
Abstract
This review emphasizes La-based metal oxides of the perovskite form and their application in heterogeneous catalytic transfer hydrogenation (CTH) reactions from year 2013. Perovskites are potential alternatives to noble metals. The possibility of induced-synergy between two cations in the ABO3 or multicationic perovskites makes them attractive for hydrogenation reactions. Herein, we look at recent developments in their synthesis protocols, and how the various physicochemical properties derived from the individual synthesis protocols affect the catalytic performances of perovskite oxides in transfer hydrogenation reactions. Furthermore, we review different type of perovskite-based catalysts and the mechanisms of the surface catalyzed transfer hydrogenation reactions.
Similar content being viewed by others
1 Introduction
Numerous catalysts with various mechanisms of action have been developed to optimise and catalyze the rapid development of the chemical industry in the twenty-first century. This review focuses on heterogeneous surface catalysis, mainly with the use of metal-oxides, with emphasis on La-based perovskites, due to their relevance in heterogeneous catalytic transfer hydrogenation reactions. Generally, hydrogenation reactions are carried out using platinum group metals (PGMs) such as iridium, ruthenium, platinum, palladium, osmium and rhodium. It is important to mention that noble metals have their own limitations too. These limitations include scarcity that leads to high cost, and susceptibility to sulphur poisoning. Therefore, it is vital to explore alternative noble-metal free catalysts to circumvent these challenges.
We focus mostly on catalytic transfer hydrogenation reaction due to its widespread application in morden catalytic technologies. This chemical conversion route is based on the reduction of unsaturated organic compounds in the presence of catalysts and hydrogen donors that generate hydrogen in-situ [8,9,10]. The in-situ production of hydrogen solves the handling, safety, and diffusion issues associated with molecular hydrogen. The reaction is also atom efficient because the sacrificial alcohol acts as both the solvent and the hydrogen donor. Overall, the catalytic transfer hydrogenation reactions are cheaper and easy to handle with reduced cost of transportation and storage. However, there are minor challenges that are often encountered in the transfer hydrogenation process. The most formidable obstacle in carrying out transfer hydrogenation processes is achieving high product yields without forming any pollutants or by-products and without using toxic solvents that are harmful to the environment.
The byproducts produced in catalytic hydrogenation reactions include partially hygrogenated substrates, esters, acetyls, chlorinated organic compounds, such as HCl, volatile organic compounds common with the hydrogenation of aldehydes, nitrous oxides and generation of heavy metal containing waste [1, 23, 99, 105]. As such, the development of sustainable, green, cost- and time-effective chemical reactions that significantly contribute to reducing contaminants of the environment is imperative. The green chemical principles accomplished by CTH reactions include atom economy as the hydrogen utilized comes from a donor molecule that is then converted to a value-added chemical, use of catalytic systems that can be recovered when the reaction is complete and reduce the need for excessive agents, and renewable and nontoxic solvents are usually utilized [99, 173]. Simulteneously, developments to establish synthetic routes that convert produced pollutants into less hazardous compounds or vaporizing the by-products are progressing [150, 151]. These involve converting abundant pollutants (such as CO2) into value-added chemicals and utilisation of bioderived materials as starting materials instead of potentially hazardous substances.
There are numerous catalysts that have been utilised to make catalytic reactions faster and more atom efficient. These include palladium on carbon, Raney catalysts, ruthenium, rhodium and platinum on carbon. These diverse catalysts have diverse benefits for different substrates offering varying reactivity and selectivity under different conditions [71, 103, 187]. For the purposes of this review, only mixed metal oxides and perovskites will be discussed further as viable substitutes of noble and expensive metal catalysts. Various mixed metal-oxides such as perovskites, cobalt-, ferrous- and ferric oxide catalysts are currently used in chemical industry. This is attributed to their controllable physicochemical properties which offer the ability to evaluate and deduce the relationship between the surface and bulk properties of the catalyst with its activity [123]. Perovskites found relevance in many reactions and different studies due to their tuneable structures. Since the evolution of nanoscience, several approaches to improve catalytic reactions were developed, driving perovskite materials into a new golden age [128, 160]. Various noble and non-noble metals are incorporated into the structures of perovskites, giving versatile perovskite-based catalysts [122].Perovskites have unique properties that potentially enhance the performance and selectivity of the catalysts. Physical properties such as electric, magnetic, and optical characteristics, and the effect of the electronic properties and electronic substitutions on catalysis with perovskites will be discussed in this review.
There are many types of perovskites discussed in literature for various applications, such as, BaMO3; (M = Ti, Zr, and Cu), bismuth-based perovskites, alkaline metal halide oxide perovskites and organic metal halide perovskites [11, 27, 61, 119]. These show remarkable advantages in catalysis and other fields. However, we will establish emphasis on La-based perovskites and their derivatives. La-based perovskites are the most used perovskites because they demonstrate remarkable catalytic performances in different reactions [66, 133, 184]. Recent advances in La-based perovskite catalytic systems will be discussed for reactions involved in the catalytic transfer hydrogenations of unsaturated organic compounds.
Overall, this literature review aims to provide a comprehensive overview of the significance of metal oxide catalysis, with focus on transition metal oxides, porous metal oxides and mostly perovskites. The subsequent discussions will emphasize more on the applications of the perovskites in catalysis, particularly in catalytic transfer hydrogenations. Furthermore, a sections will explore the other catalysts capable of catalyzing CTH reactions. This will all be done with the goal of enhancing the understanding of their roles and potential in advancing chemical reactions.
1.1 Metal-oxide catalysts
Metal-oxides are chemical species with metal–oxygen bonds as the building blocks of repeating units. They are of transition metal- or main group-oxides, depending on the type of metals used. Suitable surface, morphological, and solid-state properties of the metal oxides catalysts drive necessities and prerequisites in completing complex heterogeneous catalytic reactions [123].Therefore, it is important to cautiously study the associations and links between the structural properties and the activity of the metal oxide catalysts to develop vastly “green”, stable, active, and selective catalysts. There are various types of metal oxides and all of them have different structural makeup and properties.
1.1.1 Transition metal-oxides
Transition metal oxides (TMO) are used extensively as heterogeneous catalysts and grouped according to their physicochemical traits. The most important property is the stability, which is governed by the integrity of the metal oxidation states. For this reason, chromium (Cr), titanium (Ti), manganese (Mn), vanadium (V), zinc (Zn), and aluminium (Al) oxides with stable high oxidation states as well as those derived from metals such as iron (Fe), nickel (Ni), cobalt (Co) and lead (Pb) which produce oxides possessing intermediate stability, are the most used in catalysis. On the other hand, unstable oxides are used mostly as nanoparticles or other forms as they do not perform as stable bulk oxides under moderate temperatures. These include ruthenium (Ru), platinum (Pt), silver (Ag), palladium (Pd) and gold (Au) oxides [125].
It is known that noble metal catalysts have better activity than metal-oxides counterparts. Metal-oxides have been applied vastly in catalysis because they are more resistant to poisoning. Electrical conductivity distinguish metal-oxides since non-conducting materials have less freedom of electron movement, hence their poor catalytic capabilities. A compelling example of these is the n-type oxides that are not catalytically active in oxidation reactions. Some of the less conducting oxides are used as supports for the fabrication of other noble metal containing catalysts [125]. Metal oxides may be classified into single or mixed oxides, based on the number of metal cations in their structure.
1.1.1.1 Classification of transition metal-oxides
Single metal oxides are referred to as binary oxides because they constitute one cation, for example transition metal and oxygen anion bonded together to form an oxide compound. They are the simplest form of oxides. Binary oxides that have a ratio of different cations of the same atom with different oxidation states in their structures are called spinel oxides [20]. Some of these are used as catalysts in oxidation, ammoxidation, and reduction reactions [109]. Single metal-oxides containing a transition metal have numerous catalytic applications because of their multiple valence states and flexibility in redox reactions [163].Additionally, they are inexpensive and readily available, making them economically friendly and viable substitutes in a number of applications than their precious metal counterparts.
Apart from the general positive traits of the oxide materials, single metal-oxides do have limitations when utilised in catalysis. These include the low catalytic activity as compared to precious metals [163]. The other limitation arise when metal oxides are in their non-porous form because they only interact with molecules only at their exterior surface and not making use of the interior atoms. This served as a motivation to rather use porous oxides that can utilise the materials' interior surfaces increasing the collision frequency of the molecules within the porous network and hence, improved activity.
1.1.1.2 Porous metal-oxides
Porous metal oxides can be classified into three categories, that is, microporous (pore diameter less than 2 nm), mesoporous (diameter between 2 and 50 nm), and macroporous materials (diameter above 50 nm) [163]. Most heterogeneous reactions depend on the availability of exposed active sites on the surface of the catalytic material. Therefore, micro- and mesopores increase the material surface area while the macropores are important for transporting molecules [95, 159]. The study of porous oxides can be traced back to the mid-twentieth century. However, full characterization of these materials was not possible at the time due to lack of effective techniques, leading to some of their physical and chemical properties not being fully explored. Consequently, the development of the porous materials has recently caught the attention of scientists in order to utilize them to their full potential, especially in catalysis.
Most of the porous oxide materials synthesized at the time of their discovery had mass transfer limitations and disordered porous network. For example, Corma et al. reported that microporous and mesoporous sieves limited the transfer of reactants through the pillared clays as they had rectangular and incompletely opened pores [31].Studies were then conducted to resolve these limitations and optimise the use of porous oxide catalytic materials [38, 79, 135]. In the year 1992, the use of mesoporous structured oxides was launched, discovering Mobil Composition of Matter No. 41 (MCM-41) at Mobil company [144]. This is an ordered mesoporous silica material composed of hexagonal-ordered cylindrical pores [15]. Three years later, Santa Barbara Amorphous material (SBA-15), a three-dimensional porous material, was synthesized [60, 182]. SBA-15 has since been used extensively in catalysis as either a catalyst supports or a templating agent for the synthesis of other metal-oxide catalysts.
