
Abstract
Crop wild relatives harbor exotic and novel genetic resources, which hold great potential for crop improvement. Ipomoea imperati is a wild diploid relative of sweet potato with the capability of high salinity tolerance. We compared the transcriptomes of I. imperati under salt stress vs. control to identify candidate genes and pathways involved in salt response. De novo assembly produced 67,911 transcripts with a high depth of coverage. A total of 39,902 putative genes were assigned annotations, and 936 and 220 genes involved in salt response in roots and leaves, respectively. Functional analysis indicated a whole system response during salt stress in I. imperati, which included four metabolic processes: sensory initiation, transcriptional reprogramming, cellular protein component change, and cellular homeostasis regulation. We identified a number of candidate genes involved in the ABA signaling pathway, as well as transcription factors, transporters, antioxidant enzymes, and enzymes associated with metabolism of synthesis and catalysis. Furthermore, two membrane transporter genes, including vacuole cation/proton exchanger and inositol transporter, were considered to play important roles in salt tolerance. This study provided valuable information not only for understanding the genetic basis of ecological adaptation but also for future application in sweet potato and other crop improvements.
Similar content being viewed by others

Introduction
Soil salinity is one of the most severe constraints on global crop production. With the combined pressures of climate change and human population growth, salt tolerance is becoming an important agronomic trait to investigate for sustaining or even increasing the world’s food supply in marginal and high saline soils1. Extensive genetic and molecular studies on salt tolerance in glycophytic model plants and major crops such as Arabidopsis thaliana, rice, wheat, cotton and tomato2, 3, as well as in a number of halophytic species4,5,6, have been promising for elucidating molecular mechanisms of salt adaption. Hundreds of potential salt-responsive genes have been identified and analyzed to determine their expression patterns under salt treatments. These studies reveal molecular mechanisms in response to salt stress, including the salt overly sensitive (SOS) pathway, hormone signaling pathways, ion homeostasis, Reactive oxygen species (ROS) homeostasis, and osmotic regulation7,8,9.
Crop wild relatives typically have more genetic variations than domesticated crops, and thus provide plant breeders with a broader pool of potentially useful genetic resources10, 11. With the recent enormous progress in next generation sequencing technology, crop wild relatives have become a useful resource for developing salt and drought cultivars in many crops12,13,14. By the use of wide-hybridization and/or transgene techniques, several salt tolerance traits have been transferred from wild relatives into cultivated wheat and barley, and salt tolerance new cultivars have been successfully produced15, 16. Recently, wild halophyte rice, Porteresia coarctata, also has been used to improve rice salt tolerance17, 18.
Despite the important role of sweet potato (Ipomoea batatas) in global food and energy security, its traits improvement has been limited, including salt tolerance. Attempts to improve sweet potato salt tolerance have been made by introducing salt-tolerant candidate genes, such as, iron-sulfur cluster biosynthesis (NFU1) and pyrroline-5-carboxylate reductaserelates to proline metabolism (P5CR)19, 20. However, salt tolerance is a complex trait that has evolved independently by different mechanisms in diverse lineages21. Therefore, those salt tolerant candidate genes obtained from genetically distant species might not be effective in sweet potato. More study is needed to identify new markers and genes that can confer salt tolerance in sweet potato.
Sweet potato itself is salt sensitive, while its close wild relative, I. imperati, is a halophytic plant species22. I. imperati, known as beach morning glory, is a pantropical sandy coastal species. It grows in the sandy dune of coastal beaches and can toleratesaline and nutritionally poor soil conditions23. To our knowledge, few studies have investigated this species, and information about salt tolerance physiology, morphology, and molecular mechanism in beach morning glory is lacking. Interestingly, unlike most other halophytes, I. imperati lacks a salt gland or other specialized morphological traits to exclude salt. The adaptation to saline environments may be more exclusively attributed to the alteration in gene regulation2, 4, 24, which makes I. imperati extremely attractive for investigating its gene regulation strategies under salt stress.
In this study, we aimed to identify the candidate genes and pathways involved in response to salinity stress, using RNA-Seq analysis from the roots and leaves of I. imperati. The phenotypic and physiological responses to salt stress were studied under salt treatments and control at different time points. The transcriptomes from plants watered with saline or fresh water were compared to identify salt-responsive genes. Ultimately, the influence of crosstalk between gene expression and physiological responses were investigated to understand beach morning glory tolerance to salt stress and adaptation to saline habitats. The identified salt-responsive genes in the beach morning glory will shed light on the genetic basis of its ecological adaptation and thus they can be applied to developing salt-tolerant sweet potato cultivars.
Results
Phenotypic and physiological responses to salt stress in I. imperati
Plant growth
After the 5-days pre-treatment with serials of NaCl from 100 mM to 500 mM, the pre-treated plants were daily applied with 600 mM NaCl. There was little difference in appearance between the control and the pre-treated plants (shown as 0 h in Fig. 1), with the control plants growing healthily with 7 leaves, and the pre-treated plants growing a little slowly with 6 leaves. After one day treatment with 600 mM NaCl, we observed a slightly delayed development in the treated plants compared with control plants (Fig. 1); The new 8th leaf was developed in the control plants, while no new leaf emerged in the treated plants. After salt treatment for 72 h, in contrast to the control plants showing more new leaves and stolons, the treated plants developed relatively fewer new leaves and some were slightly curled without wilting. After salt treatment for 168 h (7 d), the control plants showed healthy growth with additional new leaves and stolons developed, while the treated-plants showed delayed development and the first-developed leaves turned yellowish and curled, but no wilting leaves were observed (Fig. 1). In summary, the treated plants showed relatively delayed growth in the treated plants than those control ones; however, no serious damage was observed under the high salinity condition (600 mM), such as leaf wilt or dehydration, suggesting a high salt tolerance capacity of beach morning glory.

Comparison of control (fresh water) and salt-treated (600 mM NaCl) plants of I. imperati at 0 h, 24 h, 72 h (3d) and 168 h (7d).
