
Abstract
Background
People living with HIV (PLWH) are at increased risk of developing Chronic Obstructive Pulmonary Disease (COPD) independent of cigarette smoking. We hypothesized that dysbiosis in PLWH is associated with epigenetic and transcriptomic disruptions in the airway epithelium.
Methods
Airway epithelial brushings were collected from 18 COPD + HIV + , 16 COPD − HIV + , 22 COPD + HIV − and 20 COPD – HIV − subjects. The microbiome, methylome, and transcriptome were profiled using 16S sequencing, Illumina Infinium Methylation EPIC chip, and RNA sequencing, respectively. Multi ‘omic integration was performed using Data Integration Analysis for Biomarker discovery using Latent cOmponents. A correlation > 0.7 was used to identify key interactions between the ’omes.
Results
The COPD + HIV −, COPD −HIV + , and COPD + HIV + groups had reduced Shannon Diversity (p = 0.004, p = 0.023, and p = 5.5e−06, respectively) compared to individuals with neither COPD nor HIV, with the COPD + HIV + group demonstrating the most reduced diversity. Microbial communities were significantly different between the four groups (p = 0.001). Multi ‘omic integration identified correlations between Bacteroidetes Prevotella, genes FUZ, FASTKD3, and ACVR1B, and epigenetic features CpG-FUZ and CpG-PHLDB3.
Conclusion
PLWH with COPD manifest decreased diversity and altered microbial communities in their airway epithelial microbiome. The reduction in Prevotella in this group was linked with epigenetic and transcriptomic disruptions in host genes including FUZ, FASTKD3, and ACVR1B.
Similar content being viewed by others

Background
Antiretroviral therapy (ART) has substantially increased the lifespan and reduced acquired immunodeficiency syndrome-related morbidity and mortality of people living with human immunodeficiency virus (PLWH, HIV) [1, 2]. The aging of PLWH nonetheless places them at higher risk for developing age-related comorbidities, including chronic obstructive pulmonary disease (COPD). PLWH have been shown to have an elevated risk for COPD [3,4,5] which appears to be independent of smoking history [3, 6] and also worsens their mortality risk [7]. As the population of PLWH ages, they are likely to bear an increasing burden of COPD.
There are many hypotheses regarding the mechanisms of HIV-associated COPD, including but not limited to longer exposure to risky behaviours [8], side effects from ART [8, 9], chronic inflammation [10], and Pneumocystis colonization [11, 12]. Speculation that microbial dysbiosis in the lung, the result of repeated pulmonary infections and antibiotic exposure, could also drive obstructive lung disease has led to multiple studies investigating the lung microbiome in PLWH [13,14,15,16]. For instance, we previously reported on decreased microbial diversity and community shifts in the airway epithelium of PLWH compared to HIV-uninfected patients [16]. The characterization of the airway epithelial microbiome specifically in PLWH with COPD, however, has yet to be reported. Moreover, although dysbiosis may conceivably lead to profound changes in host molecular mechanisms such as transcription, epigenetic regulation, metabolism, and immunity, these disruptions have not yet been fully described.
In this study, we hypothesize that microbial disruptions in the airway epithelium of PLWH with COPD are associated with key transcriptomic and epigenetic alterations that can help us gain insights into the disease pathogenesis of HIV-associated COPD. We simultaneously profile the microbiome, methylome, and transcriptome from airway epithelial cells collected via bronchoscopy in PLWH with COPD (COPD + HIV +) to (1) characterize the distinct microbiome features distinguishing them from PLWH without COPD (COPD − HIV +), HIV-uninfected patients with COPD (COPD + HIV−), and healthy controls (COPD − HIV−) and (2) link these features with epigenetic and transcriptomic alterations to better understand the host response to dysbiosis. To the best of our knowledge, no study has integrated and examined all three ’omes together in the context of the HIV airway.
Methods
Study population and design
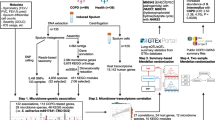
Airway epithelial cell (AEC) brushings were obtained from 76 (18 COPD + HIV +, 16 COPD− HIV + , 22 COPD + HIV− and 20 COPD – HIV −) adult patients at St. Paul’s Hospital, Vancouver, BC, who consented to undergo bronchoscopic collection of research specimens under the University of British Columbia Research Ethics Board Certificates H11-02713 and H15-02166. Bronchial brushings were obtained according to previously published methods [16,17,18]. Briefly, the bronchoscope was guided to either the right or left upper lobe segment and a cytology brush was inserted until resistance was met (approximately 2 mm in diameter) at which point gentle brushings were obtained for AEC collection. PLWH also provided background controls for the study, including oral washings, reagent controls, bronchoscope channel washings, water rinsed over unused cytology brushes, and extraction negative samples specifically to investigate for any cross-contamination with AEC samples. COPD was defined based on a pulmonologist’s diagnosis of COPD and either a pre-bronchodilator forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) ≤ lower limit of normal [19] or clear evidence of emphysema on computed tomography imaging on visual inspection. PLWH were defined as subjects with documented HIV-1 infection. An overview of the study design is provided in Additional file 1: Fig. S1. Details of the cohort and the methylation and transcriptome profiling of the samples have previously been reported [18]. Briefly, methylation profiles were obtained using the Illumina Infinium MethylationEPIC BeadChip microarray, which captures the methylation status 863,904 CpG sites across the genome. Paired end RNA sequencing was performed to a depth of 50 million reads using the Illumina Novaseq6000 platform.