1.1.1.3 Mesoporous metal-oxides
The attention towards mesoporous materials with periodic and ordered pore arrangements has increased, especially in heterogeneous catalysis, owing to their unique properties. These materials are effective as catalysts and catalyst supports in many catalytic reactions due to their robust thermal stability, high surface areas, and large pore volumes [163]. A number of studies on the use of porous oxide materials in different applications, especially catalysis have been reported [98, 120, 124, 167]. For instance, Wang et al. reported the combustion of toluene using mesoporous spherical and cubic LaMnO3. The cubic perovskites were more active obtaining 90% conversion at a minimal temperature of 110 °C [167]. This is complemented by the ordered porous materials' ability to implore exceptional mass diffusion properties, solid–gas exchange if it possesses small pores and high flow rates if with larger pores, especially with porous oxides in the meso-range. High product selectivity is also an important advantage offered by use of porous oxides as catalysts. This is achieved by either controlling the contact and progression of the reactants or regulating the reaction molecules' contact time.
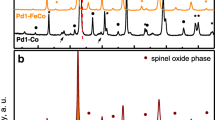
Ordered and periodic porosity in metal oxide materials is introduced into catalysts using different synthetic strategies. The most frequently used methods are the template synthetic methods. The soft templating strategy makes use of templates such as surfactants, while the hard templating method involves the use of pre-synthesised mesoporous materials such as polymers, silica, and well-ordered carbon [77, 139, 148, 177]. Biological materials are also used as soft templating agents for the synthesis of metal oxide materials. Amongst the mesoporous materials, Shen et al. [139] proved KIT-6 to be the best template compared to SBA-15, FDU-12 and SBA-16. For instance, KIT-6 templated-CeO2 catalyst displayed superior activity for the oxidation of CO compared to CeO2 templated by other mesoporous silica materials. Additionally, KIT-6 templated-CeO2 catalyst was determined to have a large surface area as compared to SBA-15 [77, 148], FDU-12 and SBA-16-templated CeO2 [177]. The average surface area of the oxide catalyst prepared using KIT-6 as a template was found to have a surface area of 852 m2/g, while an average surface are of 50–140 m2/g was measured for the SBA-15 templated oxide catalyst.
1.1.1.4 Mesoporous mixed-metal oxides
On the other hand, mixed-metal oxides have advanced properties due to the presence of more than one metal cation in their structure. These mixed-metal oxides include spinnel structures based on 3d transition metals that have robust thermal stability and that are effective in catalytic reactions. Due to their versatile properties, they are utilized in a wide range of applications in various scientific disciplines. These have a general formula given by AB2O4, where the A site is for bivalent cations and the B site for trivalent cations [25]. This review, however, will focus not on these types of mixed oxides with the formula AB2O4, but rather on the perovskite structures with the ABO3 formula. The addition of cations to the oxide structure affects the oxides' electronic structure, directly impacting its catalytic efficiency. An example that portrays this, is the study that was recently conducted, where different cations were tuned for perovskites to increase their electronic and physical properties. The A site cations were varied between methyl ammonium, formamidinium and cesium ions, whereas, the B-site cations were tailored between Sn 2+, Ge2+and Pb2+ [72]. These metals showed remarkable effects that were suitable for different reactions, showing the versatility and adaptability of perovskites.
1.2 Perovskite oxides catalysts
Perovskites are mixed-metal oxide catalysts with the general formula ABO3, a cubic lattice structure when ideal from the Pm3m space group and a tolerance factor of 1 [133]. The ideal cubic structure has the A cations located in the edges, whilst the B cations are at the center and oxygen anions in the middle of the edges [133]. Calcium titanate (CaTiO3) was the first combined oxide to be discovered with the ABO3 formula and was named a perovskite, derived from the name of the discoverer, Lev Perovski in the nineteenth century [65]. The A site cation is usually rare earth metals; however, these can be modified to make the catalyst more efficient by partial substitutions [100, 176]. The B sites are usually transition metal cations with alternating oxidation states [22]. The general structure of the perovskite consists of A and B cations and X anions arranged in an octahedral, corner sharing arrangement. The general formular of the most studied perovskite is ABX3 with a lattice occupying the corner of the lattice. The B cation is usually at the center of the octahedral cage and the X anion is usually on the face centered positions of the lattice (see Fig. 1). There are different ways of arrangement of the lattice structures which then results in a variety of perovskites with different chemical properties. The various oxidation states of the transition metals make perovskites to possess interesting properties.

Diagrammatic representation of the structure of perovkites [171]
1.2.1 Properties of perovskites
The ability of various materials to catalyze reactions is dependent on the properties the materials possess. These are properties such as electronic, steric, and magnetic properties. The multiplicity of oxidation states in the perovskite structures is the major property responsible for their catalytic activity [78, 169, 170]. However, the contribution of other important characteristics such as redox, acid–base, electron mobility, magnetic properties, and superconductivity to the catalytic activity of perovskites cannot be ignored [123, 130, 133]. Moreover, their pyro-, ferro-, thermo- and piezo-electricity give them vast applications [169]. Additionally, they have the pseudo capacity for anion intercalation and oxygen ion diffusion in devices like fuel cells that are oxide-based [65]. For these reasons, there are over a thousand papers in the literature that discuss synthesis, applications, modifications, mechanisms of action and structure clarifications of these materials.
1.2.2 Synthesis of perovskites
Synthesis procedures involving low temperatures often result in lower surface areas of the as-synthesised materials compared to those prepared at higher temperatures. For this reason, chemical synthetic methods such as the sol–gel synthesis [80], polymerizable complexations [153], co-spraying, freeze-drying [90], and spray-drying [67], are often preferred more recently as compared to the solid-state reactions used in the twentiethcentury that produced materials possessing small surface area of approximately 30 m2∙g−1 [78]. Later in the twentieth century, an improved solid reaction route that produce perovskites with higher surface area was developed. However, the limitations of this improved solid state method is that it was time-consuming [140]. Moreover, this method was not as “green” as the solution chemistry methods used for perovskites’ synthesis. As a result, the majority of twenty-first-century studies still use solution chemistry methods for the synthesis of the perovskites. However, it was necessary to note that the ratios of solvents and parameters used in the synthesis were crucial for the synthesised material's catalytic properties as discussed in the next section.
1.2.2.1 Solid state synthesis route
Compounds with different crystalline symmetries but having the same chemical formulations may be formed depending on the procedure followed to synthesise them. The ceramic route, solid-state synthesis, was the first developed technique for the synthesis of perovskites where oxides or carbonates were subjected to heat treatments. These materials, however, had poor properties, for example, the low surface area and grain growth, which negatively affected their activity in catalysis and left room for improvement [134]. As a result, in the 1980s, solution-mediated methods were developed, such as coprecipitation, sol–gel, and complexation synthesis.
These methods involve dissolving precursors in a liquid media, followed by drying and calcination. These generally reduced the temperature required for the synthesis, thus reducing the surface area and grain growth. Homogeneity is one of the advantages offered by the solution mediated methods used for perovskite synthesis. Another method used is mechano-synthesis, where high energy milling is employed at room temperature for the synthesis of perovskite and perovskite-like materials. This method has was patented by Kiliaguine et al. [133].
One of the first perovskites synthesized by the high-energy milling method is LaMnO3 [181]. The milling method is energy-saving and allows material sizes to be kept in the nano-size range. However, high-energy milling requires a longer reaction time and is, therefore, time-consuming. The use of additives was also incorporated into this technique to increase surface area. However, the time required for the milling process was still long. In addition, leaching challenges reduced its chances of being commercialised [133]. To eliminate the leaching process associated with mechano-synthetic method and increase the final product's surface area, Alamdari et al. [2] proposed a modified mechano-synthesis method for the preparation of perovskites. The process involved both the ceramic and milling routes. The modified route has since received a lot of attention as it reduced the many milling hours to 2 h.
1.2.2.2 Solution state synthesis routes
On the other hand, perovskites were synthesized from rare earth hexagonal (h) mixed oxides, such as RMnO3 using high-pressure systems. However, for some h systems, it was not possible to achieve the desired perovskites [183]. Furthermore, with an increase in perovskites’ applications over the years, the coprecipitation and sol–gel methods for synthesising perovskites were developed and extensively applied. These methods are discussed in the next sections.
1.2.2.3 Coprecipitation and Sol–gel methods for perovskite synthesis
Coprecipitation involves more than one compound precipitating out of the solution One of the compounds is usually soluble under the conditions of the reaction, but is carried down by the precipitate, that is, soluble materials may be coprecipitated by being carried together with the insoluble substance. Different parameters and precursors are used for the synthesis of perovskite as stated in the literature. Subsequently, with coprecipitation, the sintering challenge associated with distorted tetragonal perovskites was solved [39, 184]. Coprecipitation gives highly sinterable lead titanate powders of different composition using the coprecipitation of oxalates of different cations in ethanol solution. The metal precursors used are oxides and carbonates of lead, manganese, cobalt and tungsten for the B site cations. For the A site cations precursors of Ca, La, Nd, Sm and Gd nitrates are utilised. The first step to the copreciptation method involves the blending of the B-site precursors with oxalic acid and dilute ethanol. Thereafter, the A site precursors are dissolved in ethanol together with tertiary butoxide. The A site cation would react with the formed precipitate to form metal hydroxides which then react with oxalic acid to form basic oxalate. The coprecipitation is achieved by adding ammonia to the solution. Thereafter, calcination and sintering would give the desired products.
On the other hand, the sol–gel method has gained popularity in literature not only for perovskites but also for the synthesis of other various solid materials. For perovskites, the cation stoichiometric ratios should be regulated, specifically depending on the target structure. Different calcination temperatures play a role on the outcome of the final materials [51, 149]. The calcination step is carried out to remove residual carbonates from the obtained final product [145]. The citrate sol–gel process has been investigated for the synthesis of porous perovskites. The citric acid allows for the formation of acid complexes that aid with the dispersion of the metal, forming amorphous metallic organates with an extensive variability of composition. This was demonstrated in a recent study by Nzuzo et al. where high surface area perovskites where a chelating agent, citric acid and precursor solution made up of water and metal nitrates, were mixed together prior to adding ethylene glycol. The gel was then acquired through heating cycles at 120 °C and calcination at 180 to 680 °C. Perovskites of up to 28.5 m2/g were made. The perovskites had orthorhombic, cubic and hexagonal crystal systems depending on the type of A and B cation used [116].