Ion content
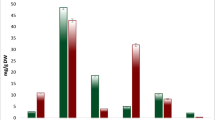
Na+ and K+ contents were measured in both roots and leaves (Fig. 2). The Na+ significantly increased in both leaves and roots during the salt stress, with equal 2-fold increases observed in the first day including 0 h, 3 h, and 24 h, and higher 4-fold increases observed at 168 h. There was no significant change in K+ content within one-day treatment in root, but there was a slight decrease after 168 h. The K+ content decreased in leaves, in equal amounts at four time points. The content of K+ did not change much during salt treatment, suggesting the uptake and transport of K+ was not significantly affected in I. imperati under high salinity conditions. There was little difference in K+/Na+ ratio between roots and leaves, with a significant decrease compared with the controls, and a dramatic decrease in 168 h. The K+/Na+ ratio did not significantly vary in 0 h, 3 h, and 24 h, suggesting that there might exist a rapidly responsive ion regulation mechanism to maintain K+ and Na+ homeostasis during early salt stress.

Ion content in control and salt-treated root and leave tissues of I. imperati. (A) Na+ content in control and salt-treatment roots and leaves. (B) K+ content in control and salt-treatment roots and leaves. (C) K+/Na+ Ratio in control and salt-treatment roots and leaves. R-C = control roots; R-T = salt-treatment roots; L-C = control leaves; L-T = salt-treatment leaves. Error bars represent SD.
Processing and mapping of Illumina reads
A total of 12 cDNA libraries were sequenced. RNA-seq analyses were performed on three independent biological replicates (each replicate represents pooled tissues collected from different time points) of each sample in this study (L-C, Leaf control; L-T, Leaf treatment; R-C, Root control; R-T, Root treatment). The reads generated by the Illumina Hiseq. 2500 were initially processed to remove adapter sequences and low-quality reads. Supplemental Table S1 summarizes the assembly metrics for each of the conditions. 252,166,154 reads from all root and leaf tissues were filtered removing ~20% of total sequence, and roughly 20 Gb of nucleotides were obtained. All sequences regardless of condition were pooled for assembly in an effort to make a suitable background reference. De novo transcript assembly produced a total of 94,728 transcripts. Redundant sequences were removed by clustering sequences via CD-HIT, resulting in 67,911 remaining sequences (Table S1).
Gene annotation
Conceptual translation of sequences produced 50,688 putative protein sequences, with 58.76% (39,902) annotated by a BLAST search against the NCBI non-redundant (NR) protein database. There are 1463 sweet potato proteins in the NCBI non-redundant (NR) protein database. We found 1297 genes (88.6%) with homologous matches (identity >60%) to sweet potato. The most commonly occurring matches were to genes from Solanum lycopersium.
Functional classification
Following their NR annotations, we mapped protein sequences onto the records from the GO database and retrieved 21,418 GO annotations. Blast2Go assigned 18,034 with terms of “Biological process”, 13,322 with terms of “cellular component”, and 17,134 with terms of “molecular functions” (Fig. S1). Among the “Biological process” category, the two most abundantly represented terms were “metabolic process” (13,120, 61.2%), and “cellular process” (11,815, 55.2%). There were also a large number of genes involved in “single-organism process” (9,608, 44.9%), “biological regulation” (4,040, 18.9%), and “response to stimulus” (3,140, 14.7%) (Figure S2). In the “Cellular component” category, most protein sequences were located in “cell” (9,099, 42.5%), “cell part” (9,095, 42.5%), “organelle” (6,826, 31.9%) and “membrane” (5,134, 24.0%). In the “Molecular function” category, “catalytic activity” (11,386, 53.2%) and “binding” (10,787, 50.4%) were predominantly represented. “Catalytic activity” was mainly represented by genes for “transferase activity”, “hydrolase activity”, and “kinase activity”, while “binding” was mainly represented by genes with “nucleotide binding” and “protein binding”.
Identification of differentially expressed genes in leaves and roots under salt stress
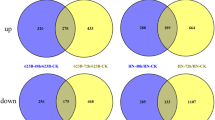
A cutoff FDR value ≤ 0.05 and |Log2 fold change (FC)| ≥ 1 was used for identifying differentially expressed genes (DEGs). 936 DEGs were found in roots and 220 DEGs in leaves under salt treatment (Fig. 3). Among those, only 36 DEGs were shared in the two tissue types, while 96.2% and 83.6% tissue-specific DEGs in roots and leaves, respectively. Interestingly, there were more DEGs in roots than in leaves, implying that roots might undergo more complicated transcriptional regulation under salt treatment. In roots, the number of upregulated DEGs was greater than downregulated DEGs (544 upregulated and 392 downregulated). In leaves, there were more downregulated DEGs than upregulated DEGs (75 upregulated and 145 downregulated).

Volcano plots of transcriptomes for salt-treated root and leaf tissues in halophyte I. imperati. For each plot, the x-axis represents log base 2 fold-change, and the y-axis represents −log base 10 FDR. (a) I. imperati leaf; (b) I. imperati root. The horizontal bars on all figures represents FDR = 0.05, red dots equate to gene expression with FDR <0.05, and blue dots are gene expression values with FDR > 0.05.
A total of 500 (53.4%) and 129 (58.6%) DEGs in roots and leaves, respectively, were annotated by Blast2Go. Although the number of DEGs differs between roots and leaves, the GO categories classifications were similar (Fig. S2). For example, some of the DEGs in roots and leaves shared GO terms, such as “Biological process”, “cellular process”, “metabolic process”, “single-organism process”, “biological regulation”, and “response to stimulus”. There were only two exceptions between the tissue types. The leave DEGs included some genes annotated with the term “signaling” but none of the DEGs from the roots had this term, while the root SEDs included genes annotated with the term “cellular component organization or biogenesis” but none of the DEGs from the leaf has this term. The significantly represented GO terms of “Cellular component” in DEGs of both roots and leaves were “intracellular”, “intracellular part”, “intracellular organelle”, “membrane-bounded organelle”, and “intrinsic component of membrane”. The only one exception was that “plasma membrane” represented in roots but not in leaves. The “Molecular function” categories were the same between roots and leaves, including “ion binding”, “organic cyclic compound binding”, “heterocyclic compound binding”, “oxidoreductase activity”, and “transferase activity binding”.