Microbiome profiling
Bacterial genomic DNA was extracted from bronchial brushings, and microbiome profiles were obtained using touchdown droplet digital polymerase chain reaction, followed by 16S amplicon sequencing using the Illumina Miseq® platform at the Sequencing and Bioinformatics Consortium at the University of British Columbia (Vancouver, Canada). Sequencing data were processed following the QIIME2™ workflow using Divisive Amplicon Denoising Algorithm (DADA2) to denoise sequences. During these steps, the sequencing reads were demultiplexed, merged and resolved into amplicon sequence variants (ASVs). Further quality filtering steps were performed to remove contaminating host mitochondrial or chloroplast sequences, ASVs present in PCR controls, ASVs with significantly fewer sequences than the majority, and ASVs present only in one sample (singletons). Taxonomy assignment was performed on ASVs using a pre-trained naive Bayes classifier artifact trained against Greengenes (138 revision) trimmed to include only the V4 hypervariable region and pre-clustered at 99% sequence identity. Phylogenetic trees were generated using the MAFFT program in QIIME 2™, which was consecutively used as input to compute different phylogenetic diversity measures.
Alpha diversity was measured using the Shannon Diversity Index, a metric of community richness and evenness. T-tests were used to identify differences in Shannon Diversity between PLWH and HIV-uninfected groups and between COPD and non-COPD groups, while analysis of variance (ANOVA) and Mann–Whitney U-tests were used to identify differences between the COPD-HIV-, COPD + HIV −, COPD −HIV + , and COPD + HIV + groups. Beta diversity was measured using Bray Curtis Dissimilarity, which measures differences in richness between two or more communities, tested with permutational multivariate analysis of variance (PERMANOVA) (adonis function in vegan R package [20]), and visualized using principal coordinate analysis (PCoA) plots. For both alpha and beta diversity, a second model was performed to adjust for age, sex, and smoking status using the following: Diversity Metric ~ Age + Sex + Smoking Status + COPD/HIV. We also examined the interaction effects between COPD and HIV on alpha and beta diversity metrics. Average relative taxon abundance comparisons were performed between the COPD + HIV + , COPD −HIV + , COPD + HIV− and COPD −HIV − groups at the phylum and genus levels. The Kruskal–Wallis and Dunn’s tests were used to identify between group differences. Significant taxon differences were identified at false discover rate (FDR) < 0.05 using the Benjamini–Hochberg method.
Multi ’omic integration
The microbiome, transcriptome, and methylome were integrated using Data Integration Analysis for Biomarker discovery using Latent cOmponents (DIABLO) implemented in the mixOmics R package. This method simultaneously identifies key ’omics features among heterogeneous datasets and their respective correlations [21, 22].
This integration analysis included features from the three datasets as input: microbiome (126 ASVs), transcriptome (28 genes) and methylome (4404 CpGs). The ASVs were obtained after quality filtering and taxonomy assignment steps described above. Top genes and CpG sites were selected based on robust linear modeling examining the interaction effect of COPD*HIV on gene expression and methylation, respectively (further methods provided in the Additional file 1: Methods). The following design matrix with values ranging between 0 (indicating no correlation between ’omics datasets) to 1 (indicating maximum correlation) was chosen, such that there was a compromise between correlation and discrimination between the features across the different datasets.

Subsequently, a DIABLO model with no variable selection was fit to evaluate the global performance and choose the number of components (ncomp) for the final model. The ncomp were chosen with considerations of centroids distance measures and the balanced error rates, after tenfold cross validation repeated 50 times. Sparse partial least squares discriminant analysis was then used to obtain the optimal number of variables of each component in the three datasets, after tenfold cross validation repeated 50 times. Using the chosen parameters, the final DIABLO models were fit to identify key interactions (using a correlation threshold of |0.7|) between the microbiome, methylome and the transcriptome. Similar methods were used to integrate just two datasets at a time—the (a) microbiome and methylome, and (b) microbiome and transcriptome—to identify correlations between the corresponding ’omes.
Results
Table 1 provides a summary of participant demographics. The study cohort was composed of 76 participants (18 COPD + HIV + , 16 COPD − HIV + , 22 COPD + HIV −, and 20 COPD – HIV −). The mean (standard deviation) age was 61.3 ± 11.6 years, with males (n = 48) making up 63.2% of the total. Of the PLWH (n = 34), 30 (88.2%) were receiving ART and 26 (76.5%) had undetectable HIV plasma viral load (< 40 copies/mL), and the mean CD4 count was 439 cells/mm3.
