
Abstract
Lucanidae (Coleoptera: Scarabaeidae) are fascinating beetles exhibiting significant dimorphism and are widely used as beetle evolutionary study models. However, lacking high-quality genomes prohibits our understanding of Lucanidae. Herein, we proposed a chromosome-level genome assembly of a widespread species, Prosopocoilus inquinatus, combining PacBio HiFi, Illumina, and Hi-C data. The genome size reaches 649.73 Mb, having the scaffold N50 size of 59.50 Mb, and 99.6% (647.13 Mb) of the assembly successfully anchored on 12 chromosomes. The BUSCO analysis of the genome exhibits a completeness of 99.6% (n = 1,367), including 1,362 (98.5%) single-copy BUSCOs and 15 (1.1%) duplicated BUSCOs. The genome annotation identifies that the genome contains 61.41% repeat elements and 13,452 predicted protein-coding genes. This high-quality Lucanidae genome provides treasured genomic information to our knowledge of stag beetles.
Similar content being viewed by others

Background & Summary
The stag beetle (Coleoptera: Lucanidae) is a family in Superfamily Scarabaeoidea, comprising around 1,500 species worldwide1. Most stag beetle species exhibit significant intraspecific or even interspecific sexual dimorphism, in which males usually tend to have extremely impressive mandibles to fight and attract females in the wild. Thus, stag beetles have received much attention since Linnaeus first described the Scarabaeus parallelipipedus from Europe (later transferred to the genus Dorcus)2. Many lucanid species have been selected as an ideal behavior and functional morphology study model, and their fascinating mandibles make them popular pets and valuably private collections3,4,5,6,7. In the wild, most stag beetles are closely related to forest ecosystems, as their carboxylic larvae usually feed on decaying logs and other litter, such as leaves or fungi8,9,10.
The major geographical distribution and species diversity of Lucanidae are associated with the Indomalayan and Palearctic regions; 33 genera and nearly 400 species are known from China11,12,13. The present research on the stag beetle primarily focuses on its taxonomy and phylogeny, including new species descriptions and mitochondrial genome studies7,11,12,13,14. Our understanding of the stag beetle genome, especially high-quality genome assembly, remains in its infancy. Only one genome, Dorcus hopei, has been reported15. Compared with other beetles’ sharply increasing genome assembly number, more high-quality genome assemblies for stag beetles have become necessary and inevitable.
To enhance the knowledge of the taxonomy, evolution, and ecology of Lucanidae, we proposed the chromosome-level genome of a widespread species, Prosopocoilus inquinatus (Westwood, 1848), with the combination of PacBio HiFi, Illumina, and Hi-C data. Genome annotation, including repeats, non-coding RNAs (ncRNAs), and protein-coding genes (PCGs) were analyzed and exhibited. The high-quality genome of P. inquinatus provides valuable genomic information for Lucanidae study.
Methods
Sample collection and sequencing
A single P. inquinatus male sample was collected for DNA and RNA sequencing data on April 30, 2023, in Motuo County, Xizang, China. Muscle tissue, including the pronotum and posterior abdomen, was extracted from the specimen and washed via phosphate-buffered saline (PBS) solution for five minutes to eliminate any possible external pollutants. The specimen was then transferred into liquid nitrogen, frozen for at least 20 minutes, and kept at −80 °C for temporary storage until sequencing.
The specimen’s genomic DNA (gDNA) was extracted using the FastPure® Blood/Cell/Tissue/Bacteria DNA Isolation Mini Kit (Vazyme Biotech Co., Ltd, Nanjing, China). High molecular weight (HMW) gDNA was sheared into 15 kb with the MegaruptorTM device (Diagenode, Liege, Belgium) and was enriched using the AMPurePB Beads. PCR-free short reads library for whole genome sequencing (WGS) was prepared using the Truseq DNA PCR-free Kit. A PacBio HiFi 15 kb library was prepared using the SMRTbellTM Express Template Prep Kit 2.0, and the resulting library was sequenced on the PacBio Sequel II platform. The Hi-C data was carried out by digesting extracted DNA with the Mbol restriction enzyme. RNA was lysed from the specimen using the TRIzoTM Reagent (Invitrogen, Carlsbad, CA, USA). RNA-seq libraries were constructed using the VAHTS mRNA-seq v2 Library Prep Kit (Vazyme, Nanjing, China). The Illumina NovaSeq. 6000 platform was used to build all short-read libraries. The Nanopore PromethlION platform constructed long reads of the RNA library. Berry Genomics (Beijing, China) carried out all library constructions and sequencing. Consequently, we obtained 272.73 Gb of sequencing data, including 109.10 Gb (152.68×) of Illumina reads, 42.50 Gb (65.41×) of PacBio HiFi reads, 101.03 Gb (155.40×) of Hi-C data, 20.10 Gb of transcriptome data, including 9.72 Gb of short reads data and 10.38 Gb of long reads data (RNA-ONT) (Table 1).