Highly ordered and homogenous metal-oxides such as perovskites are achieved from organates through thermal decomposition at low calcination temperatures [184]. The process renders the surface areas of these materials to be high, hence more favourable [73]. In some studies, low calcination temperatures are preferred when using the co-precipitation method. In contrast, solvent evaporation is increased using the freeze-drying process [89]. Recently, the in-situ carbon templating method has been developed to synthesise perovskites with high surface areas compared to those obtained from the sol–gel method. For instance, Ping et al. discovered the novel in-situ carbon templating method to prepare porous LaFeO3 catalysts with high surface areas than those obtained when using the traditional methods [173]. Colloidal crystal templating is another potential method that aids in synthesising perovskites with high surface areas [166].
1.2.2.4 The nanocasting route to mesoporous perovskites
Nanocasting is one of the procedures used to synthesize mesoporous catalysts because it results in well-defined porous structures. Over the last decades, nanocasting has been used in different applications [37, 39].This motivated the urge to explore the synthesis of mesostructured perovskites using the nanocasting method. Because materials obtained using the nanocasting method have ordered and systemized porosity in the meso-range, nanocasting is perceived as one of the powerful synthetic routes for various metal oxide materials. Different templates, such as porous silica, graphene, and carbon are used in nanocasting. However, mesostructured silica materials such as Vinyl silica [168], or KIT-6 [164], are mostly preferred because of their remarkable properties, such as high surface area and large pore sizes. The final perovskite or metal oxide structure is a replica of the pores of the template, thus producing perovskites or metal oxides of rod-like nature that are interconnected, see Scheme 1. Wang et al. are the first group to utilise mesostructured silica as hard-templates to synthesize LaFexCo1−xO3 and LaCoO3 perovskites with mesoporous pores for methane combustion [168]. To date, a few studies have been published on the use of nanocasting technique [16, 112, 165].

Nanocasting of perovskites using SBA-15 as a template [99]
The procedure involves the synthesis of the template itself before permeating the precursors previously complexed with citric acid into its pores, as shown in Scheme 1. Subsequently, the composite is subjected to heat treatments to crystalize the perovskite. An alkaline solution (such as NaOH or KOH) of high concentration is used to remove the silica template, leaving imprinted new pores on the perovskite material. Materials exceeding a surface area of 100 m2/g and reaching 270 m2/g are obtained when low-temperature heat treatments are used. The stability of nanocasted materials is exceptional [16, 111, 112]. However, the hard-template process is exceedingly expensive due to the use of a silica template and requires careful considerations with issues regarding waste and environment management. Consequently, nanocasting method is ideal for highly valued products if economic factors are to be considered.
The nanocasting method's effectiveness is portrayed in a catalytic study done using LaCoO3 by Wang et al. [168]. The nanocasted material has high thermal stability, thus effective in the combustion of natural gas. The nanocasted materials had high BET surface areas of greater than 96.7 m2/g, contributing to high catalytic activity than the LaCoO3 counterparts synthesized using the standard citrate method. However, a significant reduction of surface area (from 96.7 to 27.2 m2/g) was observed after one reaction cycle. This was attributed to the restructuring of the perovskite physical structure. In a different study by Nair et al. three different perovskites were synthesized using the hard-template method using ordered mesoporous silica (KIT-6) as a templating agent [111]. The obtained final materials had high surface areas (110 -155m2/g) than those reported by Wang et al. These materials displayed higher conversions, which reinforces that the material's surface area is one of the factors that governs nanocasted perovskites' catalytic activity. In addition, there was a minimal reduction of the surface area of the materials during recyclability tests by at most 15 m2/g. Table 1 summarizes the widely applied synthesis conditions for each protocol used in fabrication of metal oxides of the perovskite form. The table also gives surface areas and set-back associated with the reported synthesis protocols.
1.2.3 Lanthanum-based perovskites and mode of catalysis
Lanthanum based perovskites have attracted a lot of attention in the literature [7, 117, 133]. La in perovskite lattice is usually located at the A site of the material with transition metals at the B-sites. The B-sites can either induce oxygen vacancies through partial substitution, thus making the resultant perovskite suitable for oxidation reactions or precursors for nanosized catalyst preparation suitable for hydrogenation reactions. The interest in La-based perovskites is attributed to the fact that the perovskite is constantly reduced or oxidised under controlled conditions in redox reactions. The structural integrity of the catalysts depends on the metal on the A site. This is because the B metal will remain dispersed on a matrix of the A2O3 oxides. La has proven to be the most stable compared to other A site cations such as Sr, Ca, Pr, Nd, Sm, and Gd. Due to their high catalytic activities, Mn, Fe, Co, and Ni have been extensively used at the B site [122]. The Mn and Co have been proven to be the best transition metals for carbon monoxide and hydrocarbons' oxidation reactions catalyzed by La-containing perovskites [78]. Some precious metals like Pd and Pt are also used in small quantities [16]. The incorporation of these metals is done to bring unique catalytic properties to the La-containing perovskites that are not observed when cheaper and abundant transition metal cations are used.
Like all the other perovskites, La-based perovskites have the B site cations having primary roles towards catalytic activity whilst the A site is secondary. The reduction profiles of the La-containing perovskites are mostly due to the B cation, proving that it governs the material's redox catalytic properties [32, 47]. The B cations bind to oxygen in two ways; either in bulk or on the surface. Hence desorption studies (O2 − TPD) have been used to investigate the movement of oxygen in their lattice structures. The surface oxygen is usually weekly bound, and as such, it easily desorbs at a lower temperature, whereas the bulk-bound oxygen requires higher temperatures [184]. This are some of the indicators of the stability and properties of the La-based perovskites.
The interlink comprehension between solid-state and catalytic properties is essential to acquiring the most effective perovskite for any catalytic reaction. Fiero et al. has emphasized the surface structure effects on catalytic activity [42]. However, it is important to note that the exterior structure also depends on its composition, therefore, affecting its absorption properties and catalytic efficiencies. It is also important to note that perovskites tend to experience saturation in gas reactions because of loss of coordination and symmetry of the A and B sites which mask the adsorption of reactants on the surface of the catalyst, thus affecting catalytic properties of the La-based perovskite materials [42]. Therefore, for easy determination of the interlink between the two factors, stated above, knowledge about the material's reaction mechanism is important.
1.2.4 General application and mechanism of action of perovskites in catalysis
The perovskites' catalytic mechanism can interlink the solid-state and catalytic properties. The B site metals are placed at relatively large distances from each other; therefore, the substrate can only interact with one site at a time. The perovskites either catalyse the reaction intra-facially or supra-facially. In the intra-facial processes, the catalyst is used up partially in the reaction as a reagent and regenerated at the end, whereas supra-facial mode involves the reaction of the substrates on the surface of the catalyst (Fig. 2).

The (a) supra-facial and (b) intra-facial catalysis by perovskites [43]
For the supra-facial processes to occur, there must be vacant d-orbitals of correct symmetry in the perovskite catalyst. This is the reason why more often than not, transition metals occupy the catalytically active B-site [22, 24, 174]. Overall, perovskites' catalytic mechanism is demonstrated to be governed by the electron occupancy on the eg orbital. When close to unity, the intrinsic oxygen reduction or evolution reaction activity for LaBO3 (where B = transition metals) are optimized. However, the full mechanism has not been fully understood.
Overall, perovskite materials can undergo both A and B site substitutions without altering their crystal structures. However, they encounter some structural defects such as anionic or cationic vacancies which are due to lower valence substitutions on the A site. The electroneutrality of the compound is preserved by adjusting the B cation's oxidation state [117]. It has been proven that vast amounts of accessible oxygens are generated by the redox process and at minimal temperatures when the oxidation states of the B cation are higher. Furthermore, the perovskite becomes more catalytically active in oxidation reactions due to increased oxygen mobility.
Concisely, perovskites are considered potential candidates for substituting noble metal-based catalysts for extensive research in the catalysis field. Up to 90% of the metal elements in the periodic table may be used to substitute either the A or the B site of the perovskites. Thus, perovskites are tailored to meet the requirements of different reactions. However, various opportunities are explored for the development of hybrid catalysts by using the associations between various noble metals and perovskites in small quantities. This aids in reducing the costs of different reactions catalysed by perovskites, making them economically viable. This was emphasised clearly in one of the publications by Misino [107]. Therefore, these advancements in perovskite developments are in line with the requirements sustainable and green technologies.
1.2.4.1 Stability of perovskite oxides
Perovskites are extremely stable, thermally and chemically, which makes them suitable candidates for heterogeneous catalytic reactions. By simply varying the methods in which perovskites are prepared, numerous crystal phase symmetries are acquired. This includes structures such as tetrahedral, orthorhombic, monoclinic, and triclinic [78]. The stability of the perovskite is determined by the Goldschmidt’s tolerance factor. It is noteworthy to mention that, irrespective of the chosen method, the Goldschmidt tolerance factor should lie between 0.75 and 1 for the perovskite structure to be stable [52]. The Goldschmidt tolerance factor is given by Eq. 1
where rA, rB, and ro are the radii of A, B cations and O2− anion, respectively. Perovskites are potential catalysts for a number of reactions because they are reused in different cycles and reduce the amounts of precious elements from the catalysts without altering their activity and stability. In other words, for perovskites to be stable and active as well as characterized as perovskites, they should meet the required ionic radius determined by the tolerance factor calculations. This property defines the perovskite's symmetry, which in turn affects the dielectric properties associated with the material. When the tolerance factor is greater than 1, the B cation is smaller than A and has free space to move flexibly which reduces the activity and stability perovskite [110]. Furthermore, Royer et al. state that a hexagonal ilmenite structure is adopted by the perovskites when the t value goes below 0.75. However, if above 1, a hexagonal symmetry is obtained instead [133].
1.2.4.2 Overall charge distribution in perovskite oxides
Another condition to be fulfilled for the stability of the perovskites is electroneutrality. Variations of the perovskite's crystal’s stability depend on the oxygen content. For example, the A sites are usually coordinated to about 12 oxygen anions, whilst the smaller B sites form octahedral environment with oxygen atoms [78]. The valence state and non-stoichiometry of the cationic sites (A and B) and both anionic and cationic sites, respectively can be controlled using the modifications of the A and B sites based on the electroneutrality factor [170]. Overall, the A site's main function is to induce stability, whereas the B site is considered the primary active site in the perovskite structure [154]. The perovskite's electroneutrality is explained in terms of the cations' total charges in the structure (A and B metal charges) and is equal to that of oxygen anions. For example, the charge arrangement probabilities are A3+B3+O3, A2+B4+O3 and A1+B5+O3.