GO enrichment analysis in DEGs
To further understand the expression changes in these two tissues, we conducted GO enrichment analysis for the DEGs with the tomato genome database as the background using AgriGO. In roots, there were 18 enriched GO terms, including 10 of biological process, 6 of cellular components and 2 of molecular functions (Fig. 4). In leaves, there were 14 enriched GO terms, including 9 of biological process, 4 of cellular components and one of molecular functions (Fig. 4). Responses to stimulus, cell, and cell part were significantly overrepresented both in leaves and roots, while there were more categories overrepresented in root. Metabolic processes, biological regulation, catalytic activity and antioxidant activity were all activated in roots but not in leaves. Oxidoreductase activity may be activated in leaves more than roots. “Response to stimulus” (GO:0050896) was a significantly enriched GO term in both leaves and roots (Fig. S3). GO enrichment analysis reveals an enrichment of genes involved in plant responses to abiotic stimuli, external stimuli, chemical stimuli, organic substances, defense response, as well as to hormone stimuli both in leaves and roots. More significant GO terms related to “response to stimulus” were enriched in roots (Fig. S3), such as response to oxidative stress, response to inorganic substance, response to biotic stimulus, all of which imply that a more complicated physiological response occurs in roots than that in leaves during salt stress. Both roots and leaves showed a similar gene expression pattern in response to salt stress, but different gene families within the same categories were activated. For example, GO category “response to hormone stimulus” (GO:0009725) was enriched in both leaves and roots, however, only five DEGs were assigned to five GO categories in leaves, including GO:0009725, GO:0009734, GO:0009737, GO:0009738 and GO:0010104, while 22 DEGs were assigned to twelve GO categories in roots, including seven hormone stimulus GO categories (Table S2).

Enriched distributions of DEGs into GO categories (y-axis) according to GO enrichment analysis. x-axis: GO annotations; y-axis: percent of genes. Significance was determined by false discovery rate (FDR). (A) Leaf; (B) Root.
To provide further insight on which plant pathways play a role during salt stress response, we performed a pathway level analysis using GAGE. We first align the 39,902 gene sequences to the potato RefSeq proteins to identify genes with known KEGG identifiers. The reason to choose potato is that it is the most closely related species available in RefSeq. BLASTX alignment produced 22,672 unique matches (identifiers). GAGE uses Log2 transformed FPKM values as expression levels to determine which pathways were active. This type of analysis differs from a gene level analysis such as GO, as a whole pathway was considered. Table 1 shows the most significantly induced pathways in roots.
Candidate DEGs involved in response to salt stress
Based on function annotation, DEGs were allocated into several biological processes, involved in ion homeostasis, ROS scavenging, hormone metabolism, signal transduction, amino metabolism, carbohydrate metabolism, and transcription factor regulation. Table S3 highlights the most significant DEGs based on functional categories that were candidate salt responsive genes.
DEGs include components of known salt-related signaling pathways, such as Protein Phosphatase 2C (PP2Cs), serine/threonine kinases (SnRKs), which were critical components of the ABA-signaling pathway, and EIN2 and EIN3 which participate in the ethylene signaling pathway. Some genes encoding for Receptor-like Kinase (RLK) were found, such as LRR receptor-like serine/threonine-protein kinase, receptor-like protein kinase HAIKU2-like.In our study, 36 Transcription Factors (TFs) were identified. The most abundant classes included AP2/EREBP, bHLH, HD-ZIP and MYB. The existence of various TFs in roots responding to salt stress was noteworthy.
We found many transcripts that encode a diversity of ion transporters including cation/H+ antiporter, vacuolar cation/proton exchanger, and root-specific metal transporter. We also found DEGs associated with solute transporters that were either upor downregulated during salt stress. These transporters included sugar transporters, amino acid transporters, and peptide transporters.
A number of antioxidant enzymes were detected involved in ROS scavenging during salt stress. Most of these antioxidant enzymes genes were highly activated in roots but not in leaves after salt treatment. 11 members of POX family were upregulated, 9 members of the glutathione S-transferase (GST) genes were significantly upregulated, and three respiratory burst oxidase homologs (RBOHs) genes were significantly downregulated. In addition, we found highly differentially expressed genes in roots that were involved in cell wall organization or biogenesis including xyloglucan endotransglucosylase/hydrolase, cellulose synthase, expansin genes and pectin methylesterase. Genes from the biosynthesis of cutin and wax pathway were also identified, as well as various stress-responsive proteins, such as osmotin-like protein and Heat Shock Proteins. Moreover, some genes involving in plant-pathogen interaction were also identified significantly upor down regulated, such as chitinase, pathogenesis-related protein.
Validation of the RNA-Seq and gene expression analysis by real-time qRT-PCR
Fifteen DEGs were randomly selected from a range of expression values for qRT-PCR analysis using the biological replicates of the RNAs previously used for RNA-Seq (Table S4). The analysis revealed a similar expression pattern of all the selected genes in real-time PCR analysis as observed from RNA-Seq data (R2 = 0.903) (Fig. 5). The validation experiments support the reliability of the relative values provided by the RNA-Seq analysis.

Validation of differential gene expression results obtained by RNA-Seq. The correlation of gene expression results obtained from qRT-PCR analysis and RNA-Seq for 15 random selected genes is shown.
Furthermore, twenty candidate genes involved in salt tolerance were further investigated by qRT-PCR at different time points to confirm the expression profiles under salt stress in both leaves and roots (Table S4). In most cases, the expression changes were more dramatic in roots than in leaves (Fig. 6). For roots, the selected DEGs were significantly upregulated at 3 h and/or 24 h post-salt stress implying that the expression pattern in roots changed rapidly after salinity stress. Among eight transporter transcripts, the transcripts from CAX and INT genes (Fig. 6N,T), show a dramatic change in roots. High salinity results in a dramatic increase in gene expression at 24 h, with the expression of CAX and INT increasing by 22-fold and 53-fold respectively. The transcripts associated with ABA signaling pathway and ABA biosynthesis were found upregulated in roots and/or in leaves (Fig. 6A–F). Transcription factors bHLH and ILR3 all show upregulation in both leaves and roots, with the highest expression of bHLH at 3 h and the highest expression of ILR3 at 72 h (Fig. 6K,L).

Selected expression profiles were validated with quantitative qRT-PCR in roots (green line) and leaves (blue line) of I. imperati. x-axis, five yantime point. y-axis, relative expression represented by mean fold changes of expression obtained from three biological replicates. Error bars represent SD.