De novo genome assembly
Raw genomic Illumina sequencing reads for genome scan were employed as quality control using Fastp v0.23.216 to remove adaptors, duplications, and low-quality reads.
Raw PacBio HiFi reads were generated into the primary assembly using Hifiasm v0.19.817. The direct reads were then mapped with the raw HiFi reads using Minimap2 v2.2418 to calculate the mapping rate. One round of primary self-polishing assembly was performed for primary assembly by utilizing NextPolish2 v0.2.019.
Raw Hi-C data was processed under quality control to remove duplicates using Chromap v.0.2.5-r47320. Clean Hi-C data was then utilized to align the primary assembly for haplotype identification and division. Contigs were anchored and orientated onto chromosomes using YaHS v1.221 and Juicer v1.6.222. The result of the contig assembly was reviewed, and any assembly errors were corrected manually under Juicebox v.1.11.0823. To determine the autosomes and sex chromosomes, the final assembly was remapped with raw HiFi data by using MiniMap2 to determine each chromosome length. Chromosome coverage was then calculated using SAMtools v. 1.924 by dividing raw data by chromosome length. Moreover, the X chromosome was also detected by chromosome synteny between the model beetle species, Tribolium castaneum, and the relative species Trypoxylus dichotomus according to the relatively conserved feature in insect sexual chromosome X25. Syntenic blocks were identified and determined using MCScanX26 and TBtools27. Conclusively, the X chromosome was identified by exhibiting around half of the chromosome coverage compared with other chromosomes (Table 3) and re-confirmed by sharing high synteny features with other beetles’ X chromosomes (Fig. 2).
To ensure the high-quality assembly of our genome, potential contaminants were detected and eliminated by software and NCBI. In this case, we focused on Humans, Bacteria, viruses, and plant sequences. Possible contaminants were detected using MMseq. 2 v1128, which utilizes BLASTN-like searches and the UniVec database based on the NCBI nucleotide database. Potential vector contaminants were also specifically detected and identified by blastn (BLAST + v2.11.029) against the UniVec database. Sequences with over 90% hits in the database above were considered contaminants, and sequences with over 80% hits were rechecked by online BLASTN analysis in the NCBI nucleotide database. The final genome assembly was uploaded to NCBI to detect and eliminate contaminants. According to vector search, no prominent contaminant was found in our assembly, reflecting the high quality of sample preparation and accuracy of specimen sequencing.
The final P. inquinatus genome assembly eventually reached the chromosomal level with a total size of 649.73 Mb, consisting of 174 scaffolds and 195 contigs (Table 2). The scaffold and contig N50 length reached 59.5 Mb and 26.36 Mb, respectively. GC content of the P. inquinatus was 35.67%. Most contigs (612.12 Mb, 94.21%) were firmly anchored and orientated onto 12 chromosomes. All chromosome coverage was computed and exhibited (Table 3). Among these chromosomes, one particular chromosome, number 12, has a coverage of 37.02 for long-read sequencing and 88.58 for short-read sequencing, around half of the other chromosomes (Table 3). Hence, the number 12 chromosome was considered the X chromosome in P. inquinatus. All chromosomes in assembly, including 11 autosomes and X chromosome, with individual lengths ranging from 17.22 to 75.68 Mb (Tables 2, 3; Fig. 1). Compared with the assembly result of its related species, Trypoxylus dichotomus30 (Sarabaeidae) (636.37 Mb in genome size and 35.11% GC content), P. inquinatus exhibited a larger genome size and GC content (Table 4).

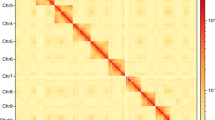
Genome-wide chromosomal heatmap of Prosopocoilus inquinatus, with each chromosome and contig framed in blue and green, respectively. “ChrX” represented the sex chromosome.

Chromosomal synteny between Tribolium castaneum, Prosopocoilus inquinatus, and Trypoxylus dichotomus. The sexual chromosome X is labeled red.