The electroneutrality allows for partial substitutions at either the A or the B site whilst maintaining the material's overall structure. However, this may result in deformed perovskites due to deficiencies of the oxygen anions and cationic sites. The former case has been reported to be more common. This deficiency is termed the non-stoichiometry in perovskites and is discussed in numerous studies in the literature [143]. To account for the partial substitutions, oxygen excess and deficiency, a more general formula for perovskites is given, that is, A1−xAx′B1−xBx′Oδ. The Ca2Fe2O5 is one example of defected oxides with one-sixth of oxygen vacancies [143]. For instance, Sayagués et al. [136] published a study where an oxygen anion deficient mixed metal oxide, La2Ni2O5, was successfully characterized, showing that the integrity of the perovskites is maintained, and its catalytic properties were conserved.
The presence of excess amount of oxygen, than required, in the perovskite structure is not common as the addition of oxygen interstitially into the framework is not thermodynamically prohibited. Nonetheless, some structures exhibit oxygen in excess, especially those that have cation non-stoichiometry. This could be due to A or B site vacancies, though not common. An example of perovskite with cation non-stoichiometry is Cu0.5TaO3 [123]. Most physical properties linked to the perovskite structure are usually for the non-defected crystal lattices. In most cases, these properties are maintained even with the defected perovskites. Small variations of these physical properties, however, may be observed. Overall, when the electroneutrality and ionic radii requirements have been met, the synthesized material had a higher probability of being a stable perovskite.
1.2.4.3 Electron arrangement in perovskite structures
The initial use of perovskites is dated to be in the mid-twentieth century by Parravano et al. [121]. During that time, numerous ferroelectrics were evaluated for carbon monoxide reduction as the reaction involves electron transfer between the gas and solid phases of the catalysts. There was an evaluation to determine if the electron arrangement contributed to this transfer. Results obtained proved this hypothesis true for sodium, potassium niobates and lanthanum ferrites (perovskites), because they had active transition points for the reaction [121]. The vitality of the electronic arrangement and properties of the perovskite in catalysis was further cemented thirteen years later by the study done by Dickens and Whittingham about the use of perovskites in the form of alkali metal tungsten bronzes [36]. In the study, the recombination of oxygen atoms on the surfaces of these bronzes was studied. The perovskites' electronic properties contributed vastly to the catalytic process. In 1970 and 1971, cobalt-based perovskites were found to be perfect substitutes in electrocatalysis processes. While manganate perovskites were found to be excellent candidates for carbon monoxide oxidation [155]. Since then, perovskites have been assessed in different reactions such as oxidation and reduction of hydrocarbons both partially or fully [133], photocatalytic processes [162], hydrogenolysis of hydrocarbons, NO removal [185], electrocatalytic processes [183], and steam reforming of bio-oil [24], all of which are accessible in the literature.
1.2.4.4 Limitations of perovskites
Despite the versatility of the perovskites, their long-term stability has not been achieved yet, which leaves room for improvement by further exploring the materials. They can be considered as viable substitutes for platinum group metals because they are easy to synthesize, cheap and can be tailored to enhance catalytic capabilities [133]. Although they have not been commercialised in most industrial applications, they are tipped to be potential candidates, especially in automobile exhaust controls [156]. It is, however, noteworthy to mention that these materials are sensitive to sulphur poisoning, which poses limitations to their potential catalytic application in automobile sector. Therefore, studies aimed at understanding the perovskite structure's mechanism are vital becauase they are the key to unlocking many possibilities in the field of catalysis.
1.2.4.5 Acid–base properties of perovskite oxides
The evaluation of perovskites' applications indicated a few other properties and parameters that stood out and contributed to the catalytic activity in various reactions. One of these is the redox property of the perovskites. It was noted that the B site of the perovskite is the main contributor to the catalytic activity observed in perovskite materials [78]. Therefore, its nature is crucial depending on the conditions required by the reaction it is intended to catalyze. The A site, on the other hand, does not bring significant changes to the outcomes of the reaction with one exception. When the partial substitution of the A site occurs, it causes a great change in the valency that influences the oxidation state of the B site, which in turn affects the catalytic properties of the overall perovskite material [157]. An example of this is the La-based perovskite, LaFeO3, where La and its oxidation state's basicity affects the B site Fe from Fe4+to Fe3+,resulting in high catalytic conversions.
Redox properties for perovskites can be evaluated using hydrogen temperature-programmed reductions (H2-TPR). A number of studies reviewing the redox properties for perovskites have been reported so far. For instance, Futai et al. [47] and Crespin et al. [32] produced similar results explaining the perovskite materials' reduction profile to be in two steps instead of the one step of their normal oxide counterparts. In these studies, it was shown that low-temperature TPR profiles were associated with the formation of an oxygen-deficient perovskite structure, whereas the high-temperature profiles indicated the complete reduction of the B cation to a zero-valent metal nanoparticle. These reduction processes are represented by Eqs. 2 and 3. The equations justify the capabilities of the perovskites to catalyse redox reactions. The position of the reduction peaks vary with calcination temperatures the material is subjected to due to the sintering process, which consequently reduces the average surface area. Similar results were also obtained for Fe-based [127], and Mn-based perovskites with small variations depending on the A site cation associated with the perovskite [127]. The Sr substitution on the A cation site was found to promote reducibility of manganese in Mn-based perovskites [68].
In contrast, perovskites' acid–base properties play a crucial role in numerous catalytic reactions such as hydrogenation reactions. The acid–base properties of perovskites are usually evaluated using desorption or adsorption studies of ammonia (NH3) and carbon dioxide (CO2) [186]. Kuhn et al. reported a study that proved the perovskite material surfaces to exhibit both Lewis acidic and basic sites [83]. The A site's changeover results in a charge balance variation that produce acidic sites. Contrarily, properties associated with basic sites dependent on the B cation. This deduction differed from that of Natile et al. [113] who showed that either of the two cations (A or/and B) contributes to the material’s acidic properties. It was also proven in a study reported by Hammami et al. [59] that the perovskites’ surface is not homogenous, leading to different acid/base strengths as shown by their ability to adsorb CO2. This adsorption possibility is made possible by oxygen-deficient sites and surface oxygen, while weak basic sites are from either the A or B cation. These parameters are dependent on the synthesis procedure used in the perovskites’ preparation and affect the morphology, crystal structure, and texture of the catalytic materials. The higher the specific surface area of the perovskite, the better its catalytic performance. This is attributed to the fact that, as the surface area is increased, there are many redox binding sites for the substrates.
Overall, the acid–base properties of perovskites have not been as vastly explored as their redox properties. However, Lewis acid–base theory allows the application of the metal cations and oxygen anions in the perovskite as acidic and basic centers respectively. This is confirmed by a study done Xiao et al., and is complimented by reviews of other studies conducted [45, 99, 126, 172, 173]. These have proven that acidity of the catalysts plays a crucial role in the conversion and selectivity of the reaction, which is a field worth exploring.
1.3 Heterogeneous catalyst supports
Immobilisation of a catalyst increases catalyst’s surface area and the catalyst's lifetime and these are the main roles of metal oxides catalytic supports. In addition, immobilization of catalytic species enhance hydrophobicity and lessen the extent of catalyst deactivation associated with sintering process. In that way, the stability of the catalyst is enhanced chemically, thermally, and hydrolytically [104]. Catalytic supports may act as co-catalysts depending on the nature of catalysed reaction, resulting in enhanced catalytic activity. The study carried out by Cooper et al. where the support improved the transportation of oxidant into the solution is a suitable example of how supports play a dual role during the catalytic reaction [30]. Porosity of the support and a reduction in the supported metal catalyst size increase the exposed active surface area of the atoms of the catalyst material. In a study by Chambers et al. it was found that the different pore sizes of silica gave varying selectivities towards cinnamyl alcohol, hydrocinnemaldehyde and phenyl propanol [19]. For instance, the overall catalysts, catalytic species and the support, with average pore sizes of 20 nm had higher selectivity toward cinnamyl alcohol than the 10 nm bimodal pore structure Cu/SiO2 materials. Therefore, it is prudent to conclude that the support's textural properties govern a material's chemo-selectivity.
Perovskites are supported on numerous supports such as metal-oxides (alumina, titania, silica to name a few) [13, 21, 54, 131], monoliths [115], zeolites [88], fibers [88], ceramic materials, pillared clay [85], red mud [97], polymers[63] and carbon black for different applications. However, this review focuses mainly on the metal-oxide supports. Silica materials are used in the immobilisation of organometallic complex catalyst [44], These are thermally stable, porous and have high surface areas. In contrast, zeolites are hydrous aluminium silicate materials that have been used extensively in oxidation and hydrogenation reactions of numerous halogenated organics [96]. These usually confine positively charged ions of sodium (Na+), calcium (Ca2+), potassium (K+), barium (Ba) and strontium (Sr) or consequent synthetic compounds. However, both the zeolites and silica are used less extensively than the metal-oxides supports because their synthesis is time consuming and require expensive reagents to synthesize as compared to other oxides [125]. The different catalysts that the perovskites are supported on will be discussed in the next sections to highlight their physical and chemical synergies to the activity of the catalyst.
1.3.1 Metal-oxides as heterogeneous catalysts supports
For the improved catalytic performance of metal-oxides, modification of their surface by base or noble metal nanoparticles is necessary. These may be on the oxide's external surface or acting as promoters embedded within the oxides’ pores [34]. In essence, metal-oxides are often utilised as supports for a noble metal catalyst, especially nanoparticles. Some of the transition metals (non-noble metal) catalysts have limitations such as low activity due to low metal-support interaction and high light-off temperatures (temperatures at which conversion reaches 50%) in catalytic reactions despite their availability and low cost. As a result, modifying their surfaces with noble metals is important in trying to overcome these setbacks.