Discussion
The overall salt tolerance of a plant is determined by integrated effects of many different mechanisms9, 25. Under high salt treatment (600 mM NaCl), I. imperati mobilizes many genes related to a variety of biological processes as part of a whole cellular responsive system. As summarized in Fig. 7, the whole responsive system is composed of four separate and dependent processes, including sensory system initiation, transcriptional reprogramming, cellular protein components change, and cellular homeostasis regulation.

Schematic summaries of the whole response system activated in response to salt stress in I. imperati. Salt stress is first perceived by ABA signaling pathway or RLKs, then the activated signaling pathway regulates transcriptional level reprogramming, resulting in gene expression and cellular protein components alteration. The activation of cellular homeostasis mechanisms for ion homeostasis, osmotic balance and redox balance: ABA, ABA signaling pathway; RLK, receptor-like kinase; TFs, transcription factors; ROS, reactive oxygen species.
Signals of salt stress can be detected by receptors and transmitted by signaling pathways that trigger transcriptional reprogramming by stress-activated transcription factors26, 27. Sensory system and signaling pathways play a key role in salt tolerance7, 28. Studies focusing on glycophytes have revealed the involvement of several signal transduction pathways in salt stress response, including SOS pathway29, 30, the ABA signaling pathway, the ethylene signaling pathway, the calcium signaling pathway, as well as the receptor-like kinase (RLK)31,32,33,34. Our study suggests that hormones might play an important role in salt signaling in I. imperati, including differentially expressed hormone receptors such as auxin receptor, ethylene receptor and ABA receptors (Table S3). The involvement of an ABA pathway also seems plausible. All four components were differentially expressed in roots, including ABA sensor PYL, negative regulator PP2C, positive regulators SnRK and AREB. Also, a number of signaling proteins, such as LRR receptor-like serine/threonine-protein kinase, were upregulated under salt stress.
Accordingly, we saw a change in cellular protein components including various enzymes involved in synthesis and catalysis processes, various transporters, and antioxidant enzymes (Fig. 7). Many metabolic processes were activated in I. imperati, including catabolic process, carbohydrate metabolic process, small molecule metabolic process, and oxidation reduction. Activations of these metabolic processes or pathways might be involved in the production of salt-induced ROS or signal molecules, and synthesis of stress-related metabolites, such as hormone, multiple osmolytes, cell wall components, etc. ref. 35. The transport system was also highly activated with transcripts encoding a diversity of transporters showing differential expression. Previous studies show antioxidative response mechanisms were effective in ROS homeostasis, especially in halophytes5, 6. The antioxidant defense system was markedly influenced in I. imperati with many genes associated with antioxidant enzyme activity showing different expression patterns under salt stress.
Many of these active proteins mobilize metabolic processes and structural changes that participate in the maintenance of osmotic, ion and redox balance (Fig. 7). For example, ion transporters including cation/H+ antiporter, vacuolar cation/proton exchanger, root-specific metal transporter, etc., may be responsible for selective accumulation of ions, control of ion uptake and compartmentalization36, 37. Cellular osmotic balance related salt tolerance relies on many kinds of compatible solutes and small molecule osmoprotectants, such as proline, inositol, sugars and betaines38. The solute transporters including sugar transporters, amino acid transporters, and peptide transporters, may be responsible for regulating cellular osmotic balance. The POX and GST gene families in roots might function in protecting I. imperati against oxidative damage.
Further investigation of the strategies used for salt tolerance in beach morning glory reveals a few halophytic-specific genes that might contribute to salt tolerance. Two genes encoding cation transporter (vacuolar cation/proton exchanger, CAX), and sugar transporter (inositol transporter, INT) were significantly upregulated (2 to 50-fold) during salt stress. These two genes thus might be important in the response to high salinity in I. imperati (Fig. 6N and T).
Cation transporters play major roles in maintaining ion homeostasis9, 39. The Na+ transporters NHX-type transporter were involved in Na+ accumulation in the vacuoles during salt exposure and have been identified in many plants40,41,42. It is interesting that NHX genes, well-studied in Arabidopsis and other species, were detected but did not show significantly differential expression between salt-treated plants vs. control plants in our study. This indicates that NHX genes might express constitutively high without a need of further activation during salt stress in this species. This can be tested by the comparison of their expression differences between beach morning glory vs. a glycophyte relative without salt stress. Further comparative gene expression investigation between I. imperati and its glycophytic relatives is needed. We found an alternative cation transporter gene encoding a putative vacuolar cation/proton exchanger, showing significant upregulation in salt-treated roots. This gene belongs to another cation transporter, cation/proton exchanger (CAX) family. CAX antiporter located in both the plasma membrane and vacuolar membrane is thought to be an important member for ion homeostasis in plant cells, but only a small number of CAX genes have been functionally characterized43. CAX antiporters transport multiple cations out of plasma membrane into the vacuole. CAX was involved in ion regulation and has been identified from a few plants44, 45. Luo et al.46 found that a soybean GmCAX1 could improve salt tolerance in transgenic Arabidopsis. We found that CAX gene expressions were maintained at high levels during salt stress, reaching a dramatic 20-fold increase (Fig. 6N). This cation transporter gene might contribute to the process of Na+ and K+ homeostasis.
The accumulation of inositols and/or its methylated derivatives were found in several halophytic species under salt stress, suggesting an important role for these compounds in salt tolerance47, 48. The importance of INT in stress signaling is known in the common ice plant (Mesembryanthemum crystallinum). In this well-studied halophyte, Myo-inositol (MI) accumulation and enhanced INT show as an adaptive osmoregulatory response to salt. Also, sodium uptake and accumulation to a high concentration in vacuoles were dependent on the presence of MI49, 50. Chauhan et al.51 found that MI transport in the tonoplast of M. crystallinumactually acts as a Na+/myo-inositol symporter system, removing Na+ from root vacuoles. We discover a putative INT gene significantly upregulated both in roots and leaves, and higher expression in roots than that in leaves. The expression of INT was 52-fold upregulated within 24 h (Fig. 6T), suggesting a rapid response to high salinity stimuli. Moreover, activation of several genes in the inositol synthesis pathway occurs during salt stress in some halophytic species49, 52. In the common ice plant, myo-inositol 1-phosphate synthase (INPS1) was upregulated in leaves49, 52. A few studies have demonstrated that engineering of some genes, such as those involved in inositol metabolic pathway in the halophytic wild rice Porteresia, can confer salinity tolerance in cultivated rice53. In contrast, Arabidopsis showed no upregulation of these genes or increases in inositol-derived compounds, which suggested that the strategy of osmoprotection was some what different between glycophyte and halophyte52. In our study, inositol-3-phosphate synthase (comp40237_c0_seq. 1, INPS3) was shown highly upregulated in leaves but not in roots based on the transcriptomes of this species, suggesting that the inositol synthesis might be activated in leaves in response to salt stress. Further studies are needed to investigate the functions of inositol transporter and inositol biosynthesis genes in I. imperati.