Genome annotation
A de novo specific repeat library for P. inquinatus was built by RepeatModeler v2.0.431. This specific repeat library was combined with RepBase-2023090932 and added to the custom library. Repeat elements in the P. inquinatus genome were recognized and masked by RepeatMasker v.4.1.433 by aligning the custom library. Repetitive elements analysis resulting from RepeatMasker demonstrated that the P. inquinatus genome contains approximately 62.19% repetitive elements, including unclassified elements (42.02%), LTR elements (8.36%), DNA transposons (7.33%), LINE (1.77%), and simple repeats (0.68%) with other elements (S Table). The density for the type of each element, including simple and TEs elements, was exhibited on each chromosome (Fig. 3). Compared with the repetitive element components in T. dichotomus, P. inquinatus showed more significant size percent of Unclassified (42.02% to 16.67%) and LTR (8.36% to 1.24%) elements; however, P. inquinatus had a significantly minor size percent of DNA transposons, LINEs, and SINEs (Table 4).

Genome characteristics of Prosopocoilus inquinatus. Circos plot showing the genomic characters of P. inquinatus from outer to inner: chromosome length (Chr) (Mb), the density of GC content (GC), the density of protein-coding genes (GENE), the density of TEs (DNA, SINE, LINE, and LTR), and simple repeats (Simple). (The sliding window size is counted for every 10 kb).
Non-coding RNAs (ncRNAs) and transfer RNA (tRNA) in P. inquinatus were detected and identified by Infernal v1.1.434 and tRNAscan-SE v2.0.935, respectively. As a result, 1,857 ncRNAs were placed in the P. inquinatus genome, including four long non-coding RNAs, six ribozymes, 55 small nuclear RNAs, 93 microRNAs, 344 other ncRNAs, 351 tRNAs, and 1,004 ribosomal RNAs (Table 4). Comparatively, the number of P. inquinatus ncRNAs was around 2.8 times more than T. dichotomus (Table 4).
Protein-coding genes (PCGs) annotation in P. inquinatus was analyzed by MAKER v3.01.0336 from transcribed RNA, ab initio gene predictions, and homologous proteins. Transcribed RNA alignment prediction was performed by HISAT2 v2.2.137. RNA-seq alignment production was then acted as a genome-guided assembly by StringTie v2.1.638. The BRAKER v3.0.339 was applied to acquire the ab inito gene predictions by employing GeneMark-ETP40 and Augustus v3.4.041 and automatically trained them based on RNA sequence alignments and reference proteins obtained from OrthoDB v11 database42. GeMoMa v1.943 analyzed protein-homology alignments from five insect species’ proteins, including two Coleopteran species, Tribolium castaneum (GCF_000002335.344) and Coccinella septempunctata (GCF_907165205.145) related to Lucanidae and three sister families of Coleoptera, including one Dipteran species, Drosophila melanogaster (GCA_000001215.446), one Hymenopteran species, Apis mellifera (GCA_003254395.247), and one Neuropteran species Chrysoperla carnea (GCA_905475395.148) (Table 5). Results from BRAKER and GeMoMA were finally combined and applied as the ab inito input for MAKER. The final result of P. inquinatus PCGs establishment indicated 13,452 genes with an average length of 17,401.8 bp (Table 6).
The functional gene annotation was proposed by searching the UniProtKB (SwissProt and TrEMBL) 20190527 database, which uses Diamond v2.0.11.149. Protein domain identifications were performed by eggNOG-mapper v2.1.950 and InterProScan 5.60–92.051 for Gene Ontology (GO) and KEGG pathway annotation analysis. Five databases, including Pfam52, SMART53, Superfamily54, Gene3D55, and CDD56, were analyzed in InterProScan. Functional annotation indicated that P. inquinatus contained 11,656 COG categories, 7,087 GO terms, 4,924 KEGG pathways, and 2,838 Enzyme Codes based on the InterProScan and eggNOG annotation integration (Table 6).
Data Records
The raw sequencing data and genome assembly of Prosopocoilus inquinatus have been deposited at the National Center for Biotechnology Information (NCBI). The Illumina, PacBio, Hi-C, transcriptome short reads, and transcriptome long reads data can be found under identification numbers SRR2712782557, SRR2724360458, SRR2712782859, SRR2712782760, and SRR2712782661, respectively, under the BioProject accession number PRJNA1015594 and BioSample accession number SAMN37358649. The assembled genome has been deposited in the GeneBank in NCBI under accession number GCA_036172665.162. The annotation results for repeated sequences, gene structure, and functional prediction have been deposited in the Figshare database63.