Most industries have adopted the use of metal oxide support materials, especially in the automotive industry as gas purifiers. Within this instance, the size of the metal oxide-supported noble metal play a crucial role. For example, methane combustion using alumina and zirconia supported-palladium and platinum-based catalysts was investigated by Robert et al. [62] to evaluate the structure sensitivity of the overall catalysts. The materials' size was the governing factor for the turnover frequency (TOF) and activation energy. In a different approach, Yang et al. investigated the performance of Ag, Pd, and Pt nanoparticles supported on three-dimensionally ordered mesoporous (3DOM) ceria and alumina (3DOM CeO2-Al2O3) for toluene oxidation. They found that smaller Au particles with homogenous dispersion and uniform porosity of 3DOM CeO2–Al2O3-support led to the lower activation energy for the toluene oxidation. These observations further confirmed that, indeed catalyst support materials play a major role during a catalytic reaction. Similar observation was reported by Arandiyan et al. [6] during methane combustion catalysed by three-dimensionally ordered macroporous La0.6Sr0.4MnO3 supported-Ag nanoparticles (Ag/3DOM La0.6Sr0.4MnO3). It was proven that the diffusion of the substrate was slow on the 3DOM La0.6Sr0.4MnO3 support, which enhanced the contact between the substrate and the Ag/3DOM La0.6Sr0.4MnO3 catalyst, hence increasing the catalytic activity. Therefore, it is safe to assume, based on these literature reports, that the promotion of metal-support interaction is one of the key factors for enhanced catalytic performance in metal oxides-supported catalytic species.
Metal-oxide supports show greater metal-support interactions than any other support. This is better revealed in the study conducted by Kwangjin et al. [5], compared to the catalytic performance concerning metal-support interactions using mesoporous Co3O4, NiO, MnO2, Fe2O3, and CeO2 as supports for platinum (Pt) nanoparticles in a CO oxidation reaction against mesoporous silica supports. It was found that the metal-oxides supported Pt catalysts were more catalytically active than when supported on mesoporous silica. Furthermore, the Co3O4 supported catalysts were substantially more catalytically active than the other metal-oxides catalysts because of the oxidation stabilisation and oxygen content of Co2+ as shown in Fig. 3.

Approximations of different activities of various metal support as reported by An et al. [5]
1.3.2 Alloying strategy on metal-oxides supports
Periodic and ordered pores in an oxide structure provide stability to the nanoparticles deposited onto its surface and make the accessibility of the active atom sites to the reactants easy [94]. In various reduction and oxidation reactions, porous oxides are used extensively to support noble metal catalysts. The wide use of porous metal oxides is attributed to the confinement of the active site and control of alternative attack pathways which subsequently promote higher selectivity [147]. Supported bimetallic noble metal alloys have better selectivity and activity than mono-metallic counterparts [94]. The alloying strategy is used vastly to produce hybrid-like catalysts on oxide supports, therefore, providing the catalytic material’s unique properties.
The alloying strategy is utilized to alter the electronic structure and properties of transition metals (such as Ag, Au, and Cu) catalysts with filled d-orbitals that are not willing to form bonds and are catalytically inactive, making them more catalytically active [101]. The first bimetallic noble metal alloy reported was comprised of Au and Pd catalysts in 1980 which was utilised in hydrogen fuel cells, pollution control, [161] and other various applications [152]. Thereafter, the study on acetoxylation of ethylene to vinyl acetate conducted by Chen et al. over palladium-gold alloy (Pd-Au alloy) catalysts in the year 2005 validated the alloying strategy proof that the formation rate of vinyl acetate was enhanced for non-contiguous and suitable spaced monomer Pd sites than for just single Pd atoms [26]. To date, numerous studies have been reported on bimetallic catalysts supported on metal-oxides with ordered pores and large surface area, thus promoting effective dispersion of the metal alloys on the oxide support’s surface.
1.3.3 Metal oxides as supports for other metal oxides
Just like metallic nanoparticle catalysts, metal-oxides may too be supported on solid supports. The metal-oxides and the support are bonded via surface hydroxyl groups to form M–O–S bonds [132]. This, however, varies vastly depending on the metal-oxides used. For instance, molybdenum species offer Mo=O on their terminal side and four bridging oxygen to the support while the vanadium species offers three bridging instead, see Fig. 4. In aqueous conditions, the M–O–S bond can be hydrolysed, forming aqueous solutions of the supported oxides. However, some dehydrated species have been used in other studies, and it was found that the structure is conserved during catalytic reactions [14].

Proposed environment of vanadium (V) oxide and manganese on silica supports [33]
Species spread out on dehydrated supported oxides are stabilized through surface functional group interactions such as hydroxyl groups, which control the oxide's monolayer coverage on the support [14]. These are usually isolated or bi-dimensional polymeric species. As Wachs reported, the monolayer coverage of many oxides has been determined to be more than that of silica supports [158]. The interactions amongst the supported oxide materials are directly proportional to the monolayer loading resulting in the formation of M–O–M bonds for polymeric species less pronounced in silica supports. For this reason, supported metal oxides have vast applications in many catalytic reactions [14].
1.4 Immobilised perovskite type oxides catalysts on metal oxide supports
Despite the advantages stated about perovskites as catalysts such as their ability to be tailored, high absorption coefficient, excellency as transport materials, robust stability, and low cost, they are limited by small surface areas, more especially if their porous structures collapse due to harsh reaction conditions [28, 74, 76]. To counteract this problem, the use of supports with large surface areas that expose the perovskites' active sites has been explored [180, 184]. It is, however, still challenging to model an ideal perovskite/support hybrid type catalysts.
Achieving the ideal hybrid catalyst that conserves the perovskite structure upon immobilisation on a support and maximising the perovskite's uniform dispersion is the main goal driving many researchers to explore the preparation and application of these types of catalysts. For this reason, there are numerous studies carried out on metal oxide-supported perovskites catalysts [3, 4]. Attempts are made to immobilize perovskites on silica and alumina by coating the perovskites onto the supports. However, non-active silicate and aluminate materials were formed, therefore, these materials were found to display poor catalytic activity as supported perovskites [108]. Despite the inactivity of silicate and aluminates, silica- and alumina-centred supports are frequently used because they ensure well-defined perovskites due to the inert characteristics of the supports. According to the discoveries made by Kaliaguine et al. MCM-41 mesopores are highly advantageous for supporting LaCoO3 with 10–50 wt% of the perovskites within the pores [114]. When analysing the supported perovskites’ physicochemical properties such as surface areas and pore diameters, they are far improved than their unsupported counterparts [102]. However, the supported perovskite materials suffered from the reduction of the MCM-41 support weight ratio of the catalyst. Therefore, ceria and zirconia oxides counterparts are more ideal candidates for this purpose. This is due to the oxygen storing properties associated with the low reduction potential amongst Ce3+and Ce4+ [46, 184]. This was proven when LaCoO3 and LaFeO3showed high conversions in the CO oxidation when supported on ZrO2 [29]. Even better conversions were obtained when LaCoO3 was supported on mixed oxides support, Ce1−xZrxO2, via wet impregnation method. Overall, it suggested that enhanced oxygen mobility and high active surface area govern the catalytic performance of supported perovskite. The next sections place emphasis on the use of supported and unsupported perovskites as catalysts in catalytic transfer hydrogenations. To do so, catalytic transfer hydrogenation in general will be discussed prior to explaining the advances of perovskites with this type of chemical transformations.
1.5 Catalytic transfer hydrogenations
Hydrogenation reactions involving organic compounds play a crucial role to play both small-scale laboratories and industries. Reduction reactions can be classified into (i) hydrogen addition to unsaturated compounds and (ii) addition of hydrogen across single bonds leading to bond cleavage, known as hydrogenolysis. It can also be the removal of oxygen from organic compounds [75]. This review, however, will solely focus on the hydrogenation reaction aspects of reduction, especially catalytic transfer hydrogenations (CTH). During the twentiethcentury, CTH reactions were under-utilized. However, with the progress of research in the twenty-first century, there are advances to explore their use. For example, Braude et al. [17] conducted one of the first CTH reactions using cyclohexane and similar hydroaromatic compounds as hydrogen donors for the reduction of ethylenic and acetylenic bonds in hydrocarbons and carboxylic acids in the year 1954 although it was overlooked due to long reaction times and poor product yields. However, with advances in the mordern techniques, the yield and the reaction times are significantly improved and shortened, respectively.
The advantages of CTH reactions are vast as compared using molecular hydrogen. Molecular hydrogen is an easily diffusible gas with low molecular weight, which contacts the catalyst-solvent mixture easily. However, it is easily ignitable and presents hazards, especially in large-scale processes. Moreover, it has low solubility in many solvents. Therefore, high pressures are often required in order to achieve optimum yields and selectivity. This consequently increases the costs associated with the reaction processes, especially when considering large-scale production in industrial setups [49]. The use of bioderived hydrogen-containing molecules as sources of hydrogen for hydrogenation reactions helps reduce these drawbacks and opens an opportunity to control the reaction's selectivity using the hydrogen donor's properties [75].
Consequently, the best hydrogen donors are made of hydrogen-bonded to compounds, groups or elements with similar electronegativities, such as hydrogen itself. Therefore, alcohols, formic acids, hydrazine, hydrides of boron, and hydrocarbons can be utilised as hydrogen donors [49]. The transfer of hydrogen can be through different pathways, for example, the intermolecular hydride transfer which can be swayed in favour of better selectivity control. However, the intermolecular hydrides were overshadowed by the use of molecular hydrogen until a few decades ago. Low yields and reaction rates are also some of the major drawbacks but vast progress has been made in reducing various functional groups such as alkenes, alkynes, carbonyls, nitrous, cyano, and N=N bonds through the CTH reactions.
There were concerns about the use of bio-derived materials as hydrogen donors for catalytic transfer hydrogenation reactions because the process increase the steps of separation and recycling processes. This concerns both the alcohol that remains untransformed, which act as internal hydrogen donor and aldehydes produced, which can subsequently be separated and utilised as substrates in Aldol condensation reactions to make value-added chemicals [27, 49]. Therefore, this serves as one of the major setbacks of catalytic transfer hydrogenations. To add to the setback, the identification of active sites on solid catalysts in catalytic transfer hydrogenations poses a challenge because the catalyst's surface is usually unstable.
Extremely effective method for the catalytic generation of 2,5-di-methylfuran from 5-(hydroxymethyl)furfural was reported by Thananatthanachon et al. [146], using formic acid as an internal hydrogen donor and as an acid catalyst together with Pd/C. High conversions of up to 95% were obtained. One vital aspect to note from the conclusions drawn from this study was that formic acid as a hydrogen donor in catalytic transfer hydrogenations reduces handling and transport issues posed by hydrogen but presents difficulties in maintaining the material and durability of the catalyst. This is due to the corrosive nature of the acid. Because of their non-corrosiveness, alcohols are extensively studied for catalytic transfer hydrogenations. For example, in another related study, the same reaction was investigated using 2-propanol as a hydrogen donor. A lower yield of 80% was achieved, compared to when formic acid is used [70].
