
Abstract
The Stag beetle (Coleoptera: Lucanidae) is a fascinating group, often considered one of the most primitive within the Scarabaeoidea. They are valuable models for studying beetle evolution. However, the lack of high-quality genomes hinders our understanding of the evolution and ecology of Lucanidae. In this study, we present a chromosome-level genome of Serrognathus titanus by combining PacBio HiFi long reads, Illumina short reads, and Hi-C data. The genome spans 384.07 Mb, with a scaffold N50 size of 75.81 Mb, and most contigs (97.45%, 374.30 Mb) were anchored into six chromosomes. Our BUSCO analysis of the assembly indicates a completeness of 97.6% (n = 1,367), with 92.8% single-copy BUSCOs and 4.8% duplicated BUSCOs identified. Additionally, we found that the genome contains 43.87% (168.50 Mb) repeat elements and identified 14,263 predicted protein-coding genes. The high-quality genome of S. titanus provides valuable genomic information for comprehending the evolution and ecology of Lucanidae.
Similar content being viewed by others

Background & Summary
The Stag beetle (Coleoptera: Lucanidae) constitutes a relatively modest taxonomic cluster within the superfamily Scarabaeoidea, encompassing approximately 1,500 species1. Notably, male stag beetles are distinguished by their oversized mandibles, utilized in assertive interactions to establish dominance over favored mating territories, a behavior evocative of the competitive sparring seen among stags2. Their striking size and intricate mandibles have long captivated the interest of both professional taxonomists and amateur collectors3. The larvae of stag beetles primarily subsist on decaying wood, while adults from various species typically demonstrate nocturnal tendencies and consume decaying fruits, logs, and plant fluids4,5. Species of Lucanidae are distributed across all major zoogeographical regions, excluding Antarctica, and hold significant importance as a focal point in evolutionary processes6. Historically, the Lucanidae family has been regarded as one of the most primitive groups within the Scarabaeoidea, with scarabaeoid classifications and evolutionary hypotheses typically placing Lucanidae at the basal position within the superfamily7,8. Research on the Lucanidae family has predominantly focused on systematic evolutionary studies, leveraging data from nuclear gene fragments, mitochondrial multi-gene fragments, and mitochondrial genomes9,10,11,12. However, these datasets have proven insufficient in providing comprehensive insights into the evolutionary status of Lucanidae within the Scarabaeoidea10. Deciphering more high-quality reference genomes has emerged as a crucial step in inferring the phylogeny of insects. The limited availability of genomic data has impeded systematic evolutionary research on the Lucanidae family.
To enhance our understanding of Lucanidae evolution and ecology, we propose assembling a chromosome-level genome of Serrognathus titanus (Boisduval, 1835), combining PacBio HiFi, Illumina, and Hi-C data. We annotated repeats, non-coding RNAs, and protein-coding genes. The high-quality genome of S. titanus represents a significant advancement in Lucanidae research, offering valuable insights into Scarabaeoidea evolution and ecology.
Methods
Sample collection and sequencing
A single male sample of S. titanus was collected for DNA and RNA sequencing on July 26, 2023, from Guiyang, China. Muscle tissue, specifically from the pronotum and posterior abdomen, was extracted from the specimen. The tissue was thoroughly washed with a phosphate-buffered saline (PBS) solution for five minutes to eliminate potential external contaminants. The sample was then put into flash-frozen condition within a liquid nitrogen container for 20 minutes and finally stored at −80 °C in the laboratory before sequencing.
Genomic DNA and RNA were extracted from the specimen using the FastPure® Blood/Cell/Tissue/Bacteria DNA Isolation Mini Kit (Vazyme Biotech Co., Ltd, Nanjing, China) and TRIzol reagent (YiFeiXue Tech, Nanjing, China), respectively. PCR-free short-read libraries for whole genome sequencing (WGS) were generated using the Truseq DNA PCR-free Kit. The libraries consisted of 150 bp paired-end reads with a 350 bp insert size. The Hi-C experiment was conducted on a previously published protocol13, which involved several steps: DNA cross-linking, chromatin digestion using the restriction enzyme MboI, end repair, and DNA purification. All short-read libraries were sequenced using the Illumina NovaSeq 6000 platform. A 20 kb insert size library was prepared using the SMRTbellTM Express Template Prep Kit 2.0, and the resulting library was sequenced on the PacBio Sequel II platform with HiFi mode. Berry Genomics (Beijing, China) conducted all library constructions and sequencing procedures. In total, we obtained 167.62 Gb of sequencing data, which included 54.30 Gb (141.37×) of PacBio HiFi reads, 57.89 Gb (150.71×) of Illumina reads, 44.09 Gb (114.80×) of Hi-C data, and 11.34 Gb of transcriptome data (Table 1). The PacBio HiFi long reads had a scaffold N50 of 17.76 kb and an average length of 17.82 kb.