Technical Validation
Berry Genomics (Beijing, China) carried out the DNA extraction. Two quantities, including the NanoDrop and Qubit, were mentioned during the extraction process (Table 7). Our extraction yielded a NanoDrop of 86 ng/μl and a 44.65 ng/μl Qubit. The 280/260 and the 260/230 of our stag beetle are 1.78 and 1.85, respectively.
Two methods were used to evaluate the quality of the genome assembly. Firstly, BUSCO v5.4.464 was applied for assembly completeness calculation with the reference Insecta gene set (n = 1,367) with the euk_genome_met mode. The final genome assembly showed a BUSCO completeness of 99.6%, including 1,362 (98.5%) single-copy BUSCOs, 15 (1.1%) duplicated BUSCOs, 1 (0.1%) fragmented BUSCOs, and 4 (0.3%) missing BUSCOs. To investigate the quality of the de novo assembly, Merqury v1.365 was performed to identify possible assembly sequence errors based on efficient k-mer set operations and QV score calculation. Consequently, the k-mer completeness value of the stag beetle is 94.2%, and the QV score is 46.60. Both the k-mer value and the QV score reflect the high accuracy of the base pairs, combined with the BUSCOs, which exhibit the high completeness and accuracy of our genome assembly. The final annotation validation was also calculated by BUSCOs with a protein mode with the reference Insecta gene set (n = 1,367). The final annotation genome exhibited a BUSCO completeness of 99.6%, including 1,079 (78.9%) single-copy BUSCOs, 283 (20.7%) duplicated BUSCOs, 1 (0.1%) fragmented BUSCOs, and 4 (0.3%) missing BUSCOs. The mapping rate was also measured to determine the assembly accuracy. The mapping rates for PacBio, Illumina, RNA short reads, and RNA long reads were 99.6%, 96.51%, 96.93%, and 97.59%, respectively. These evaluations altogether reflected the high-quality value of the genome assembly.
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software. The settings and parameters of software were listed below: (1) Fastp v0.23.2: ‘-D’ (drop the duplicated reads), ‘-g’ (tail trimming), ‘-x’ (polymer trimming on 3′ ends), ‘-5’ (move a sliding window from 5′ tail to tail), ‘-u 10’ (unqualified percentage limit), ‘-c’ (overlapped bases correction); (2) Hifiasm v0.19.8: ‘-l2’ (strongly remove haplotig duplications); (3) Minimap2 v2.24: default parameters; (4) NextPolish2 v0.2.0: default parameters; (5) YaHS v1.2: default parameters; (6) Juicer v1.6.2: default parameters; (7) Juicebox v.1.11.08: default parameters; (8) MMseq2 v11: default parameters with ‘--search-type 3’, ‘—min-seq-id 0.8’ for potential contaminants; (9) SAMtools v. 1.9: default parameters; (10) RepeatModeler v2.0.4: ‘-LTRStruct’ LTR discovery pipeline; (11) RepeatMasker v.4.1.4: default parameters; (12) Infernal v1.1.4: default parameters; (13) tRNAscan-SE v2.0.9: ‘EukHighConfidenceFilter’ script with default parameters; (14) MAKER v3.01.03: default parameters; (15) HISAT2 v2.2.1: default parameters; (16) StringTie v2.1.6: default parameters; (17) BRAKER v3.0.3: default parameters; (18) GeneMark-ETP: default parameters; (19) Augustus v3.4.0: default parameters; (20) GeMoMa v1.9: ‘GeMoMa.m = 15000’, ‘ERE.c = false’ with default parameters; (21) Diamond v2.0.11.1: default parameters; (22) eggNOG-mapper v2.1.9: default parameters; (23) InterProScan 5.60–92.0: default parameters.
References
Fujita, H. The Lucanid Beetles of the World. Mushi-sha, Tokyo. (2010).
Linnaeus, C. Systema Naturae per regna tria naturae, secundum classes, ordines, genera, species cum characteribus, differntiis, synonymis locis. Tomus I. Editio X. Laurentii Salvi, Holmiae. (1758).
Kojima, H. Breeding Technique of Lucanid Beetles. Mushi-sha, Tokyo (1996).
New, T. R. Beetles in Conservation. Oxford University Press (2010).
Gotoh, H. et al. Developmental link between sex and nutrition; doublesex regulates sex-specific mandible growth via juvenile hormone signaling in stag beetles. PLoS Genet. 10, e1004098 (2014).
Goyen, J., Dirckx, J. & Aerts, P. Costly sexual dimorphism in Cyclommatus metallifer stag beetles. Funct. Ecol. 29, 35–43 (2015).
