. Methods in lung microbiome research. Am. J. Respir. Cell Mol. Biol. 62, 283–299 (2020).
Marsh, R. L. et al. How low can we go? The implications of low bacterial load in respiratory microbiota studies. Pneumonia 10, 7 (2018).
Païssé, S. et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion (Paris) 56, 1138–1147 (2016).
Whittle, E., Leonard, M. O., Harrison, R., Gant, T. W. & Tonge, D. P. Multi-method characterization of the human circulating microbiome. Front. Microbiol. 9, 3266 (2019).
Qiu, J., Zhou, H., Jing, Y. & Dong, C. Association between blood microbiome and type 2 diabetes mellitus: A nested case-control study. J. Clin. Lab. Anal. 33, e22842 (2019).
Li, Q. et al. Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Front. Cell Infect. Microbiol. 8, 5 (2018).
Olde Loohuis, L. M. et al. Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl. Psychiatry. 8, 96 (2018).
Lagier, J.-C. et al. Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 16, 540–550 (2018).
Shah, N. B. et al. Blood microbiome profile in CKD: A pilot study. Clin. J. Am. Soc. Nephrol. 14, 692–701 (2019).
Lelouvier, B. et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 64, 2015–2027 (2016).
Poore, G. D. et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 (2020).
Castillo, D. J., Rifkin, R. F., Cowan, D. A. & Potgieter, M. The healthy human blood microbiome: Fact or fiction?. Front. Cell Infect. Microbiol. 9, 148 (2019).
Mangul, S. et al. ROP: dumpster diving in RNA-sequencing to find the source of 1 trillion reads across diverse adult human tissues. Genome Biol. 19, 36 (2018).
Regan, E. A. et al. Genetic epidemiology of COPD (COPDGene) study design. COPD J. Chronic Obstr. Pulm. Dis. 7, 32–43 (2011).
Stringer, W. W. et al. Physiologic insights from the COPD genetic epidemiology study. Chronic Obstr. Pulm. Dis. COPD Found. 6, 256–266 (2019).
Bowler, R. P. et al. Prediction of acute respiratory disease in current and former smokers with and without COPD. Chest 146, 941–950 (2014).
Parker, M. M. et al. RNA sequencing identifies novel non-coding RNA and exon-specific effects associated with cigarette smoking. BMC Med. Genomics 10, 58 (2017).
Walker, M. A. et al. GATK PathSeq: A customizable computational tool for the discovery and identification of microbial sequences in libraries from eukaryotic hosts. Bioinformatics 34, 4287–4289 (2018).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Mallick, H. et al. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 18, 228 (2017).
Dewey, M. R package metap: Meta-analysis of significance values (2020).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Karstens, L. et al. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems 4, e0029019-19 (2019).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
Jervis-Bardy, J. et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 3, 19 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 14, 7 (2013).
Platig, J., Castaldi, P. J., DeMeo, D. & Quackenbush, J. Bipartite community structure of eQTLs. PLOS Comput. Biol. 12, e1005033 (2016).
Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal. 2006;Complex Systems:1695.
Putcha, N. et al. A simplified score to quantify comorbidity in COPD. PLoS ONE 9, e114438 (2014).
Zhang, D. & Frenette, P. S. Cross talk between neutrophils and the microbiota. Blood 133, 2168–2177 (2019).
Wong, D. et al. Clinical and pathophysiological overview of acinetobacter infections: A century of challenges. Clin Microbiol Rev. 30, 409–447 (2017).
Su, J. et al. Sputum bacterial and fungal dynamics during exacerbations of severe COPD. PLoS ONE 10, e0130736 (2015).
Huang, Y. J. et al. A Persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS J. Integr. Biol. 14, 9–59 (2010).
Huang, W.-C. et al. Dynamics of the lung microbiome in intensive care patients with chronic obstructive pulmonary disease and community-acquired pneumonia. Sci. Rep. 10, 11046 (2020).