In recent years, various halophytic species have been used as sources to improve salt tolerance in many crops3, 12. However, conventional breeding aimed at trait improvement has been limited in sweet potato due to its large genome size, hexaploidy, male sterility, and self-incompatibility. There is potential to deliver substantial improvement in salt tolerance from its close wild halophytic relatives. In this study, we utilized a closely-related diploid wild-relative of sweet potato, I. imperati, which offers distinct salt tolerance capacity as a genetic resource to study salt adaptation and improve salt tolerance in sweet potato. Using transcriptome analysis by RNA-Seq, we successfully identified 67,911 putative genes in this species. Among these sequences, 1120 genes were differentially induced by salt stress. These salt-responsive genes with various functions such as ion transport, osmolytes, signal transduction, antioxidant enzymes, biosynthesis enzymes (Table S3), as we discussed above, might play key roles in conferring salt tolerance in I. imperati. Most of those genes involved in salt tolerance mechanisms may be evolutionarily conserved and important for conferring salt tolerance in different plant lineages54. The strategies used for salt tolerance in beach morning glory are partly in accordance with the hypothesis that salt tolerance mechanisms are largely conserved in halophytes and glycophytes, and that the large variations in their tolerance arise from subtle differences in the regulation of the same basic set of genes2. On the other hand, this de novo RNA sequencing assembly approach allowed us to explore novel roles of known genes as well as assess the roles of new genes, so-called orphan genes, taxonomically distinct genes that lack a distinguishable homology to unrelated species, in salt tolerance55. Orphan genes often contribute novel, lineage specific functionality under extreme abiotic and biotic stresses56. In the present study, there were 28,009 (41.24%) putative genes identified in I. imperati that have no matches to the NCBI sequence database. Of these unknown sequences, 129 were upregulated and 148 were downregulated, respectively. Although it is difficult to discern if these sequences encode functional proteins without further investigation, the high levels of expression following salt stress suggest that these orphan genes may play important roles in conferring salt tolerance57. This highlights the potential to discover novel species-specific genes and pathways in beach morning glory.
In conclusion, we characterized a comprehensive transcriptome of a wild halophytic sweet potato, I. imperati, using the Illumina sequencing technology. The results suggest that extensive genetic regulation under salt stress is responsible for salt tolerance in this halophytic species. The candidate salt-responsive genes identified in I. imperati contain both previously reported salt-responsive genes and novel genes that can be used as a new resource for genetic engineering in sweet potato or other crops. Meanwhile, a great number of orphan genes might have the great potential to discover novel species-specific or lineage specific salt tolerance mechanisms. The transcriptome analyses presented in this study not only provide plenty of bioinformatics data but also expand the vision of the genetic basis underlying adaptation for beach sandy dune.
Materials and Methods
Plant material and salt stress conditions
Seeds of I. imperati were collected from St. George Island, Florida, USA. All seeds were scarified before germination. The germinated seeds were transferred to soil in pots, and grown at 28 °C under long day conditions in a 16 h light/8 h dark cycle in an environmental chamber (Model E-41L2, Percival Scientific, USA) with 50% humidity and 600 micromoles/m2/s light intensity. For salinity stress treatment, two–weeks-old seedlings were watered with NaCl solution from 100 mM to 500 mM for 5 days to acclimate to salt soil. On day 6, 600 mM NaCl was applied daily for 7 days. Meanwhile, seedlings watered with freshwater were used as controls. Three biological replicates of roots and leaves from both treated (600 mM NaCl) and control (fresh water) plants were harvested at 0 h, 3 h, 24 h, 3 days, and 7 days after the treatment, respectively. All samples were flash frozen in liquid nitrogen and stored at −80 °C for RNA extraction.
Ion content measurement
Control and salt-treated leaves and roots were harvested at 0 h, 3 h, 24 h and 7 days. Tissues were rinsed with deionized water and dried at 65 °C for 2 days.50 milligrams of dried leavesandrootsmaterial was milled to powder, then extracted with 5 ml of 0.1 M HNO3 for 90 min at 95 °C and then filtered through Whatman filter paper. Na+ and K+ contents in the solutions were determined with a Varian Spectra AA-10 atomic absorption spectrophotometer.
RNA extraction, cDNA library preparation, and RNA-Seq
Tissues from all time points (0 h, 3 h, 24 h, 3 days, and 7 days) were powdered and equal amounts of tissue (by weight) were pooled as one biological replicate for RNA preparation; three replicates were prepared for treatment and control resulting in 12 RNA libraries. The pooled samples were denoted as L-C (Leaves control), L-T (Leaves treated), R-C (Roots control) and R-T (Roots treated) according to the leaves and roots with salt or water treatments. Total RNA was extracted using RNeasy Mini Kit (Qiagen, USA) following the supplied protocol and treated with DNase I (Thermo Scientific, USA). The quantity and integrity of the total RNA were determined using an Agilent 2100 bioanalyzer (Agilent, USA), and found to be RIN >9. The 12 RNA libraries were prepared using Illumina TruSeqRNA sample Preparation Kit (Illumina, USA). The quality of cDNA libraries were determined by applying the DNA 1000 chip on the Bioanalyzer 2100 (Agilent, USA). Twelve normalized cDNA libraries were constructed and sequenced using the Illumina Hiseq. 2500 platform (North Carolina State University) to generate 100 bp paired-end reads.