Genome assembly
To perform quality control on the raw Illumina data, we utilized BBTools v38.8214. The quality control process involved using the “clumpify.sh” script to remove duplicate reads. In addition, we utilized the “bbduk.sh” script to trim the reads to a high standard. This involved removing sequences with quality scores lower than 20, filtering out sequences containing more than 5 Ns, trimming poly-A/G/C tails longer than 10 bp, and correcting overlapping paired reads.
The primary assembly of PacBio HiFi long reads was generated using Hifiasm v0.19.815 with the default parameters. To remove heterozygous regions, we employed Purge_Dups v1.2.516 with a haploid cutoff set at 70 for identifying contigs as haplotigs (“-s 70”). After conducting quality control, we aligned the Hi-C reads to the assembly using Juicer v1.6.217. The process of anchoring primary contigs into chromosomes was carried out using 3D-DNA v.18092218. The contig assembly result was thoroughly reviewed, and any assembly errors were manually corrected using Juicebox v.1.11.0818. To identify potential contaminants, we utilized MMseqs. 2 v1119 to perform BLASTN-like searches against the NCBI nucleotide and UniVec databases. In addition, we employed blastn (BLAST + v2.11.0)20 to identify vector contaminants against the UniVec database. Sequences demonstrating a match of over 90% in the databases above were considered potential contaminants. Furthermore, sequences with over 80% hits underwent a subsequent verification step through online BLASTN analysis in the NCBI nucleotide database. The potential bacterial and human contamination were removed from the assembled scaffolds. To identify the autosomes and sex chromosomes, the final assembly was remapped with raw PacBio HiFi long reads using MiniMap2 v2.1721. Subsequently, the coverage of each chromosome was calculated using SAMtools v1.922 by dividing raw data by chromosome length. The X chromosome was distinguished by displaying approximately half of the coverage compared to the other chromosomes. The final assembly of the S. titanus genome reached the chromosome-level assembly with a size of 384.09 Mb, consisting of 83 scaffolds and 99 contigs and a GC content of 34.31% (Table 2). The scaffold and contig N50 sizes of 75.81 Mb and 37.83 Mb, respectively. Most contigs (97.45%, 374.30 Mb) were anchored into six chromosomes with lengths ranging from 10.11 to 84.98 Mb (Table 3; Figs. 1, 2). The chromosome coverage was computed and displayed in Table 3. Among these chromosomes, chromosome 6 exhibited a PacBio HiFi long-read sequencing coverage of 63.59, which is approximately half of the coverage observed for the other chromosomes. Consequently, chromosome 6 was designated as the X chromosome (Table 3).

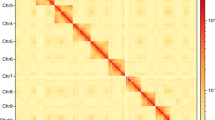
Genome-wide chromosomal heatmap of Serrognathus titanus, with each chromosome framed in blue and each contig framed in green.

Genome characteristics of Serrognathus titanus. From the outer ring to the inner ring are the distributions of chromosome length, GC content, gene density, transposable elements: DNA transposon, short interspersed nuclear elements (SINE), long interspersed nuclear element (LINE), and long terminal repeats (LTR), and simple repeats.
Genome annotation
A de novo repeat library of S. titanus was constructed using RepeatModeler v2.0.423, employing the “-LTRStruct” LTR discovery pipeline. This specific repeat library was merged with RepBase-2023090924 and Dfam 3.525, resulting in a custom library. To identify and mask repeat elements in the S. titanus genome, RepeatMasker v.4.1.426 was employed, aligning the genome against the custom library. The analysis conducted by RepeatMasker revealed that the S. titanus genome comprises approximately 43.87% repetitive elements, including unclassified elements (24.29%), DNA transposons (10.58%), LINE transposons (5.09%), LTR transposons (2.84%), simple repeats (0.68%), and other elements (Table 4).