Mantero, M. et al. Role of Streptococcus pneumoniae infection in chronic obstructive pulmonary disease patients in Italy. Ther. Adv. Respir. Dis. 11, 403–407 (2017).
Shimizu, K. et al. Pathogens in COPD exacerbations identified by comprehensive real-time PCR plus older methods. Int. J. Chron. Obstruct. Pulmon Dis. 10, 2009–2016 (2015).
Estirado, C. et al. Microorganisms resistant to conventional antimicrobials in acute exacerbations of chronic obstructive pulmonary disease. Respir. Res. 19, 119 (2018).
Zakharkina, T. et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by T-RFLP and clone sequencing. PLoS ONE 8, e68302 (2013).
Bouquet, J. et al. Microbial burden and viral exacerbations in a longitudinal multicenter COPD cohort. Respir. Res. 21, 77 (2020).
Celli, B. et al. The 6-minute-walk distance test as a chronic obstructive pulmonary disease stratification tool. Insights from the COPD biomarker qualification consortium. Am. J. Respir. Crit. Care Med. 194, 1483–1493 (2016).
Sapkota, A. R., Berger, S. & Vogel, T. M. Human pathogens abundant in the bacterial metagenome of cigarettes. Environ. Health Perspect. 118, 351–356 (2010).
Falagas, M. E., Karveli, E. A., Kelesidis, I. & Kelesidis, T. Community-acquired Acinetobacter infections. Eur. J. Clin. Microbiol. Infect. Dis. 26, 857–868 (2007).
Wos-Oxley, M. L. et al. A poke into the diversity and associations within human anterior nare microbial communities. ISME J. 4, 839–851 (2010).
Yan, M. et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 14, 631–640 (2013).
Yu, G. et al. The effect of cigarette smoking on the oral and nasal microbiota. Microbiome. 5, 3 (2017).
Charlson, E. S. et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE 5, e15216 (2010).
Huang, C. & Shi, G. Smoking and microbiome in oral, airway, gut and some systemic diseases. J. Transl. Med. 17, 225 (2019).
Feldman, C. & Anderson, R. Cigarette smoking and mechanisms of susceptibility to infections of the respiratory tract and other organ systems. J. Infect. 67, 169–184 (2013).
Brook, I. & Gober, A. E. Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. Chest 127, 2072–2075 (2005).
Garmendia, J., Morey, P. & Bengoechea, J. A. Impact of cigarette smoke exposure on host–bacterial pathogen interactions. Eur. Respir. J. 39, 467–477 (2012).
Dang, A. T. & Marsland, B. J. Microbes, metabolites, and the gut–lung axis. Mucosal. Immunol. 12, 843–850 (2019).
Ma, Y., Yang, X., Chatterjee, V., Wu, M. H. & Yuan, S. Y. The Gut-Lung axis in systemic inflammation. Role of mesenteric lymph as a conduit. Am. J. Respir. Cell Mol. Biol. 64, 19–28 (2020).
Tilg, H., Cani, P. D. & Mayer, E. A. Gut microbiome and liver diseases. Gut 65, 2035–2044 (2016).
Borgo, F. et al. Body mass index and sex affect diverse microbial niches within the Gut. Front. Microbiol. 9, 213 (2018).
de la Cuesta-Zuluaga, J. et al. Age- and sex-dependent patterns of gut microbial diversity in human adults. mSystems. 4, e00261-19 (2019).
Vázquez-Martínez, E. R., García-Gómez, E., Camacho-Arroyo, I. & González-Pedrajo, B. Sexual dimorphism in bacterial infections. Biol. Sex Differ. 9, 27 (2018).
Chen, R. et al. Sex effects in the association between airway microbiome and asthma. Ann. Allergy Asthma Immunol. 125, 652-657.e3 (2020).