Armstrong et al. conducted a study on transfer hydrogenolysis of aromatic alcohols using Raney Co catalysts and 2-propanol as a sacrificial alcohol [53]. The dehydromethylation was found in the hydrogenolysis of primary alcohols, and Raney Co in refluxing 2-propanol is an effective system for deoxygenating α-substituted alcohols only. A study was conducted by Scholz et al. that initially focused on the catalytic transfer hydrogenation of furfural and 5-hydroxymethylfurfural to 2-methylfuran and 2, 5-dimethylfuran, respectively over in-situ reduced, Fe2O3-supported Cu, Ni, and Pd catalysts using 2-propanol as an internal hydrogen donor [137]. The expected primary product in this reaction was furfural alcohol. However, successive hydrogenolysis and decarbonylating of the furfural were secondary reactions. Therefore, it was necessary for further studies to be conducted to circumvent these secondary reactions. Further investigations towards the development of selective perovskites and other potential catalytic candidates were conducted. Remarkably, the studies validated that 2-propanol acted as a hydrogen donor by monitoring the produced acetone concentration, a ketone by-product.
1.5.1 Donor capabilities of alcohols in CTH
In terms of the hydrogen donor capabilities of alcohols, secondary alcohols display a better donor potential than primary alcohols [35, 50, 64, 86]. The strong donor capabilities of secondary alcohols is attributed to the stabilisation of the secondary carbocation that forms upon donation of hydrogen. Scholz et al. [137] upon transfer hydrogenation of furfural and 5-hydroxymethylfurfural to 2-methylfuran and 2, 5-dimethylfuran, respectively over in-situ reduced Fe2O3-supported Cu, Ni, and Pd catalysts also reported that the substitution of the alcohol plays a part in its donor capabilities. Therefore, a decrease in carbon chain length results in a decrease in the donor capabilities. Of the studied alcohols such as 2-butanol, ethanol, pentanol and 2-propanol, the best hydrogen donor capability was displayed by 2-propanol. Similarly, Johnstone et al. reported that alcohols could be used as both donors and solvents in the catalytic transfer hydrogenation reactions of aromatic compounds [75]. Tertiary alcohols, although stabilised, cannot serve as donors because they lack the α-hydrogen (hydrogen on the carbon next to the hydroxyl group).
The effect of the structure of alcohols in catalytic transfer hydrogenations was reported by Vlachos et al. who discovered that secondary alcohols were superior to primary counterparts in converting furfural to 2-methylfuran utilising a carbon supported-Ru based catalyst [118]. Furthermore, they proved that whether in primary or secondary alcohols, the length of the alkyl chain affected the reaction. The longer the chain, the better the donor capabilities resulting in higher catalytic activity. However, there was one challenge encountered when the side chain (alkyl group) had more than two carbon atoms, the donor became weak. This could be attributed to steric hindrance or a stronger C-H bond. The study's findings further proved that the alcohols' activity was in the order 2-pentanol, 2-butanol, 2-propanol, 1-butanol, 1-propanol, and lastly, ethanol.
Unsaturated alcohols have numerous applications in industry, for example in fragrances and drug synthesis. It is, therefore, important for cheaper, greener, and sustainable procedures to be established in this endeavour. Acquiring desirable selectivity during these CTH reactions has proven to be challenging for unsaturated carbonyl compounds, especially if the target site is the carbonyl site because it is less thermodynamically favoured than the C=C [142]. The use of homogenous catalysis for the synthesis of these alcohols is possible. However, heterogeneous counterparts are more environmentally friendly as they are easy to recycle and separate from the products [100]. The catalysts' ability to activate the C=C and C=O bond of the unsaturated aldehydes is the key towards CTH reactions of unsaturated aldehydes.
Hydrogen generation from liquid sources has gained so much interest because it is known in organic synthesis. Methanol, ethanol, propanol, and formic acid are promising hydrogen donors produced in vast amounts from fossil-based feed. Base-free catalytic transfer hydrogenations of aldehydes and ketones using 2-propanol as a hydrogen source were evaluated by Dubey et al. [37]. In the study, Pd, Ru and Rh complexes were used as catalysts stabilized by organochalcogen ligand. They were successfully characterised using NMR, HR-MS and single-crystal X-ray diffraction. The base free hydrogenation was done upon optimum loading of the catalyst (0.2–0.5 mol %) at ambient conditions. The reaction was proven to be a homogenous reaction with the alkoxides, and M-H bond formation proposed in an uncertain proposed mechanism based on the 1H NMR and ESI–MS at reaction times of 1–2 h.
Most CTH reactions use biomass-derived chemicals with high oxygen moieties and high boiling points and molecules that decompose easily. Therefore, the use of alcohols as donors proves advantageous because they serve as hydrogen donors and solvents at the same time. Self-etherification of alcohols is a challenge because it is a side reaction in catalytic transfer hydrogenations, especially in the presence of Lewis acid sites on the catalyst surface. However, these side reactions have not received much attention in literature because their yields are negligible. Supported metal nanoparticles such as Pd, Ru, Au, and Pt are the most frequently employed catalysts for catalytic transfer hydrogenations and the supports of choice include activated carbon, Al2O3, zeolites, and porous metal-oxides.
1.5.2 Catalysts for catalytic transfer hydrogenation reactions
Zeolites are effective catalysts in catalytic transfer hydrogenations, especially tetrahedrally coordinated d0 or d10 metal centres. This is attributed to their Lewis acid character [69]. Little is known about long-term stability and the deactivation of zeolites in transfer hydrogenations. During zeolite catalysed CTH reactions, the synthesis of diether is favourable but challenging as it requires carbonyl group hydrogenation before etherification in unsaturated aldehydes [92]. There are numerous catalysts that are utilised for this reaction. Noble metal catalysts like palladium, rhodium and platinum have also been used in CTH reactions, however, they are expensive which increases the cost of the research. Therefore, perovskites serve as viable counterparts which are cheaper and can be tailored to suit the properties essential for the catalytic transformation [41]. This review will focus on the use of mixed oxides, in particular, perovskites oxides. Table 2 compares the activity and selectivity of different catalysts on the CTH reaction of cinnamaldehyde.
1.6 Perovskites metal oxides as catalysts for catalytic transfer hydrogenation reactions
The selection of metals to use during heterogeneous catalytic transfer reactions can prove complex because of the need to evaluate their geometric and electronic properties. Firstly, metals exist in different crystal phases, influencing the catalytic properties. The face cantered cubic metals with Miller indices (111) have lower adsorption steps as compared to (100) [101]. It is imperative to perform an X-ray diffraction analysis of the as-synthesized materials to estimate the reaction pathway that the reaction will follow. Secondly, the metal's electronic properties are crucial in selecting the catalyst because they determine the reactivity and adsorption of substrates onto the catalysts' surface.
Hydrogenation transformations of various compounds into value-added chemicals using perovskites as heterogeneous catalysts have been a success. Bewana et al. studied the selective catalysis of cinnamaldehyde to cinnamyl alcohol using selective nanoparticles embedded in perovskite materials [16]. In extremely short reaction times, conversions of approximately 25% were achieved when pure perovskites were used, owing to the materials' porous nature and their remarkable electronic properties. Moreover, the catalyst was recyclable, achieving up to 100% selectivity towards hydrocinnemaldehyde. The perovskite materials had been synthesized using the nanocasting method and induced a synergistic effect on Pd nanoparticles to obtain conversions greater than 80%.
Similarly, perovskites in catalytic transfer hydrogenation reaction are advantageous in terms of the ease of separation upon completion of the reaction and show robust activity during the reduction of nitro compounds to the desired amines. This was shown in a study of selective transfer hydrogenation of aromatic nitro compounds into aromatic amines under microwave heating by Farhadi et al. where ferromagnetic BiFeO3 nano-powdered catalysts were synthesized from the Bi[Fe(CN)6].5H2O complex through thermal decomposition [40]. Particles of up to 20 nm and with surface areas of 48.5 m2/g produced yields and selectivities of up to 96% using propanol as a hydrogen donor under microwave radiation. The perovskite catalysts were magnetically recoverable and recyclable, and this proved faster and more suitable for large scale anilines production. The catalytic transfer hydrogenation of these compounds is cheap, regio- and chemo-selective, and compatible with the reducible substrates.
Conversely, Pt, Pd, Ru and Raney Ni are classical catalysts employed in catalytic transfer hydrogenation reactions [40]. This provides a promising method for generating pure nanosized perovskite materials with relatively high surface areas for catalysis. This method is the thermal decomposition of heteronuclear complexes and provides high circumvention of strong acidic media superior to its noble metal catalyst counterparts. The strong acidic media must be circumvented to prevent corrosion and to give better control of the outcomes of the reaction.
This method validates the findings made by Kulkarni et al. who used microwave radiation to synthesize La-based perovskites, LaMO3, (where M = Mn, Fe, Co, Cr or Al) [84]. Just like with Farhadi et al. the method was found to be exceedingly fast and cost-effective for the synthesis of specifically tailor-made perovskites. The perovskites were synthesized in just 15 min and were successfully used to reduce aromatic nitro compounds using isopropanol, in this instance, with potassium hydroxide as a promoter. High conversions and selectivities were obtained. This was further validated by the study published a year later, where the influence of substitutions at A sites in the perovskite structure was evaluated in the nitrobenzene reduction, using the same conditions and a series of La1−xSrxFeO3 (x = 0.0–1.0) oxides as catalysts [84]. There have been numerous other reports on the catalytic transfer hydrogenation of nitro compounds using different hydrogen donors over the past two decades [58, 138].
1.6.1 Metal oxides as heterogeneous catalyst supports for CTH
The use of metal-oxides supports is studied in catalytic hydrogenation reactions because its nature affects different catalysts' motion during the catalysts' pre-treatment. The acidic supports favour side reactions [82], whilst basic supports favour unsaturated alcohols due to their ability to present properties that are electron-rich to the active metal. Due to its ability to enhance chemoselectivity in carbonyl compounds' hydrogenation, iron oxide (Fe2O3) has been used extensively as supports [101]. In the studies done by Milone et al. the purpose of introduction of ferric sites was to enhance the electronic properties of the Au NPs [106]. Therefore, catalysts support plays a vital role in governing the electronic and adsorption properties of the catalytic material, enhancing conversions and selectivities in CTH reactions. Therefore, the addition of inexpensive metals often reduces the occurrence of side reactions. Similarly, in a study done by Scholz et al. it was found that the hydrogenation of furfural using Pd/FeO3 as a catalyst was attributed to strong metal-support interactions, whilst the selectivity has been shown to depend strongly on Pd loading.