Transcriptome de novo assembly, annotation and differentially gene expression analysis
Sequence reads were filtered using the Fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) for quality and adapter sequences were removed using the fastq_quality_trimmer. Trimmed reads were combined across all conditions and de novo assembled via Trinity58 using default settings in order to build a suitable set of reference contigs. Trinity contigs of high similarity were clustered into groups with CD-HIT-EST and a single representative from each cluster was used as a reference sequence for read alignment. For annotation purposes, sequences from each CD-HIT cluster were transdecoded into predicted proteins using Transdecoder (http://transdecoder.github.io)59. Only proteins greater than 100 amino acids in length were retained for further annotating. Sequences were initially annotated by blasting nucleotide sequences against the NCBI NR database using BLASTX. Sequences were annotated with Gene Ontology categories (GO, http://www.geneontology.org) using Blast2Go60. Read count tables were produced via SAMtools61, from each biological replicate, the transcript abundance of each gene was estimated by fragments per kilobase of exon per million fragments mapped (FPKM). FPKM values were calculated and RSEM was used for counting the mapped reads. Expression levels of each tissue were compared between the treated and control samples to find DEGs using the edgeR R package62. A false-discovery rate (FDR) of 0.05 and |Log2 fold change| ≥ 1were used as the threshold to identify differentially expressed genes (DEGs).
Differentially expressed genes abundance analysis
The DEGs were subjected to GO enrichment analysis using AgriGO software (http://bioinfo.cau.edu.cn/agriGO/)63 (cutoffs: FDR <0.05) with Singular Enrichment Analysis (SEA) performing. DEGs from the roots or leaves were the input list, while the reference background was tomato genome database (Solanum lycopersicum iTAG2.3), which is a closely related species.
To identify relevant KEGG pathways, we aligned the sequences to an annotated NCBI RefSeq dataset of a closely related species, potato (Solanum tuberosum, downloaded from NCBI’s FTP site ftp://ftp.ncbi.nih.gov/genomes/refseq/plant/Solanum_tuberosum/representative/GCF_000226 075.1_SolTub_3.0/). Sequences were aligned via Blastx to assign a Refseq ID annotation. These IDs along with their corresponding read count data (FPKM values) were subjected to GAGE analysis after log transformation64 to identify potential differentially expressed pathways. GAGE performs gene set enrichment by assigning KEGG IDs to each matching Ref-Seq hit and then identifies and ranks the most enriched pathways based on fold change differences between conditions. Identified pathways were then visualized using Pathview65.
Validation of RNA-seq and gene expression analysis by real-time PCR (qRT-PCR)
In order to prove the reliability of the RNA-Seq data, qRT-PCR was performed on the same RNA pools that had been previously used for RNA-Seq. Fifteen DEGs, which were shown significantly differential expression in roots and/or leaves, were randomly selected for qRT-PCR analysis (Table S4). Furthermore, to verify the differential expression, qRT-PCR was performed on a new set of 3 replicates for each sample at different time points. A set of 20 DEGs in response to salt stress was chosen from different categories, including eight transcripts encoding transporters, six transcripts belonged to components of ABA signaling pathway and ABA biosynthesis, three transcripts belong to components of ethylene signaling pathway, two transcripts encoding transcription factors, and one transcript encoding a responsive protein (Table S4). The gene-specific primers were designed based on the sequencing data using the Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast). The actin gene of I. imperati was used as an internal control. The gene description and abbreviation and primer sequences used in this study were listed in Table S4.
The first-strand cDNAs were synthesized from 1 ug of DNase-treated total RNA using RevertAid RT Reverse Transcription Kit (Thermo Scientific, USA). qRT-PCR was performed using ABI 7500 fast Real-Time PCR system (Applied Biosystems, USA) and SYBR Green FastMix (Quanta Biosciences, USA). Reactions were performed in triplicate and contained 200 ng of cDNA, 10 uM of each primer. Amplifications were performed under the following conditions: 95 °C for 5 min, followed by 40 cycles of 95 °C for 3 s, 60 °C for 30 s. Relative expression for each sample was calculated using the 2−ΔΔCt methods with normalization to the internal control genes.
References
Flowers, T. J. & Muscolo, A. Introduction to the Special Issue: Halophytes in a changing world. AoB Plants 7, doi:10.1093/aobpla/plv020 (2015).
Zhu, J. K. Plant salt tolerance. Trends Plant Sci 6, 66–71, doi:10.1016/S1360-1385(00)01838-0 (2001).
Flowers, T. J. Improving crop salt tolerance. J Exp Bot 55, 307–319, doi:10.1093/jxb/erh003 (2004).
Flowers, T. J. & Colmer, T. D. Plant salt tolerance: adaptations in halophytes. Ann Bot-London 115, 327–331, doi:10.1093/aob/mcu267 (2015).
Jithesh, M. N., Prashanth, S. R., Sivaprakash, K. R. & Parida, A. K. Antioxidative response mechanisms in halophytes: their role in stress defence. J Genet 85, 237–254, doi:10.1007/Bf02935340 (2006).
Bose, J., Rodrigo-Moreno, A. & Shabala, S. ROS homeostasis in halophytes in the context of salinity stress tolerance. J Exp Bot 65, 1241–1257, doi:10.1093/jxb/ert430 (2014).
Zhu, J. K. Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53, 247–273, doi:10.1146/annurev.arplant.53.091401.143329 (2002).
Munns, R. & Tester, M. Mechanisms of salinity tolerance. Annu Rev Plant Biol 59, 651–681, doi:10.1146/annurev.arplant.59.032607.092911 (2008).
Deinlein, U. et al. Plant salt-tolerance mechanisms. Trends Plant Sci 19, 371–379, doi:10.1016/j.tplants.2014.02.001 (2014).
Hajjar, R. & Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 156, 1–13, doi:10.1007/s10681-007-9363-0 (2007).
Zhang, H., Mittal, N., Leamy, L. J., Barazani, O. & Song, B. H. Back into the Wild – Apply untapped genetic diversity of wild relatives for crop improvement. Evolutionary Applications (2016).
Colmer, T. D., Flowers, T. J. & Munns, R. Use of wild relatives to improve salt tolerance in wheat. J Exp Bot 57, 1059–1078, doi:10.1093/jxb/erj124 (2006).