PLWH with COPD feature reduced airway epithelial microbiome diversity and microbial community shifts
There were no significant differences in 16S rRNA gene copy levels between the COPD + HIV + , COPD + HIV −, COPD − -HIV + and COPD – HIV − groups (overall Kruskal–Wallis p = 0.300, Additional file 1: Fig. S2). Alpha diversity measured using the Shannon Diversity Index is represented in Fig. 1A–C. We observed significant differences between the (a) COPD + and COPD − groups (median[interquartile range] 2.80[1.73] vs. 3.99[1.29]; p = 0.003, adjusted p = 0.046), (b) HIV + and HIV − groups (2.66[1.74] vs. 3.83[1.15]; p = 0.002, adjusted p = 0.021), and (c) combined COPD and HIV groups: COPD + HIV + , COPD + HIV −, COPD − HIV + and COPD – HIV − (2.56[1.61] vs. 3.19[1.33] vs. 3.48[1.94] vs. 4.13[0.91]; p = 1.5e−04, adjusted p = 0.016). PLWH with COPD featured the lowest alpha diversity of the four groups. There were no statistically significant differences in alpha or beta diversity measures by sex groups.

Shannon Diversity Index differences between (a) COPD + and COPD− groups, (b) HIV + and HIV− groups, and (c) COPD + HIV + , COPD + HIV−, COPD−HIV + and COPD−HIV− groups. The COPD + HIV + group featured the lowest Shannon Diversity Index of all four groups. P-values were adjusted for age, sex, and smoking status. Microbial community structures in AECs according to (d) COPD status (COPD− (N): red circles; COPD + (Y): blue circles), (e HIV status (HIV− (Negative): red circles; HIV + (Positive): blue circles), and (f) combined COPD + HIV status (COPD + HIV +: purple circles; COPD + HIV−: blue circles; COPD-HIV +: green circles; COPD−HIV−: red circles) based on Bray–Curtis distances; the centroids for each group are also shown. Permutational multivariate ANOVA (PERMANOVA) was used for comparisons of microbial community structures between groups, with adjustment for age, sex, and smoking status
Beta diversity measured between the COPD −HIV −, COPD + HIV −, COPD −HIV + , and COPD + HIV + groups are shown in Fig. 1D–F. The principal coordinate plots and PERMANOVA analysis show that there were significant differences between the resident microbial communities of the COPD + and COPD − groups (p = 0.001, adjusted p = 0.009), HIV + and HIV − groups (p = 0.009, adjusted p = 0.041), and the combined COPD and HIV groups (p = 0.001, adjusted p = 0.006). Consistent with these findings, smoking status was also associated with significant differences in resident microbial communities (p = 0.037, Additional file 1: Fig. S3). There were no significant interaction effects between COPD and HIV on alpha and beta diversity metrics (Additional file 1: Fig. S4). Removal of patients using inhaled corticosteroids (n = 4) did not significantly change relationships in alpha and beta diversity (Additional file 1: Figs. S5, S6).
Relative phyla and genera abundance are shown in Fig. 2 and in Additional file 1: Tables S1 and S2. The most abundant phylum in airway epithelial cells was Firmicutes, followed by Bacteroidetes and Proteobacteria. At the genus level, Prevotella, Veillonella, and Streptococcus were the most abundant. COPD − showed higher relative abundance of phyla Bacteroidetes and Fusobacteria, and genera Prevotella, Veillonella, Megasphaera, Neisseria, Selenomonas, and Fusobacterium compared to the COPD + group. Similarly, the HIV − group had higher abundance of phyla Fusobacteria, and genera Prevotella, Neisseria, Selenomonas and Fusobacterium compared to the HIV + group. Phyla Fusobacteria and Bacteroidetes, and genera Prevotella, Megasphaera, Neisseria, Selenomonas and Fusobacterium showed significant differences between the 4 groups (COPD-HIV-, COPD-HIV +, COPD + HIV −, and COPD + HIV +). There were no individual genera or phyla, however, that were significantly correlated with FEV1 percent predicted.

Average relative taxa abundance comparisons at the phylum level between (a) COPD + and COPD− groups, (b) HIV+ and HIV− groups, and (c) COPD + HIV + , COPD + HIV−, COPD−HIV + and COPD−HIV− groups. Average relative taxa abundance comparisons at the genus level between (d) COPD + and COPD− groups, (e) HIV + and HIV− groups, and (f) COPD + HIV + , COPD + HIV−, COPD−HIV + and COPD−HIV− groups
To address possible microbial contamination in AEC brushings, oral washings, reagent controls, bronchoscope channel washings, water rinsed over unused cytology brushes, and extraction negative samples were collected from the PLWH group. 16S rRNA gene copies were significantly elevated in the AEC brushings and oral wash controls compared to the background environmental samples (Additional file 1: Fig. S7A). PCA plots demonstrated that each sample type had a distinct community profile (Additional file 1: Fig. S7B, Table S3), thus suggesting that there was minimal cross-contamination.
Multi ‘omic integration using DIABLO
The microbiome, transcriptome and methylome were integrated using DIABLO to identify any ‘between’ and ‘within’ ’ome correlations based on (a) COPD, (b) HIV and (c) COPD*HIV statuses (Additional file 1: Fig. S8). The top three ASV-Gene, ASV-CpG and Gene-CpG pairs and their respective correlation values are shown in representative Table 2. The balanced error rate, used as an estimate of model performance, was ∼30–40% in each component. Network interactions between the microbiome, methylome, and transcriptome for the strongest correlations (> 0.75) are shown in Fig. 3. Complete tables of pairwise correlations for COPD, HIV, and COPD*HIV are provided in Additional files 2, 3, and 4, respectively.