Huttenhower, C. et al. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Brooks, A. W., Priya, S., Blekhman, R. & Bordenstein, S. R. Gut microbiota diversity across ethnicities in the United States. PLOS Biol. 16, e2006842 (2018).
Cloonan, S. M. & Choi, A. M. K. Mitochondria in lung disease. J. Clin. Invest. 126, 809–820 (2016).
Puente-Maestu, L. et al. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur. Respir. J. 33, 1045–1052 (2009).
Saint-Georges-Chaumet, Y. & Edeas, M. Microbiota–mitochondria inter-talk: consequence for microbiota–host interaction. Pathog. Dis. 74, 096 (2016).
Houssaini, A. et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 3, 9e93203 (2018).
Wang, Y. et al. MTOR Suppresses cigarette smoke-induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J. Immunol. 200, 2571–2580 (2018).
Mitani, A., Ito, K., Vuppusetty, C., Barnes, P. J. & Mercado, N. Restoration of corticosteroid sensitivity in chronic obstructive pulmonary disease by inhibition of mammalian target of rapamycin. Am. J. Respir. Crit. Care Med. 193, 143–153 (2016).
Powell, J. D., Pollizzi, K. N., Heikamp, E. B. & Horton, M. R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68 (2012).
Thomson, A. W., Turnquist, H. R. & Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337 (2009).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012).
Jung, M.-J. et al. Chronic repression of mTOR complex 2 induces changes in the gut microbiota of diet-induced obese mice. Sci. Rep. 6, 30887 (2016).
Noureldein, M. H. & Eid, A. A. Gut microbiota and mTOR signaling: Insight on a new pathophysiological interaction. Microb. Pathog. 118, 98–104 (2018).
Wang, R. et al. Down-regulation of the canonical Wnt β-catenin pathway in the airway epithelium of healthy smokers and smokers with COPD. PLoS ONE 6, e14793 (2011).
Heijink, I. H. et al. Role of aberrant WNT signalling in the airway epithelial response to cigarette smoke in chronic obstructive pulmonary disease. Thorax 68, 709–716 (2013).
MacDonald, B. T., Tamai, K. & He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 17, 9–26 (2009).
Rogan, M. R., Patterson, L. L., Wang, J. Y. & McBride, J. W. Bacterial manipulation of Wnt signaling: A host-pathogen Tug-of-Wnt. Front. Immunol. 10, 2390 (2019).
Silva-García, O., Valdez-Alarcón, J. J. & Baizabal-Aguirre, V. M. Wnt/β-catenin signaling as a molecular target by pathogenic bacteria. Front. Immunol. 10, 2135 (2019).
Kogut, M. H., Lee, A. & Santin, E. Microbiome and pathogen interaction with the immune system. Poult. Sci. 99, 1906–1913 (2020).
Acknowledgements
COPDGene Phase 3
COPDGene® Investigators – Core Units
Administrative Center: James D. Crapo, MD (PI); Edwin K. Silverman, MD, PhD (PI); Barry J. Make, MD; Elizabeth A. Regan, MD, PhD.
Genetic Analysis Center: Terri H. Beaty, PhD; Peter J. Castaldi, MD, MSc; Michael H. Cho, MD, MPH; Dawn L. DeMeo, MD, MPH; Adel El Boueiz, MD, MMSc; Marilyn G. Foreman, MD, MS; Auyon Ghosh, MD; Lystra P. Hayden, MD, MMSc; Craig P. Hersh, MD, MPH; Jacqueline Hetmanski, MS; Brian D. Hobbs, MD, MMSc; John E. Hokanson, MPH, PhD; Wonji Kim, PhD; Nan Laird, PhD; Christoph Lange, PhD; Sharon M. Lutz, PhD; Merry-Lynn McDonald, PhD; Dmitry Prokopenko, PhD; Matthew Moll, MD, MPH; Jarrett Morrow, PhD; Dandi Qiao, PhD; Elizabeth A. Regan, MD, PhD; Aabida Saferali, PhD; Phuwanat Sakornsakolpat, MD; Edwin K. Silverman, MD, PhD; Emily S. Wan, MD; Jeong Yun, MD, MPH.