Flowers, T. J., Flowers, S. A., Hajibagheri, M. A. & Yeo, A. R. Salt Tolerance in the Halophytic Wild Rice, Porteresia-Coarctata Tateoka. New Phytol 114, 675–684, doi:10.1111/j.1469-8137.1990.tb00439.x (1990).
Nevo, E. & Chen, G. X. Drought and salt tolerances in wild relatives for wheat and barley improvement. Plant Cell Environ 33, 670–685, doi:10.1111/j.1365-3040.2009.02107.x (2010).
Chen, S. Y. et al. Introgression of salt-tolerance from somatic hybrids between common wheat and Thinopyrum ponticum. Plant Sci 167, 773–779, doi:10.1016/j.plantsci.2004.05.010 (2004).
Garthwaite, A. J., von Bothmer, R. & Colmer, T. D. Salt tolerance in wild Hordeum species is associated with restricted entry of Na+ and Cl− into the shoots. J Exp Bot 56, 2365–2378, doi:10.1093/jxb/eri229 (2005).
Sengupta, S. & Majumder, A. L. Insight into the salt tolerance factors of a wild halophytic rice, Porteresia coarctata: a physiological and proteomic approach. Planta 229, 911–929, doi:10.1007/s00425-008-0878-y (2009).
Garg, R. et al. Deep transcriptome sequencing of wild halophyte rice, Porteresia coarctata, provides novel insights into the salinity and submergence tolerance factors. DNA Res 21, 69–84, doi:10.1093/dnares/dst042 (2014).
Wang, L. J. et al. Molecular Cloning and Functional Characterization of a Salt Tolerance-Associated Gene IbNFU1 from Sweetpotato. J Integr Agr 12, 27–35, doi:10.1016/S2095-3119(13)60202-6 (2013).
Liu, D. G. et al. Overexpression of IbP5CR enhances salt tolerance in transgenic sweetpotato. Plant Cell Tiss Org 117, 1–16, doi:10.1007/s11240-013-0415-y (2014).
Bromham, L. Macroevolutionary patterns of salt tolerance in angiosperms. Ann Bot 115, 333–341, doi:10.1093/aob/mcu229 (2015).
Miller, R. E., Rausher, M. D. & Manos, P. S. Phylogenetic systematics of Ipomoea (Convolvulaceae) based on ITS and waxy sequences. Syst Bot 24, 209–227, doi:10.2307/2419549 (1999).
Lonard, R. I. & Judd, F. W. The biological flora of coastal dunes and wetlands. Ipomoea imperati (Vahl) Griseb. J Coastal Res 15, 645–652 (1999).
Shu, S. et al. De novo sequencing and transcriptome analysis of Wolfiporia cocos to reveal genes related to biosynthesis of triterpenoids. Plos One 8, e71350, doi:10.1371/journal.pone.0071350 (2013).
Hey, S. J., Byrne, E. & Halford, N. G. The interface between metabolic and stress signalling. Ann Bot-London 105, 197–203, doi:10.1093/aob/mcp285 (2010).
Agarwal, P. K. & Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol Plantarum 54, 201–212, doi:10.1007/s10535-010-0038-7 (2010).
Cui, M. H. et al. An Arabidopsis R2R3-MYB transcription factor, AtMYB20, negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance. Febs Lett 587, 1773–1778, doi:10.1016/j.febslet.2013.04.028 (2013).
Golldack, D., Li, C., Mohan, H. & Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Frontiers in plant science 5, 151, doi:10.3389/fpls.2014.00151 (2014).
Qiu, Q. S. et al. Regulation of vacuolar Na+/H+exchange in Arabidopsis thaliana by the salt-overly-sensitive (SOS) pathway. J Biol Chem 279, 207–215, doi:10.1074/jbc.M307982200 (2004).
Martinez-Atienza, J. et al. Conservation of the salt overly sensitive pathway in rice. Plant Physiol 143, 1001–1012, doi:10.1104/pp.106.092635 (2007).
Raghavendra, A. S., Gonugunta, V. K., Christmann, A. & Grill, E. ABA perception and signalling. Trends Plant Sci 15, 395–401, doi:10.1016/j.tplants.2010.04.006 (2010).
Cao, Y. R., Chen, S. Y. & Zhang, J. S. Ethylene signaling regulates salt stress response: An overview. Plant Signal Behav 3, 761–763 (2008).
Knight, H., Trewavas, A. J. & Knight, M. R. Calcium signalling in Arabidopsis thaliana responding to drought and salinity. Plant J 12, 1067–1078 (1997).
Ouyang, S. Q. et al. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J 62, 316–329, doi:10.1111/j.1365-313X.2010.04146.x (2010).
Sanchez, D. H., Siahpoosh, M. R., Roessner, U., Udvardi, M. & Kopka, J. Plant metabolomics reveals conserved and divergent metabolic responses to salinity. Physiol Plantarum 132, 209–219, doi:10.1111/j.1399-3054.2007.00993.x (2008).
Serrano, R. & Rodriguez-Navarro, A. Ion homeostasis during salt stress in plants. Curr Opin Cell Biol 13, 399–404 (2001).
Zhu, J. K. Regulation of ion homeostasis under salt stress. Current opinion in plant biology 6, 441–445 (2003).
Gagneul, D. et al. A reassessment of the function of the so-called compatible solutes in the halophytic Plumbaginaceae Limonium latifolium. Plant Physiol 144, 1598–1611, doi:10.1104/pp.107.099820 (2007).
Shabala, S. & Mackay, A. Ion Transport in Halophytes. Adv Bot Res 57, 151–199, doi:10.1016/B978-0-12-387692-8.00005-9 (2011).
Apse, M. P., Aharon, G. S., Snedden, W. A. & Blumwald, E. Salt tolerance conferred by overexpression of a vacuolar Na+/H+antiport in Arabidopsis. Science 285, 1256–1258, doi:10.1126/science.285.5431.1256 (1999).
Shi, H. Z., Ishitani, M., Kim, C. S. & Zhu, J. K. The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. P Natl Acad Sci USA 97, 6896–6901, doi:10.1073/pnas.120170197 (2000).
Blumwald, E. Sodium transport and salt tolerance in plants. Curr Opin Cell Biol 12, 431–434 (2000).