Non-coding RNAs (ncRNAs) in S. titanus were detected using Infernal v1.1.427 against the Rfam v14.10 database28, while transfer RNAs (tRNAs) were identified using tRNAscan-SE v2.0.929. The analysis revealed a total of 1,186 ncRNAs in the S. titanus genome, including 4 long non-coding RNAs, 21 ribozymes, 51 small nuclear RNAs, 67 microRNAs, 359 other ncRNAs, 406 tRNAs, and 278 ribosomal RNAs (Table 4).
Protein-coding gene annotation in S. titanus was analyzed by MAKER v3.01.0330 from transcribed RNA, ab initio gene predictions, and homologous proteins. Transcribed RNA alignment prediction was performed by HISAT2 v2.2.131. RNA-seq alignment production was then acted as a genome-guided assembly by StringTie v2.1.632. To obtain ab initio gene predictions, BRAKER v2.1.633 was utilized, employing GeneMark-ES/ET/EP 4.68_lic34 and Augustus v3.4.035 and automatically trained them based on RNA sequence alignments and reference proteins mined from the OrthoDB v11 database36. GeMoMa v1.937 was used to predict genes based on protein homology and intron position conservation, employing the parameters “GeMoMa.c = 0.4” “GeMoMa.p = 10” and protein sequences from five species (Drosophila melanogaster (GCF_000001215.4)38, Prosopocoilus inquinatus (GCA_036172665.1), Tribolium castaneum (GCF_000002335.3)39, Apis mellifera (GCF_003254395.2)40, and Coccinella septempunctata (GCF_907165205.1))41. The results obtained from BRAKER and GeMoMa were combined and utilized as the ab initio input for MAKER. In the S. titanus genome, we predicted a total of 14,263 protein-coding genes, with an average length of 9,487.6 bp. Each gene exhibited an average of 6.2 exons, 5.0 introns, and 6.0 coding sequences (CDS). The mean lengths of the exons, introns, and CDS were 314.4 bp, 1,599.3 bp, and 271.4 bp, respectively (Table 4). The completeness of the protein sequences was assessed using BUSCO, resulting in a high score of 98.0% (n = 1,367). This encompassed 63.3% (865) single-copy, 34.7% (474) duplicated, 0.1% (2) fragmented, and 1.9% (26) missing BUSCOs, indicating high-quality predictions.
We performed gene functional annotation searches against the UniProtKB database using Diamond v2.0.11.142 in sensitive mode with the parameters “–more-sensitive -e 1e-5”. Additionally, we utilized eggNOGmapper v2.0.143 and InterProScan 5.53–87.044 to assign Gene Ontology (GO) terms and identify (KEGG and Reactome) pathways, as well as protein domains. The InterProScan analyses included five databases: Pfam45, SMART46, Superfamily47, Gene3D48, and CDD49. The results obtained from these tools were integrated to derive the final gene function predictions. Functional annotation indicated that S. titanus contained 12,126 COG categories, 10,592 GO terms, 5,055 KEGG pathways, and 2,863 Enzyme Codes based on the InterProScan and eggNOG annotation integration. Additionally, we generated visualizations of repeat content, gene density, and GC content on individual pseudochromosomes using TBtool50.
Data Records
The raw sequencing data and genome assembly of Serrognathus titanus have been deposited at the National Center for Biotechnology Information (NCBI). The Hi-C, transcriptome, Illumina, and PacBio HiFi data can be found under identification numbers SRR2899952551, SRR2899952652, SRR2899952753, and SRR2899952854. The assembled genome has been deposited in the NCBI assembly with the accession number GCA_039766575.155. The annotation results for repeated sequences, gene structure, and functional prediction have been deposited in figshare56.
Technical Validation
Two methods were utilized to evaluate the quality of the genome assembly. Initially, assembly completeness was assessed using BUSCO v5.0.457 with the reference Insecta gene set (n = 1,367). The final genome assembly demonstrated a BUSCO completeness of 97.6%, with 92.8% single-copy BUSCOs, 4.8% duplicated BUSCOs, 0.5% fragmented BUSCOs, and 1.9% missing BUSCOs. As a measure of assembly accuracy, the mapping rate was calculated by aligning the reads from PacBio, Illumina, and RNA sequencing to the final assembly using Minimap2 and SAMtools. The mapping rates for PacBio, Illumina, and RNA reads were 99.85%, 87.64%, and 87.70%, respectively. These assessments collectively validate the high quality of the genome assembly generated in this study.