Imaging Center: Juan Pablo Centeno; Jean-Paul Charbonnier, PhD; Harvey O. Coxson, PhD; Craig J. Galban, PhD; MeiLan K. Han, MD, MS; Eric A. Hoffman, Stephen Humphries, PhD; Francine L. Jacobson, MD, MPH; Philip F. Judy, PhD; Ella A. Kazerooni, MD; Alex Kluiber; David A. Lynch, MB; Pietro Nardelli, PhD; John D. Newell, Jr., MD; Aleena Notary; Andrea Oh, MD; Elizabeth A. Regan, MD, PhD; James C. Ross, PhD; Raul San Jose Estepar, PhD; Joyce Schroeder, MD; Jered Sieren; Berend C. Stoel, PhD; Juerg Tschirren, PhD; Edwin Van Beek, MD, PhD; Bram van Ginneken, PhD; Eva van Rikxoort, PhD; Gonzalo Vegas Sanchez-Ferrero, PhD; Lucas Veitel; George R. Washko, MD; Carla G. Wilson, MS;
PFT QA Center, Salt Lake City, UT: Robert Jensen, PhD.
Data Coordinating Center and Biostatistics, National Jewish Health, Denver, CO: Douglas Everett, PhD; Jim Crooks, PhD; Katherine Pratte, PhD; Matt Strand, PhD; Carla G. Wilson, MS.
Epidemiology Core, University of Colorado Anschutz Medical Campus, Aurora, CO: John E. Hokanson, MPH, PhD; Erin Austin, PhD; Gregory Kinney, MPH, PhD; Sharon M. Lutz, PhD; Kendra A. Young, PhD.
Mortality Adjudication Core: Surya P. Bhatt, MD; Jessica Bon, MD; Alejandro A. Diaz, MD, MPH; MeiLan K. Han, MD, MS; Barry Make, MD; Susan Murray, ScD; Elizabeth Regan, MD; Xavier Soler, MD; Carla G. Wilson, MS.
Biomarker Core: Russell P. Bowler, MD, PhD; Katerina Kechris, PhD; Farnoush Banaei-Kashani, Ph.D
COPDGene® Investigators – Clinical Centers
Ann Arbor VA: Jeffrey L. Curtis, MD; Perry G. Pernicano, MD.
Baylor College of Medicine, Houston, TX: Nicola Hanania, MD, MS; Mustafa Atik, MD; Aladin Boriek, PhD; Kalpatha Guntupalli, MD; Elizabeth Guy, MD; Amit Parulekar, MD;
Brigham and Women’s Hospital, Boston, MA: Dawn L. DeMeo, MD, MPH; Craig Hersh, MD, MPH; Francine L. Jacobson, MD, MPH; George Washko, MD.
Columbia University, New York, NY: R. Graham Barr, MD, DrPH; John Austin, MD; Belinda D’Souza, MD; Byron Thomashow, MD.
Duke University Medical Center, Durham, NC: Neil MacIntyre, Jr., MD; H. Page McAdams, MD; Lacey Washington, MD.
HealthPartners Research Institute, Minneapolis, MN: Charlene McEvoy, MD, MPH; Joseph Tashjian, MD.
Johns Hopkins University, Baltimore, MD: Robert Wise, MD; Robert Brown, MD; Nadia N. Hansel, MD, MPH; Karen Horton, MD; Allison Lambert, MD, MHS; Nirupama Putcha, MD, MHS.
Lundquist Institute for Biomedical Innovationat Harbor UCLA Medical Center, Torrance, CA: Richard Casaburi, PhD, MD; Alessandra Adami, PhD; Matthew Budoff, MD; Hans Fischer, MD; Janos Porszasz, MD, PhD; Harry Rossiter, PhD; William Stringer, MD.