Liu, J., Chenggong, L. I., You, S., Wan, X. & Ecology, D. O. The first complete mitogenome of Cyclommatus stag beetles (Coleoptera: Lucanidae) with the phylogenetic implications. Entomotaxonomia. 39, 294–299 (2017).
Araya, K. Relationship between the decay types of dead wood and occurrence of lucanid beetles (Coleoptera: Lucanidae). Appl. Entomol. Zool. 28, 27–33 (1993).
Tanahashi, M., Matsuchita, N. & Togshi, K. Are stag beetles fungivorous? J. Insect Physiol. 55, 983–988 (2009).
Songvorawit, N., Butcher, B. A. & Chaisuekul, C. Decaying Wood preference of stag beetles (Coleoptera: Lucanidae) in a tropical dry-Evergreen Forest. Environ. Entomol. 46, 1322–1328 (2017).
Huang, H. & Chen, C. C. Stag Beetles of China I. Formosa Press, Taipei. (2010).
Huang H. & Chen, C. C. Stag Beetles of China II. Formosa Press, Taipei. (2013).
Huang H. & Chen, C. C. Stag Beetles of China III. Formosa Press, Taipei. (2017).
Zhou, L. Y., Zhan, Z. H., Zhu, X. L. & Wan, X. Multilocus phylogeny and species delimitation suggest synonymies of two Lucanus Scopoli, 1763 (Coleoptera, Lucanidae) species names. Zookeys. 1135, 139–155 (2023).
Li, X. et al. The first chromosome-level genome of the stag beetle Dorcus hopei Saunders, 1854 (Coleoptera: Lucanidae). Sci Data. 11, 396 (2024).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. Fastp: an ultra-fast all-in-one FASTAQ preprocessor. Bioinformatics. 34(17), 884–890 (2018).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Hu, J. et al. Nextpolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. bioRxiv. 2023.04.26.538352 (2023).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat Communications. 12(1), 1–6 (2021).
Zhou, C. X., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics. 39(1), btac808 (2023).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Dudchenko, O. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
Toups, A. M. & Vicoso, B. The X chromosome of insects likely predates the origin of class Insecta. Evolution. 77(11), 2504–2511 (2023).
Wang, Y. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Chen, C. et al. Tbtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
Steinegger, M. & Soding, J. MMseqs 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Wang, Q. Y., Liu, L. W., Zhang, S. J., Wu, H. & Huang, J. H. A chromosome-level genome assembly and intestinal transcriptome of Trypoxylus dichotomus (Coleoptera: Scarabaeidae). GigaScience. 11, giac059 (2022).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 14 October 2023) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP: automatic gene finding in eukaryotic genomes in consistency with extrinsic data. bioRxiv. 13.524024. (2023).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res. 51(D1), D445–D451 (2023).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics. 21, 47 (2020).
Crowley, L. et al. The genome sequence of the seven-spotted ladybird, Coccinella septempunctata Linnaeus, 1758. Welcome Open Res. 6, 319 (2021).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015).
Wallberg, A. et al. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics. 20(1), 275 (2019).
Wang, Y. et al. The first chromosome-level genome assembly of a green lacewing Chrysopa pallens and its implication for biological control. Mol Ecol Resour. 22(2), 755–767 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27127825 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27243604 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27127828 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27127827 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27127826 (2024).
NCBI GeneBank https://identifiers.org/ncbi/insdc.gca:GCA_036172665.1 (2024).
Bo, P. Genome Annotation. figshare https://doi.org/10.6084/m9.figshare.24635889.v1 (2024).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Rhie, A., Walenz, P. B., Koren, S. & Philippy, M. A. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Acknowledgements
This research was supported by the key research and development program of the Tibet Autonomous Region, investigating invasive species in Xizang agricultural fields and developing key prevention and control techniques for important invasive species under project XZ202201ZY0002N, and Plan Project of Zunyi Science and Technology NO.ZSKHZ [2023]148.
Author information
Authors and Affiliations
Contributions
B.P. contributed to the research design. B.P. collected the samples. B.P. analyzed the data. B.P. and Z.Z.H. wrote the draft manuscript, and W.Y.C. revised it. All co-authors contributed to this manuscript and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pang, B., Zhan, Z. & Wang, Y. A chromosome-level genome assembly of Prosopocoilus inquinatus Westwood, 1848 (Coleoptera: Lucanidae). Sci Data 11, 808 (2024). https://doi.org/10.1038/s41597-024-03647-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03647-9
- Springer Nature Limited