1.6.2 Heterogeneous metal oxide catalysts pre-treatment for CTH reactions
Considering the origins of the metals intensively, catalysts need to undergo pre-treatment as they might not be active as metal precursors. There are many reasons for pre-treatment, these include changing the material’s size, increasing activity and selectivity [87, 178], removing residual chlorides and influencing alloy formation [101]. Furthermore, it reduces the reducibility and increase oxygen sites depending on the treatment [12]. Depending on the material's origins, it is important to calcine the material in pre-treatment [54, 55]. Catalytic metal dispersion of the metal is one of the attributes of pre-treated material. Other techniques that may be utilized for the enhancement of catalytic material are ageing and the use of modifiers [56, 57].
1.6.3 Effect of acidity and basicity of metal oxide catalysts in CTH reactions
The transformation of furfural to a value-added chemicals such as furfural alcohol using CTH requires the carbonyl and alkene functional groups' activation. This is like sugar hydrogenation, in which many catalysts have proved to be selective and highly active. Metal–acid or base pairs are remarkable with CTH reactions [73]. Catalytic activity in terms of turnover frequencies of 60 h−1 has been obtained for Ru and Pd catalysts with high selectivities. However, this does not exclude non-noble metals like Ni, Cu, Co and Fe, especially when infused in the perovskite structure. It has been proven that the Lewis acidity outpaces the basicity when it comes to methyl furan during the CTH of furfural. This is true for all the other unsaturated carbonyl compounds. The formation of the side products requires further investigations. Therefore, it is vital to note that morphology and metal support interactions are crucial for CTH reactions, with the acid/base-H bond determining the intrinsic kinetics for C-O cleavage.
Acid–Base catalysts accelerate catalytic hydrogenation reactions mostly through direct hydrogen transfer between a donor and substrate molecule. This is attributed to the fact that hydrides' formation is not thermodynamically favoured on these acid–base pairs and it is extremely difficult. Both the acid and the base sites are crucial for the reaction, although they are unlikely to be on the same catalysts. The electron-deficient metal centre of the Lewis acid bonds to the carbonyl and hydroxyl oxygen of both the substrate and the donor, respectively. On the other hand, the base attracts the H of the O–H bond, weakening it. Therefore, the acidity depends on the Lewis acid’s strength and the hydroxyl interaction, facilitating the proton's base abstraction. If the base is too strong, it initiates a hydride transfer mechanism as it abstracts the proton leaving an alkoxide [49]. This mechanism is similar to the one described by Xiao et al. as illustrated in Fig. 5.

Catalytic transfer hydrogenation mechanism as proposed by Ping et al. [173]
1.6.4 Carbon -doped perovskites in CTH reactions
Carbon composites improve perovskites’ performance when prepared in-situ as far as CTH reactions are concerned. Xiao et al. studied the design of perovskites to improve the activity and selectivity of LaFeO3 perovskites towards the CTH reaction of furfural-to-furfuryl alcohol [172]. The basic notion is to ensure the timely delivery of the reactants onto catalysts' active sites for the reaction to occur at its optimum, manipulating the holes made between the catalyst particles. The perovskites with the carbon material have an enhanced reaction rateS, six times faster than that of perovskites without the carbon material, of 5.17 mmol g−1 h−1. Thus, from the results of this study, it is evident that carbon's absorptive characteristics increase the migration of reactants from the solution onto the catalyst's active sites. In addition, the area of the material seemed to be positively affected by the presence of carbon. This opens the channels for improving perovskites and all other low surface area catalysts, providing a better future for these types of catalytic materials. Moreover, the mechanism of the reaction was fully explored. This mechanism was similar to the discoveries they made the previous year, as illustrated in Fig. 4 [173]. Proposed mechanism of catalytic transfer hydrogenation reactions on metal oxides.
Xiao et al. described the catalytic hydrogen transfer mechanism of furfural and cinnamaldehyde over perovskites to be an acid–base rather than the redox mechanism [173]. The isopropanol binds to the perovskite's metal cations, either the A or B cations, using the oxygen atom on the hydroxyl group. The hydrogen atom of the hydroxyl group, in turn, interacts with the adjacent oxygen anion. The carbonyl oxygen of the substrate binds to the oxygen vacancy on the perovskite, from which the reducing electron comes. This makes the acyl carbon susceptible to nucleophilic attack, initiating enolization. This way, the catalyst acts as an interface for the hydrogen donor and the substrate. Enolization occurs with this arrangement, resulting in the transfer of two hydrogen atoms from the hydrogen donor to the carbonyl compound (Fig. 4).
Catalytic transfer hydrogenation reactions described by Gilkey et al. have two main mechanisms: direct hydrogen transfer or the metal hydride route in the homogenous systems [49]. These use deuterated hydrogens as tracking atoms. Upon utilising alcohols as hydrogen donors, the direct hydrogen pathway is employed where the –H is transferred from the –carbon of the donor to that of the substrate without forming the metal hydride. The mechanism is believed to go through a concerted step involving forming a six-membered ring intermediate: the Meerwein-Ponndorf-Verley (MPV) mechanisms. The presence of a base enhances reactivity. The MPV mechanism has also been proven to apply to heterogeneous catalysts. With solid catalysts, the possibility of the MPV mechanism is brought by a Lewis acid site that has a deficit of electrons and a neighbouring base site.
However, some heterogeneous catalysts undergo the metal hydride route, split into two: the monohydride and dihydride mechanisms, as shown in Scheme 2. The difference between the two lies in whether the hydrogen atoms in either the hydroxyl or C-H are preserved in the product. Thus, when alcohols and formic acid loose the hydrogen from the –carbon to the metal, the mechanism is referred to as the monohydride process. If both the hydrogen from the –carbon and the hydroxyl group are transferred, it is called the dihydride mechanism [49]. Nevertheless, the study of the mechanisms of catalytic transfer hydrogenation reactions has proven to be complex when using heterogeneous catalysts. As a result, more studies are conducted to try and rectify this challenge. However, it is believed that the mechanism with perovskites is an acid–base catalysed reaction.

Direct hydrogen transfer and metal hydride route of catalytic transfer hydrogenation [49]
Overall, it is vital to note that despite all the benefits of CTH reactions, there is a gap in the fundamental and systematic studies on the hydrogen generation's kinetics on bimetallic and metal acid–base pair catalysts. Furthermore, the changes that occur to the catalyst surface during the reaction are not yet fully understood. However, postulations have been made on vital parameters such as hydrogen spill-over, hydrogen donor efficiency, and uptake capabilities. This may be attributed to the fact that most studies concentrated on catalyst composition and performance instead of the nanostructures and interfacial characteristics. Therefore, there is a need for more research to be conducted in this regard.
1.7 Synopsis and outlook
Due to the reliability of heterogeneous and electro-chemical catalysis on the exposure of metal cations to electrons to accommodate the reactants and intermediates, metal oxides, more especially perovskites have been proven by different sources to be suitable candidates to replace noble metals. This is due to their tunable unique structural properties. In attaining purity and suitable textural properties for catalysis, the metal oxides' synthesis protocol is vital. The sol–gel citrate and nanocasting methods gained the popularity among the reported protocols. The use of perovskite structured oxide in heterogeneous catalysis is progressing, especially in applications involving high temperatures and oxygen steam atmospheres where their stability comes to play.
However, there are challenges that emanate when synthesizing metal-oxides. These include high coordination numbers of transition metal ions, which pose challenges in terms of control during the assembly with surfactants and destruction of the micropores by heat treatment required for template removal or structure crystallization. Therefore, there is a need to either use easily removable templates or less coordinated oxides. The costs of the synthesis of perovskites is high due to expensive templates such as biopolymers [18, 163]. Ideally, natural sources like wood and porous minerals would be used to substitute expensive templates, improving the synthesis steps economically. The template removal step would be eliminated in terms of structure retainment, although they minimise architectural control. Perovskites require high temperatures and long calcination times during their synthesis, especially when comprising two or more metals. This challenge makes them thermally stable with extremely high boiling points; however, it results in low surface areas, limiting their surface reactions. Sintering, poisoning, and leaching are the main causes of catalyst deactivation on heterogeneous catalysis. However, these can be controlled using catalytic supports. Perovskites on catalyst supports are promising alternatives that sort the surface area problem associated with heterogeneous catalysts. Perovskites with high surface areas need to be synthesized by focusing on improving synthesis methods by advancing porous structures or nanomaterials. This should be done considering the associations between the physical and chemical properties and catalytic activity. Secondly, due to their diverse, interconnected channels that maximise contact with substrates, there is a need to synthesise 3D perovskites. Lastly, there must be an extensive improvement of perovskites’ electronic conductivity before implementing them on catalysis.
Catalytic transfer hydrogenation reactions are sophisticated processes that entail the complex design of interfacial structures for controllable kinetics. Numerous research efforts have concentrated on the composition of the catalysts for reactions other than interfacial characteristics and nanostructures. Therefore, future research is recommended to focus on enhancing the atom efficiency of catalytic transfer hydrogenations and understanding electrons’ reconfigurations due to morphological changes and structural manipulation. Furthermore, understanding the characterization of substrate interactions with in-situ hydrogen is vital for unsaturated aldehydes hydrogenation on the catalyst’s surface. Therefore, intrinsic properties need to be considered for complete optimisation, such as metal hydrogen bonding and activation barriers.
Overall, catalytic transfer hydrogenation reactions are among the green and cheaper reactions that make chemical synthesis more sustainable. This reaction eliminates the need for precious metal catalysts and the use of hazardous molecular hydrogen. This decreases costs as the need for high-pressure reactors is eliminated. Moreover, sustainable development is achieved as these readily available bioderived hydrogen donors replace their non-renewable counterparts, reducing the carbon footprint. There are limitations with this reaction, including the price of external alcohols, hence reducing this reaction’s applicability compared to hydrogen-based hydrogenations. Incidentally, cheap primary and secondary alcohols have been utilized as hydrogen donors and solvents.