Network visualization of correlations between the microbiome, methylome and transcriptome in (a) COPD, (b) HIV, and (c) COPD*HIV analyses. Nodes represent multiomic features (ASVs—Pink triangle, CpGs—Purple circle, and genes—Yellow square), and edges connecting any two nodes corresponds to their correlation (Positive correlation—Green; Negative correlation—Red). Correlation cutoff = 0.75
In both our COPD effect and combined COPD*HIV effect analyses, we identified Bacteroidetes Prevotella to be a top ASV correlated with features of the transcriptome and the epigenome. In the former analysis, Bacteroidetes Prevotella was correlated with genes WDR72, AKR1C2 and SETDB1, and methylation sites CpG-TIMP3;SYN3, CpG-UTP11L and CpG-PHACTR2; CpG-UTP11L was in turn correlated with genes WDR72 and AKR1C2, and CpG-TIMP3; SYN3 was correlated with gene WDR72. In the COPD*HIV analysis, Bacteroidetes Prevotella was correlated with genes FASTKD3, FUZ, and ACVR1B, and CpG-FUZ and CpG-PHLDB3.
Discussion
In support of the hypothesis that HIV infection is associated with alterations in the airway epithelial microbiome, our analysis showed several novel conclusions: (1) PLWH with COPD had significantly lower microbial diversity and a distinct microbial community in their airway epithelium compared to PLWH without COPD, HIV-negative COPD patients, and patients with neither HIV nor COPD; (2) microbial features that appeared disrupted in PLWH with COPD were correlated with methylation and transcriptomic alterations along genes not previously recognized to be part of the pathogenesis of HIV-associated COPD.
Our analysis of the microbiome revealed that relatively “healthier” individuals were enriched in characteristic phyla Fusobacteria (in COPD −, HIV − and COPD – HIV − groups) and Bacteroidetes (in the COPD − and COPD −HIV − groups), and that decreases in these phyla may be associated with disease. However, we found no significant differences in the relative abundance of other characteristic phyla Proteobacteria and Firmicutes, contrary to the findings of Sze et al. [23], Xu et al.[16], and Ramsheh et al. [24]. Lower in the taxonomic hierarchy, we noted differences in relative abundance of genera Prevotella, Selenomonas, Neisseria, Fusobacterium, Streptococcus and Veillonella in the group with both COPD and HIV. These microbes are known to be oral commensals and have previously been implicated in lower airway colonization and driving severity of disease in COPD [25,26,27]. Our results reinforce the notion that diversity and composition are important components of a “healthy” microbiome [28]. However, how exactly lung microbial dysbiosis translates to the clinical disease presentations observed in patients with HIV and COPD is still unclear. This relationship is likely multifactorial, with variations in host processes such as transcription, metabolism, and immunity contributing to a certain extent.
In light of this uncertainty, we carried out multi ‘omic integration to effectively combine information from three ‘omes (microbiome, methylome and transcriptome) and identified highly correlated ‘omic features that may be relevant in the combined COPD and HIV states. Our integrative analyses consistently identified the single microbiome feature Bacteroidetes Prevotella, reduced in relative abundance in PLWH with COPD. Although a widely studied microbe, the exact role of Prevotella in the respiratory system is still poorly understood. Certain pathobiontic strains have been implicated as promoters of subclinical inflammation, particularly through increased Th-17 cytokine expression [29]. Twigg et al.observed that long-term ART use (> 3 years), which would normally be associated with a “healthier” phenotype, was associated with decreased Prevotella abundance in nine PLWH [14]. On the other hand, Prevotella has also been described in terms of healthy microbial ecosystems. Within HIV-uninfected patients with COPD, airway epithelial abundance of Prevotella has been associated with increased lung function, reduced dyspnea scores and inflammation, and expression of epithelial genes involved in tight junction promotion [24]. Many of these properties of Prevotella may be due to its interactions with other microbes and its dynamic role within the respiratory ecosystem. Recent studies have shown that Prevotella may exert its anti-inflammatory effects by inhibiting cytokine production by other gram-negative bacteria like Haemophilus influenzae or may co-aggregate and form heterotrophic biofilms with microbes like Porphyromonas [30, 31]. Future research based on cell culture models can help determine the strain-specific phenotypic response of the host.