Shigaki, T., Rees, I., Nakhleh, L. & Hirschi, K. D. Identification of three distinct phylogenetic groups of CAX cation/proton antiporters. J Mol Evol 63, 815–825, doi:10.1007/s00239-006-0048-4 (2006).
Hirschi, K. D., Zhen, R. G., Cunningham, K. W., Rea, P. A. & Fink, G. R. CAX1, an H+/Ca2+ antiporter from Arabidopsis. P Natl Acad Sci USA 93, 8782–8786, doi:10.1073/pnas.93.16.8782 (1996).
Ueoka-Nakanishi, H. & Maeshima, M. Quantification of Ca2+/H+ antiporter VCAX1p in vacuolar membranes and its absence in roots of mung bean. Plant Cell Physiol 41, 1067–1071, doi:10.1093/pcp/pcd023 (2000).
Luo, G. Z. et al. A putative plasma membrane cation/proton antiporter from soybean confers salt tolerance in Arabidopsis. Plant Mol Biol 59, 809–820, doi:10.1007/s11103-005-1386-0 (2005).
Liu, X. A. & Grieve, C. Accumulation of Chiro-inositol and Other Non-structural Carbohydrates in Limonium Species in Response to Saline Irrigation Waters. J Am Soc Hortic Sci 134, 329–336 (2009).
Slama, I., Abdelly, C., Bouchereau, A., Flowers, T. & Savoure, A. Diversity, distribution and roles of osmoprotective compounds accumulated in halophytes under abiotic stress. Ann Bot-London 115, 433–447, doi:10.1093/aob/mcu239 (2015).
Nelson, D. E., Rammesmayer, G. & Bohnert, H. J. Regulation of cell-specific inositol metabolism and transport in plant salinity tolerance. Plant Cell 10, 753–764 (1998).
Nelson, D. E., Koukoumanos, M. & Bohnert, H. J. Myo-inositol-dependent sodium uptake in ice plant. Plant Physiol 119, 165–172 (1999).
Chauhan, S. et al. Na+/myo-inositol symporters and Na+/H+ -antiport in Mesembryanthemum crystallinum. Plant J 24, 511–522, doi:10.1046/j.1365-313x.2000.00903.x (2000).
Ishitani, M. et al. Coordinate transcriptional induction of myo-inositol metabolism during environmental stress. Plant J 9, 537–548, doi:10.1046/j.1365-313X.1996.09040537.x (1996).
Das-Chatterjee, A. et al. Introgression of a novel salt-tolerant L-myo-inositol 1-phosphate synthase from Porteresia coarctata (Roxb.) Tateoka (PcINO1) confers salt tolerance to evolutionary diverse organisms. Febs Lett 580, 3980–3988, doi:10.1016/j.febslet.2006.06.033 (2006).
Hasegawa, P. M., Bressan, R. A., Zhu, J. K. & Bohnert, H. J. Plant cellular and molecular responses to high salinity. Annu Rev Plant Phys 51, 463–499, doi:10.1146/annurev.arplant.51.1.463 (2000).
Khalturin, K., Hemmrich, G., Fraune, S., Augustin, R. & Bosch, T. C. G. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet 25, 404–413, doi:10.1016/j.tig.2009.07.006 (2009).
Donoghue, M. T. A., Keshavaiah, C., Swamidatta, S. H. & Spillane, C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. Bmc Evol Biol 11, doi:10.1186/1471-2148-11-47 (2011).
Dong, X. et al. Global Gene-Expression Analysis to Identify Differentially Expressed Genes Critical for the Heat Stress Response in Brassica rapa. Plos One 10, doi:10.1371/journal.pone.0130451 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–U130, doi:10.1038/nbt.1883 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512, doi:10.1038/nprot.2013.084 (2013).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676, doi:10.1093/bioinformatics/bti610 (2005).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, doi:10.1093/bioinformatics/btp352 (2009).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi:10.1093/bioinformatics/btp616 (2010).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. H. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38, W64–W70, doi:10.1093/nar/gkq310 (2010).
Luo, W., Friedman, M. S., Shedden, K., Hankenson, K. D. & Woolf, P. J. GAGE: generally applicable gene set enrichment for pathway analysis. BMC bioinformatics 10, 161, doi:10.1186/1471-2105-10-161 (2009).
Luo, W. & Brouwer, C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831, doi:10.1093/bioinformatics/btt285 (2013).
Acknowledgements
We thank the Editorial Board Member Dr. A. Albert and the anonymous reviewers for their helpful suggestions. We also thank C. Brower and S. Guo for their help with the improvement of the manuscript. We thank the funding support from the Charlotte Research Institute, Award Number: 1603–020; North Carolina Biotechnology Center, Award number: 2014-CFG-8005; the National Institute of General Medical Sciences of the National Institutes of Health, Award Number: R15GM12202; and the National Natural Science Foundation of China (31270283).
Author information
Authors and Affiliations
Contributions
B.S. and A.L. initiated the project and designed the experiment; Y.L. and J.W. collected the phenotypic data; Y.L., R.R., and D.F. performed the data analyses; C.L., H.S., H.Z., C.B., and S.G. assisted with the data analyses; Y.L., R.R., B.S., and A.L. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, Y., Reid, R., Freese, D. et al. Salt tolerance response revealed by RNA-Seq in a diploid halophytic wild relative of sweet potato. Sci Rep 7, 9624 (2017). https://doi.org/10.1038/s41598-017-09241-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09241-x
- Springer Nature Limited
This article is cited by
-
Identification of QTNs, QTN-by-environment interactions, and their candidate genes for salt tolerance related traits in soybean
BMC Plant Biology (2024)
-
Integrated single-molecule real-time sequencing and RNA sequencing reveal the molecular mechanisms of salt tolerance in a novel synthesized polyploid genetic bridge between maize and its wild relatives
BMC Genomics (2023)
-
Tapping the potential of Solanum lycopersicum L. pertaining to salinity tolerance: perspectives and challenges
Genetic Resources and Crop Evolution (2021)
-
Transcriptome sequencing and whole genome expression profiling of hexaploid sweetpotato under salt stress
BMC Genomics (2020)
-
miRNAs involved in transcriptome remodeling during pollen development and heat stress response in Solanum lycopersicum
Scientific Reports (2020)