Code availability
The scripts used for genome assembly and annotation in this article were uploaded to figshare56. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Fujita, H. The Lucanid Beetles of the World. Mushi-sha, Tokyo (2010).
Inoue, A. & Hasegawa, E. Effect of morph types, body size and prior residence on food-site holding by males of the male-dimorphic stag beetle Prosopocoilus inclinatus (Coleoptera: Lucanidae). J Ethol. 31, 55–60 (2013).
Kim, S. I. & Farrell, B. D. Phylogeny of world stag beetles (Coleoptera: Lucanidae) reveals a Gondwanan origin of Darwin’s stag beetle. Mol Phylogenet Evol. 86, 35–48 (2015).
Kubota, K. et al. Evolutionary relationship between Platycerus stag beetles and their mycangium-associated yeast symbionts. Front Microbiol. 11, 1436 (2020).
Hendriks, P. Life cycle length of the lesser stag beetle (Coleoptera: Lucanidae: Dorcus parallelipipedus). Entomol Ber. 79, 208–216 (2019).
Zhang, M. et al. Geometric morphometric analysis of the pronotum and elytron in stag beetles: insight into its diversity and evolution. Zookeys. 833, 21–40 (2019).
Howden, H. F. Larval and adult characters of Frickius Germain, its relationship to the Geotrupini, and a phylogeny of some major taxa in the Scarabaeoidea (Insecta: Coleoptera). Canadian Journal of Zoology. 10, 2713–2724 (1982).
Iablokoff-Khnzorian, S. M. Über die Phylogenie der Lamellicornia (Insecta, Coleoptera) Entomologische Abhandlungen, Staatliches Museum für Tierkunde in Dresden. 41:135–200 (1977).
Kubota, K. et al. Diversification process of stag beetles belonging to the genus Platycerus Geoffroy (Coleoptera: Lucanidae) in Japan based on nuclear and mitochondrial genes. Entomol Sci. 14, 411–427 (2011).
Yuan, J. J., Chen, D. & Wan, X. A multilocus assessment reveals two new synonymies for East Asian Cyclommatus stag beetles (Coleoptera, Lucanidae). ZooKeys. 1021, 65–79 (2021).
Lin, Z. Q., Song, F., Li, T., Wu, Y. Y. & Wan, X. New mitogenomes of two Chinese stag beetles (Coleoptera, Lucanidae) and their implications for systematics. J Insect Sci. 17, 63 (2017).
Zeng, L. et al. Comparative mitochondrial genomics of five Dermestid beetles (Coleoptera: Dermestidae) and its implications for phylogeny. Genomics. 113, 927–934 (2021).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Dudchenko, O. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–124 (2005).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome research. 25, 445–458 (2015).
Tribolium Genome Sequencing Consortium. The genome of the model beetle and pest Tribolium castaneum. Nature. 452, 949–955 (2008).
Gibbs, R. A. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 443, 931–949 (2006).
Crowley, L. The genome sequence of the seven-spotted ladybird, Coccinella septempunctata Linnaeus, 1758. Wellcome open research. 6, 319 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Chen, C. et al. Tbtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR28999525 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR28999526 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR28999527 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR28999528 (2024).
NCBI Assembly. https://identifiers.org/ncbi/insdc.gca:GCA_039766575.1 (2024).
Jin, J. Genome annotation of Serrognathus titanus. figshare. Dataset. https://doi.org/10.6084/m9.figshare.25792383 (2024).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (U1704111 and 31401732).
Author information
Authors and Affiliations
Contributions
S.J. contributed to the research design. J.J., Z.Z. and M.Y. collected the samples. J.J., and S.J. analyzed the data. J.J. and Z.Z. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jin, J., Zhan, Z., Ye, M. et al. A chromosomal-level genome assembly of Serrognathus titanus Boisduval, 1835 (Coleoptera: Lucanidae). Sci Data 11, 888 (2024). https://doi.org/10.1038/s41597-024-03727-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03727-w
- Springer Nature Limited