The power of multi ‘omics integration allows for greater insight into what impact microbial dysbiosis might have on the host airway response. For instance, Bacteroidetes Prevotella was highly correlated with the methylation and expression of specific genes in the COPD and COPD*HIV interaction effect multi ‘omic analyses. Moreover, we found that in PLWH with COPD, reduced Prevotella abundance was correlated with the increased methylation of CpG sites along the genes PHLDB3 and FUZ, and the decreased expression of gene FUZ and the increased expression of gene FASTKD3. The consistent appearance of FUZ in relation to Prevotella abundance in our multi ‘omic integration analyses suggests a strong relationship between these two features along both epigenetic and transcription pathways. FUZ encodes a planar cell polarity protein that plays a prominent role in ciliogenesis [32]. Within the lung, higher methylation and lower expression of FUZ has previously been associated with tumour promotion and poor prognosis in lung adenocarcinoma [33], a compelling finding given the known increased rate of lung cancer in PLWH [34]. In our analysis, FUZ expression was also positively correlated ACVR1B and PTPRF and inversely correlated with FASTKD3. ACVR1B, part of the transforming growth factor-beta family, has established associations with COPD pathogenesis, as an identified expression quantitative trait loci in COPD blood, sputum, and lung [35, 36] and as a causal gene in emphysema distribution [37]. While the associations of PTPRF and FASTKD3 with COPD are not as clear, links between these two genes and lung cancer prognosis [38, 39] suggest some degree of activity within the lung through their roles in apoptosis, cell growth and differentiation, and oncogenesis. Interestingly, PTPRF has been noted to have a role in regulating the assembly and contraction of actin and actomyosin, and formation of tight junctions, with potential barrier function against HIV entry into target cells [40]. Whether modulation of Prevotella abundance within the airway epithelium can subsequently affect the activity of these downstream genes in PLWH with COPD is unknown, but may be a worthy area for future study.
Although this study is one of the largest to evaluate the airway epithelial microbiome in HIV-associated COPD, it has several limitations. We showed that microbial disruptions are evident in the airways of PLWH with COPD, however this study was not designed to prove causation of airway injury. Future experimentation using cell culture models or germ-free animals mimicking HIV infection may provide greater insight into the direction of the microbe-gene relationships identified in this study. Second, since the majority of our cohort were receiving ART and had undetectable HIV plasma viral loads, these results may not reflect microbiome and gene changes observed in PLWH not on ART. Third, we collected brushings from only one upper lobe lung segment per patient and acknowledge that there may be regional variation in the microbiome which we would not have been able to detect. Fourth, we did not have independently verified records of the cohort’s previous exacerbation and antibiotic use history. Fifth, there were demographic differences between our study groups and future studies with greater numbers of female PLWH would be welcomed. Greater balance of concurrent asthma and bronchiectasis, as well as of lung function amongst the COPD subgroups, would also be beneficial. Finally, while a promising field to uncover novel biologic relationships, multi ‘omic integration brings further challenges. Different ‘omics datasets are often generated via varied technologies and platforms and the search for a “gold-standard” workflow for data filtering, normalization, and integration continues [21]. “Over-fitting” the data in these workflows is often a concern which can cause the predictive performance to suffer in other cohorts. Future work using a validation dataset can improve these shortcomings and provide better assurance of model performance [41].
Conclusions
In this study, we are able to demonstrate that multi ‘omics integration can yield new insights into the impact microbial dysbiosis can have in the airway epithelium of PLWH, identifying novel genes in the pathogenesis of HIV-associated COPD. These genes could be further explored as potential biomarkers or drug targets specific to PLWH.
Availability of data and materials
Microbiome sequencing data have been deposited into the National Center for Biotechnology Information’s Sequence Read Archive (SRA) (BioProject PRJNA965797). Methylation and transcriptome data have been deposited into the GEO database (GSE178809).
Abbreviations
- AEC:
-
Airway epithelial cell
- ART:
-
Antiretroviral therapy
- ANOVA:
-
Analysis of variance
- ASV:
-
Amplicon sequence variants
- COPD:
-
Chronic obstructive pulmonary disease
- DIABLO:
-
Data Integration Analysis for Biomarker discovery using Latent cOmponents
- FDR:
-
False discovery rate
- FEV1 :
-
Forced expiratory volume in one second
- FVC:
-
Forced vital capacity
- HIV:
-
Human immunodeficiency virus
- PCoA:
-
Principal coordinate analysis
- PERMANOVA:
-
Permutational multivariate analysis of variance
- PLWH:
-
People living with HIV
References
Cheung CC, Ding E, Sereda P, Yip B, Lourenco L, Barrios R, Montaner J, Hogg RS, Lima V, Moore DM. Reductions in all-cause and cause-specific mortality among HIV-infected individuals receiving antiretroviral therapy in British Columbia, Canada: 2001–2012. HIV Med. 2016;17(9):694–701.
Edwards JK, Cole SR, Breger TL, Rudolph JE, Filiatreau LM, Buchacz K, Humes E, Rebeiro PF, D’Souza G, Gill MJ, Silverberg MJ, Mathews WC, Horberg MA, Thorne J, Hall HI, Justice A, Marconi VC, Lima VD, Bosch RJ, Sterling TR, Althoff KN, Moore RD, Saag M, Eron JJ. Mortality among persons entering HIV care compared with the general U.S. population: an observational study. Annal Intern Med. 2021;174(9):1197–206.
Crothers K, Butt AA, Gibert CL, Rodriguez-Barradas MC, Crystal S, Justice AC, Veterans Aging Cohort 5 Project T. Increased COPD among HIV-positive compared to HIV-negative veterans. Chest. 2006;130(5):1326–33.
Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, Oursler KK, Rimland D, Gibert CL, Butt AA, Justice AC. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med. 2011;183(3):388–95.
Drummond MB, Kirk GD. HIV-associated obstructive lung diseases: insights and implications for the clinician. Lancet Respir Med. 2014;2(7):583–92.