Michael E. DeBakey VAMC, Houston, TX: Amir Sharafkhaneh, MD, PhD; Charlie Lan, DO.
Minneapolis VA: Christine Wendt, MD; Brian Bell, MD; Ken M. Kunisaki, MD, MS.
Morehouse School of Medicine, Atlanta, GA: Eric L. Flenaugh, MD; Hirut Gebrekristos, PhD;
Mario Ponce, MD; Silanath Terpenning, MD; Gloria Westney, MD, MS.
National Jewish Health, Denver, CO: Russell Bowler, MD, PhD; David A. Lynch, MB.
Reliant Medical Group, Worcester, MA: Richard Rosiello, MD; David Pace, MD.
Temple University, Philadelphia, PA: Gerard Criner, MD; David Ciccolella, MD; Francis Cordova, MD; Chandra Dass, MD; Gilbert D’Alonzo, DO; Parag Desai, MD; Michael Jacobs, PharmD; Steven Kelsen, MD, PhD; Victor Kim, MD; A. James Mamary, MD; Nathaniel Marchetti, DO; Aditi Satti, MD; Kartik Shenoy, MD; Robert M. Steiner, MD; Alex Swift, MD; Irene Swift, MD; Maria Elena Vega-Sanchez, MD.
University of Alabama, Birmingham, AL: Mark Dransfield, MD; William Bailey, MD; Surya P. Bhatt, MD; Anand Iyer, MD; Hrudaya Nath, MD; J. Michael Wells, MD.
University of California, San Diego, CA: Douglas Conrad, MD; Xavier Soler, MD, PhD; Andrew Yen, MD.
University of Iowa, Iowa City, IA: Alejandro P. Comellas, MD; Karin F. Hoth, PhD; John Newell, Jr., MD; Brad Thompson, MD.
University of Michigan, Ann Arbor, MI: MeiLan K. Han, MD MS; Ella Kazerooni, MD MS; Wassim Labaki, MD MS; Craig Galban, PhD; Dharshan Vummidi, MD.
University of Minnesota, Minneapolis, MN: Joanne Billings, MD; Abbie Begnaud, MD; Tadashi Allen, MD.
University of Pittsburgh, Pittsburgh, PA: Frank Sciurba, MD; Jessica Bon, MD; Divay Chandra, MD, MSc; Joel Weissfeld, MD, MPH.
University of Texas Health, San Antonio, San Antonio, TX: Antonio Anzueto, MD; Sandra Adams, MD; Diego Maselli-Caceres, MD; Mario E. Ruiz, MD; Harjinder Singh.
Funding
Supported by NIH Grants: K25 HL136846, R01HL130512, R01HL125583, U01HL089856, U01HL089897, R01HL124233, R01HL147326. COPDGene is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion.
Author information
Authors and Affiliations
Contributions
J.D.M.: analysis and interpretation of data, manuscript preparation and approval of the final version; P.J.C.: acquisition of data, review of manuscript and approval of the final version; R.P.C.: analysis of data, review of manuscript and approval of the final version; J.H.Y.: interpretation of data, review of manuscript and approval of the final version; S.L.: analysis of data, review of manuscript and approval of the final version; Y.Y.L.: interpretation of data, manuscript preparation and approval of the final version; C.P.H.: acquisition of data, analysis and interpretation of data, manuscript preparation and approval of the final version.
Corresponding author
Ethics declarations
Competing interests
Dr. Hersh has received grant support from Bayer, Boehringer-Ingelheim, Novartis, and Vertex. Dr. Castaldi has received consulting fees and grant support from GSK. All other authors declare that they have no competing interests related to this manuscript.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morrow, J.D., Castaldi, P.J., Chase, R.P. et al. Peripheral blood microbial signatures in current and former smokers. Sci Rep 11, 19875 (2021). https://doi.org/10.1038/s41598-021-99238-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-99238-4
- Springer Nature Limited