Bigna JJ, Nansseu JR, Um LN, Noumegni SR, Sime PS, Aminde LN, Koulla-Shiro S, Noubiap JJ. Prevalence and incidence of pulmonary hypertension among HIV-infected people in Africa: a systematic review and meta-analysis. BMJ Open. 2016;6(8): e011921.
Triplette M, Justice A, Attia EF, Tate J, Brown ST, Goetz MB, Kim JW, Rodriguez-Barradas MC, Hoo GWS, Wongtrakool C, Akgün K, Crothers K. Markers of chronic obstructive pulmonary disease are associated with mortality in people living with HIV. AIDS (London, England). 2018;32(4):487–93.
Gingo MR, George MP, Kessinger CJ, Lucht L, Rissler B, Weinman R, Slivka WA, McMahon DK, Wenzel SE, Sciurba FC, Morris A. Pulmonary function abnormalities in HIV-infected patients during the current antiretroviral therapy era. Am J Respir Crit Care Med. 2010;182(6):790–6.
George MP, Kannass M, Huang L, Sciurba FC, Morris A. Respiratory symptoms and airway obstruction in HIV-infected subjects in the HAART era. PLoS ONE. 2009;4(7): e6328.
Fitzpatrick ME, Singh V, Bertolet M, Lucht L, Kessinger C, Michel J, Logar A, Weinman R, McMahon D, Norris KA, Vallejo AN, Morris A. Relationships of pulmonary function, inflammation, and T-cell activation and senescence in an HIV-infected cohort. AIDS (London, England). 2014;28(17):2505–15.
Norris KA, Morris A. Pneumocystis infection and the pathogenesis of chronic obstructive pulmonary disease. Immunol Res. 2011;50(2–3):175–80.
Norris KA, Morris A, Patil S, Fernandes E. Pneumocystis colonization, airway inflammation, and pulmonary function decline in acquired immunodeficiency syndrome. Immunol Res. 2006;36(1–3):175–87.
Lozupone C, Cota-Gomez A, Palmer BE, Linderman DJ, Charlson ES, Sodergren E, Mitreva M, Abubucker S, Martin J, Yao G, Campbell TB, Flores SC, Ackerman G, Stombaugh J, Ursell L, Beck JM, Curtis JL, Young VB, Lynch SV, Huang L, Weinstock GM, Knox KS, Twigg H, Morris A, Ghedin E, Bushman FD, Collman RG, Knight R, Fontenot AP. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am J Respir Crit Care Med. 2013;187(10):1110–7.
Twigg HL 3rd, Knox KS, Zhou J, Crothers KA, Nelson DE, Toh E, Day RB, Lin H, Gao X, Dong Q, Mi D, Katz BP, Sodergren E, Weinstock GM. Effect of advanced HIV infection on the respiratory microbiome. Am J Respir Crit Care Med. 2016;194(2):226–35.
Beck JM, Schloss PD, Venkataraman A, Twigg H 3rd, Jablonski KA, Bushman FD, Campbell TB, Charlson ES, Collman RG, Crothers K, Curtis JL, Drews KL, Flores SC, Fontenot AP, Foulkes MA, Frank I, Ghedin E, Huang L, Lynch SV, Morris A, Palmer BE, Schmidt TM, Sodergren E, Weinstock GM, Young VB. Multicenter comparison of lung and oral microbiomes of HIV-infected and HIV-uninfected Individuals. Am J Respir Crit Care Med. 2015;192(11):1335–44.
Xu S, Tsai A, Sze MA, Vucic EA, Shaipanich T, Harris M, Guillemi S, Yang J, Sinha S, Nislow C, Montaner J, Lam W, Lam S, Sin DD, Paul Man SF, Leung JM. Decreased microbiome diversity in the HIV small airway epithelium. Respir Res. 2018;19(1):140.
Leung JM, Yang CX, Tam A, Shaipanich T, Hackett TL, Singhera GK, Dorscheid DR, Sin DD. ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur Respir J. 2020;55(5):2000688.
Hernández Cordero AI, Yang CX, Yang J, Horvath S, Shaipanich T, MacIsaac J, Lin DT, Kobor MS, Guillemi S, Harris M, Lam W, Lam S, Montaner J, Man SFP, Sin DD, Leung JM. Airway aging and methylation disruptions in HIV-associated chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2022;206:150.
Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, Enright PL, Hankinson JL, Ip MS, Zheng J, Stocks J. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–43.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. vegan: Community Ecology Package. 2016 [cited; Available from: https://rdrr.io/rforge/vegan/.
Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ, Le Cao KA. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics. 2019;35(17):3055–62.
Lee AH, Shannon CP, Amenyogbe N, Bennike TB, Diray-Arce J, Idoko OT, Gill EE, Ben-Othman R, Pomat WS, van Haren SD, Cao KL, Cox M, Darboe A, Falsafi R, Ferrari D, Harbeson DJ, He D, Bing C, Hinshaw SJ, Ndure J, Njie-Jobe J, Pettengill MA, Richmond PC, Ford R, Saleu G, Masiria G, Matlam JP, Kirarock W, Roberts E, Malek M, Sanchez-Schmitz G, Singh A, Angelidou A, Smolen KK, Brinkman RR, Ozonoff A, Hancock REW, van den Biggelaar AHJ, Steen H, Tebbutt SJ, Kampmann B, Levy O, Kollmann TR. Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nat Commun. 2019;10(1):1092.
Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, Erb-Downward JR, Huffnagle GB, Hayashi S, Elliott WM, Cooper J, Sin DD, Lenburg ME, Spira A, Mohn WW, Hogg JC. The host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438.
Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Muller-Quernheim J, Greulich T, Nowinski A, Barta I, Stendardo M, Boschetto P, Korzybski D, Prasse A, Parr DG, Hohlfeld JM, Dome B, Welte T, Heath S, Gut I, Morrissey JA, Ziegler-Heitbrock L, Barer MR, Singh D, Brightling CE. Lung microbiome compsition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancert Microbe. 2021;2(7):E300–10.
Haldar K, George L, Wang Z, Mistry V, Ramsheh MY, Free RC, John C, Reeve NF, Miller BE, Tal-Singer R, Webb AJ, Brookes AJ, Tobin MD, Singh D, Donaldson GC, Wedzicha JA, Brown JR, Barer MR, Brightling CE. The sputum microbiome is distinct between COPD and health, independent of smoking history. Respir Res. 2020;21(1):183.
Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, Magid-Slav M, Miller BE, Ostridge KK, Patel R, Sathe G, Simola DF, Staples KJ, Sung R, Tal-Singer R, Tuck AC, Van Horn S, Weynants V, Williams NP, Devaster JM, Wilkinson TMA. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018;73(5):422–30.
Leitao Filho FS, Alotaibi NM, Ngan D, Tam S, Yang J, Hollander Z, Chen V, FitzGerald JM, Nislow C, Leung JM, Man SFP, Sin DD. Sputum microbiome is associated with 1-year mortality after chronic obstructive pulmonary disease hospitalizations. Am J Respir Crit Care Med. 2019;199(10):1205–13.
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–10.
Segal LN, Alekseyenko AV, Clemente JC, Kulkarni R, Wu B, Gao Z, Chen H, Berger KI, Goldring RM, Rom WN, Blaser MJ, Weiden MD. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome. 2013;1(1):19.
Larsen JM, Steen-Jensen DB, Laursen JM, Søndergaard JN, Musavian HS, Butt TM, Brix S. Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS ONE. 2012;7(2): e31976.
Engel M, Endesfelder D, Schloter-Hai B, Kublik S, Granitsiotis MS, Boschetto P, Stendardo M, Barta I, Dome B, Deleuze JF, Boland A, Müller-Quernheim J, Prasse A, Welte T, Hohlfeld J, Subramanian D, Parr D, Gut IG, Greulich T, Koczulla AR, Nowinski A, Gorecka D, Singh D, Gupta S, Brightling CE, Hoffmann H, Frankenberger M, Hofer TP, Burggraf D, Heiss-Neumann M, Ziegler-Heitbrock L, Schloter M, Zu CW. Influence of lung CT changes in chronic obstructive pulmonary disease (COPD) on the human lung microbiome. PLoS ONE. 2017;12(7): e0180859.
Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM, Wallingford JB, Finnell RH. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat Cell Biol. 2009;11(10):1225–32.
Chen ZS, Lin X, Chan TF, Chan HYE. Pan-cancer investigation reveals mechanistic insights of planar cell polarity gene Fuz in carcinogenesis. Aging. 2021;13(5):7259–83.
Sigel K, Wisnivesky J, Gordon K, Dubrow R, Justice A, Brown ST, Goulet J, Butt AA, Crystal S, Rimland D, Rodriguez-Barradas M, Gibert C, Park LS, Crothers K. HIV as an independent risk factor for incident lung cancer. AIDS (London, England). 2012;26(8):1017–25.
Castaldi PJ, Cho MH, Zhou X, Qiu W, McGeachie M, Celli B, Bakke P, Gulsvik A, Lomas DA, Crapo JD, Beaty TH, Rennard S, Harshfield B, Lange C, Singh D, Tal-Singer R, Riley JH, Quackenbush J, Raby BA, Carey VJ, Silverman EK, Hersh CP. Genetic control of gene expression at novel and established chronic obstructive pulmonary disease loci. Hum Mol Genet. 2015;24(4):1200–10.
Morrow JD, Cho MH, Platig J, Zhou X, DeMeo DL, Qiu W, Celli B, Marchetti N, Criner GJ, Bueno R, Washko GR, Glass K, Quackenbush J, Silverman EK, Hersh CP. Ensemble genomic analysis in human lung tissue identifies novel genes for chronic obstructive pulmonary disease. Hum Genomics. 2018;12(1):1.
Boueiz A, Pham B, Chase R, Lamb A, Lee S, Naing ZZC, Cho MH, Parker MM, Sakornsakolpat P, Hersh CP, Crapo JD, Stergachis AB, Tal-Singer R, DeMeo DL, Silverman EK, Zhou X, Castaldi PJ. Integrative genomics analysis identifies ACVR1B as a candidate causal gene of emphysema distribution. Am J Respir Cell Mol Biol. 2019;60(4):388–98.
Soulières D, Hirsch FR, Shepherd FA, Bordogna W, Delmar P, Shames DS, Klughammer B. PTPRF expression as a potential prognostic/predictive marker for treatment with erlotinib in non-small-cell lung cancer. J Thorac Oncol. 2015;10(9):1364–9.
Magraner-Pardo L, Gobelli D, de la Fuente MA, Pons T, Simarro M. Systematic analysis of FASTK gene family alterations in cancer. Int J Mol Sci. 2021;22(21):11337.
Xu WW, Han MJ, Chen D, Chen L, Guo Y, Willden A, Liu DQ, Zhang HT. Genome-wide search for the genes accountable for the induced resistance to HIV-1 infection in activated CD4+ T cells: apparent transcriptional signatures, co-expression networks and possible cellular processes. BMC Med Genomics. 2013;6:15.
Picard M, Scott-Boyer MP, Bodein A, Périn O, Droit A. Integration strategies of multi-omics data for machine learning analysis. Comput Struct Biotechnol J. 2021;19:3735–46.
Acknowledgements
The authors would like to thank the participants enrolled in this study for their generous contributions of bronchoscopic specimens.
Previous presentations
Portions of this work were presented at the American Thoracic Society 2020 and 2021 Meetings.
Funding
This study was funded by the Canadian Institutes of Health Research and the British Columbia Lung Association. CN hold a Canada Research Chair in Translational Genomics. AIHC is supported by MITACS, the Providence Airway Centre, the St. Paul’s Foundation, and the Michael Smith Foundation for Health Research trainee award. CC holds a Canada Research Chair in Occupational and Environmental Lung Disease. MSK holds a Canada Research Chair in Social Epigenomics. DDS holds a Canada Research Chair in COPD and the de Lazzari Family Chair at the Centre for Heart Lung Innovation. JML is supported by the Michael Smith Foundation for Health Research Health Professional Investigator Award and holds a Canada Research Chair in Translational Airway Biology.
Author information
Authors and Affiliations
Contributions
Study Design and Concept: MSJ, FSLF, TS, SL, WL, SFPM, DDS, JML. Data Acquisition: FSLF, JY, TS, DL, JMacIsaac, MSK, SS, CN, SG, MH, JMontaner, SFPM, DDS, JML. Data Analysis: MSJ, CXY, AIHC, JY, DL, JMacIsaac, MSK, SS, CN, SG, MH, RTN, CC, JMontaner, SFPM, DDS, JML. Manuscript Writing and Editing: MSJ, CXY, FSLF, AIHC, JY, TS, DL, JMacIsaac, MSK, SL, WL, SS, CN, SG, MH, JMontaner, CC, RTN, SFPM, DDS, JML. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed under the University of British Columbia Research Ethics Board Certificates H11-02713 and H15-02166. All participants provided written, informed consent.
Consent for publication
Not applicable.
Competing interests
These authors report the following disclosures: JML reports honoraria from the University of British Columbia and research grants from Genome BC, the Canadian Institutes of Health Research, the BC Lung Association, and the Michael Smith Foundation for Health Research. DDS reports speaking honoraria from AstraZeneca, Boehringer Ingelheim, and GSK. JMontaner reports research grants from Health Canada, the Public Health Agency of Canada, the BC Ministry of Health, Gilead Sciences, Merck, ViiV Healthcare, Genome BC, and the Canaidan Institues of Health Research. The remainder of the authors report no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Figure S1:
Multi ‘omic integration study design. Figure S2: 16S RNA gene copies/μL measured in airway epithelial cells. Figure S3: Principal coordinate plot by smoking status. Figure S4: Plot showing potential interactive effects of COPD and HIV status on Shannon Index, Faith PD, Bray Curtis PC1, and Bray Curtis PC2. Figure S5: Shannon Diversity Index differences between the COPD and HIV groups after removing 4 COPD- subjects administered with inhaled corticosteroids from the analysis. Figure S6: Principal coordinate plot showing microbial community structures among subject groups based on the Bray-Curtis metric, after removing 4 COPD- subjects administered with inhaled corticosteroids from the analysis. Figure S7: 16S RNA gene copies/μL measured in bronchial brushings and control specimens of HIV+ subjects. Table S1 Relative taxa abundance comparisons at the phylum level between the COPD+ and COPD-,HIV+ and HIV-, andCOPD-HIV-, COPD-HIV+, COPD+HIV- and COPD+HIV+ groups in AEC samples. Table S2: Relative taxa abundance comparisons at the genus level between the COPD+ and COPD-, HIV+ and HIV-, and COPD-HIV-, COPD-HIV+, COPD+HIV- and COPD+HIV+ groups in AEC samples. Table S3: Pairwise PERMANOVA comparisons between the different specimen types obtained from HIV+ subjects.
Additional file 2.
Containing DIABLO results by COPD status.
Additional file 3.
Containing DIABLO results by HIV status.
Additional file 4.
Containing DIABLO results by COPD and HIV status.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jude, M.S., Yang, C.X., Filho, F.S.L. et al. Microbial dysbiosis and the host airway epithelial response: insights into HIV-associated COPD using multi’omics profiling. Respir Res 24, 124 (2023). https://doi.org/10.1186/s12931-023-02431-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02431-4