References
Alam MI, Khan TS, Haider MA (2019) Alternate biobased route to produce Î-decalactone: elucidating the role of solvent and hydrogen evolution in catalytic transfer hydrogenation. ACS Sustain Chem Eng 7:2894–2898. https://doi.org/10.1021/ACSSUSCHEMENG.8B05014/ASSET/IMAGES/LARGE/SC-2018-050145_0004.JPEG
Alamdari H (2007) United States US 20090324470A1 (12) Patent Application Publication (10) Pub
Alifanti M, Blangenois N, Florea M, Delmon B (2005) Supported co-based perovskites as catalysts for total oxidation of methane. Appl Catal A Gen 280:255–265. https://doi.org/10.1016/j.apcata.2004.11.005
Alifanti M, Florea M, Pârvulescu VI (2007) Ceria-based oxides as supports for LaCoO3 perovskite; catalysts for total oxidation of VOC. Appl Catal B Environ 70:400–405. https://doi.org/10.1016/j.apcatb.2005.10.037
An K, Alayoglu S, Musselwhite N, Plamthottam S, Melaet G, Lindeman AE, Somorjai GA (2013) Enhanced CO oxidation rates at the interface of mesoporous oxides and Pt nanoparticles. J Am Chem Soc 135:16689–16696. https://doi.org/10.1021/ja4088743
Arandiyan H, Dai H, Deng J, Wang Y, Sun H, Xie S, Bai B, Liu Y, Ji K, Li J (2014) Three-dimensionally ordered macroporous La0.6Sr0.4MnO3 supported Ag nanoparticles for the combustion of methane. J Phys Chem C 118:14913–14928. https://doi.org/10.1021/jp502256t
Arjun N, Pan GT, Yang TCK (2017) The exploration of Lanthanum based perovskites and their complementary electrolytes for the supercapacitor applications. Results Phys 7:920–926. https://doi.org/10.1016/j.rinp.2017.02.013
Asiedu A, Barbera E, Naurzaliyev R, Bertucco A, Kumar S (2019) Waste cooking oil to jet-diesel fuel range using 2-propanol via catalytic transfer hydrogenation reactions. Biofuels 12:723–736. https://doi.org/10.1080/17597269.2018.1532754
Asiedu A, Davis R, Kumar S (2020) Catalytic transfer hydrogenation and characterization of flash hydrolyzed microalgae into hydrocarbon fuels production (jet fuel). Fuel 261:116440. https://doi.org/10.1016/J.FUEL.2019.116440
Asiedu A, Kumar S (2019) Kinetics and optimization of catalytic transfer hydrogenation of WCO using 2-propanol as a hydrogen donor over NiOx–MoOx–CoOx/zeolite. Ind Eng Chem Res 58:15787–15802. https://doi.org/10.1021/ACS.IECR.9B00648/SUPPL_FILE/IE9B00648_SI_001.PDF
Assirey EAR (2019) Perovskite synthesis, properties and their related biochemical and industrial application. Saudi Pharm J SPJ 27:817. https://doi.org/10.1016/J.JSPS.2019.05.003
Bachiller-Baeza B, Guerrero-Ruiz A, Rodríguez-Ramos I (2000) Role of the residual chlorides in platinum and ruthenium catalysts for the hydrogenation of α, β-unsaturated aldehydes. Appl Catal A Gen 192:289–297. https://doi.org/10.1016/S0926-860X(99)00411-1
Bagheri S, Muhd Julkapli N, Bee Abd Hamid S (2014) Titanium dioxide as a catalyst support in heterogeneous catalysis. Sci World J 2014:1–21. https://doi.org/10.1155/2014/727496
BanÄares MA (1999) Supported metal oxide and other catalysts for ethane conversion: a review. Catal today 51:319–348
Beck JS, Vartuli JC, Roth WJ, Leonowicz ME, Kresge CT, Schmitt KD, Chu CTW, Olson DH, Sheppard EW, McCullen SB, Higgins JB, Schlenker JL (1992) A new family of mesoporous molecular sieves prepared with liquid crystal templates. J Am Chem Soc 114:10834–10843. https://doi.org/10.1021/ja00053a020
Bewana S, Ndolomingo MJ, Carleschi E, Doyle BP, Meijboom R, Bingwa N (2019) Inorganic perovskite-induced synergy on highly selective Pd-catalyzed hydrogenation of cinnamaldehyde. ACS Appl Mater Interfaces 11:32994–33005. https://doi.org/10.1021/acsami.9b10820
Braude EA, Linstead RP, Mitchell PW (1954) Hydrogen transfer. Part VI. Metal-catalysed transfer-hydrogenation of ethylenic compounds. J Chem Soc 3578–3585
Burch R, Harris PJF, Pipe C (2001) Preparation and characterization of supported La0.8Sr0.2MnO3+x. Appl Catal A Gen 210:63–73. https://doi.org/10.1016/S0926-860X(00)00790-0
Chambers A, Jackson SD, Stirling D, Webb G (1997) Selective hydrogenation of cinnamaldehyde over supported copper catalysts. J Catal 168:301–314. https://doi.org/10.1006/jcat.1997.1683
Chapelle A, Oudrhiri-Hassani F, Presmanes L, Barnabé A, Tailhades P (2010) CO2 sensing properties of semiconducting copper oxide and spinel ferrite nanocomposite thin film. Appl Surf Sci 256:4715–4719. https://doi.org/10.1016/j.apsusc.2010.02.079
Chatterjee M, Ikushima Y, Zhao FY (2002) Highly efficient hydrogenation of cinnamaldehyde catalyzed by Pt-MCM-48 in supercritical carbon dioxide. Catal Lett 82:141–144. https://doi.org/10.1023/A:1020644213929
Chen D, Chen C, Baiyee ZM, Shao Z, Ciucci F (2015) Nonstoichiometric oxides as low-cost and highly-efficient oxygen reduction/evolution catalysts for low-temperature electrochemical devices. Chem Rev 115:9869–9921
Chen H, Lin T, Zhang S, Xu H, Tao H, Chen W (2021) Novel FeII/EDDS/UV/PAA advanced oxidation process: mechanisms and applications for naproxen degradation at neutral pH and low FeII dosage. Chem Eng J 417:127896. https://doi.org/10.1016/J.CEJ.2020.127896
Chen J, Sun J, Wang Y (2017) Catalysts for steam reforming of bio-oil: a review. Ind Eng Chem Res. https://doi.org/10.1021/acs.iecr.7b00600
Chen J, Zhang X, Arandiyan H, Peng Y, Chang H, Li J (2013) Low temperature complete combustion of methane over cobalt chromium oxides catalysts. Catal Today 201:12–18. https://doi.org/10.1016/j.cattod.2012.03.026
Chen M, Kumar D, Yi CW, Goodman DW (2005) Chemistry: the promotional effect of gold in catalysis by palladium-gold. Science 310:291–293. https://doi.org/10.1126/science.1115800
Chen Y, Zhang L, Zhang Y, Gao H, Yan H (2018) Large-area perovskite solar cells—a review of recent progress and issues. RSC Adv 8:10489–10508. https://doi.org/10.1039/C8RA00384J
Chiang C-H, Tseng Z-L, Wu C-G, Wu CG (2014) Planar heterojunction perovskite/PC 71 BM solar cells with enhanced open-circuit voltage via (2/1)-step spin-coating process. J Mater Chem A 2:15897–15903
Colonna S, De Rossi S, Faticanti M, Pettiti I, Porta P (2002) XAS characterization and CO oxidation on zirconia-supported LaFeO3 perovskite. J Mol Catal A Chem 187:269–276. https://doi.org/10.1016/S1381-1169(02)00231-5
Cooper C, Burch R (1999) An investigation of catalytic ozonation for the oxidation of halocarbons in drinking water preparation. Water Res 33:3695–3700. https://doi.org/10.1016/S0043-1354(99)00091-3
Corma A (1997) From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem Rev 97:2373–2419. https://doi.org/10.1021/cr960406n
Crespin M, Hall WK (1981) The surface chemistry of some perovskite oxides. J Catal 69:359–370. https://doi.org/10.1016/0021-9517(81)90171-8
Das N, Eckert H, Hu H, Wachs IE, Walzer JF, Feher FJ (1993) Bonding states of surface vanadium(V) oxide phases on silica: structural characterization by 51V NMR and Raman spectroscopy. J Phys Chem 97:8240–8243. https://doi.org/10.1021/j100133a020
Davis ME (2002) Ordered porous materials for emerging applications. Nature 417:813–821
Díaz-álvarez AE, Cadierno V (2013) Glycerol: a promising green solvent and reducing agent for metal-catalyzed transfer hydrogenation reactions and nanoparticles formation. Appl Sci 3:55–69. https://doi.org/10.3390/APP3010055
Dickens PG, Whittingham MS (1965) Recombination of oxygen atoms on oxide surfaces: part 2. Catalytic activities of the alkali metal tungsten bronzes. Trans Faraday Soc 61:1226–1231. https://doi.org/10.1039/TF9656101226
Dubey M, Wadhwa S, Mathur A, Kumar R (2022) Progress in mesoporous ceria: a review on synthesis strategies and catalytic applications. Appl Surf Sci Adv 12:100340. https://doi.org/10.1016/J.APSADV.2022.100340
Enibe SO, Iloeje OC (2000) Heat and mass transfer in porous spherical pellets of CaCl2 for solar refrigeration. Renew Energy 20:305–324. https://doi.org/10.1016/S0960-1481(99)00105-6
Erika D, Mardiana S, Rasrendra CB, Khalil M, Kadja GTM (2022) Nanocasting nanoporous nickel oxides from mesoporous silicas and their comparative catalytic applications for the reduction of p-nitrophenol. Chem Phys Lett 803:139809. https://doi.org/10.1016/J.CPLETT.2022.139809
Farhadi S, Rashidi N (2012) Perovskite-type ferromagnetic BiFeO3 nanopowder: A new magnetically recoverable heterogeneous nanocatalyst for efficient and selective transfer hydrogenation of aromatic nitro compounds into aromatic amines under microwave heating. J Iran Chem Soc 9:1021–1031. https://doi.org/10.1007/s13738-012-0149-5
Feng ZV, Lyon JL, Croley JS, Crooks RM, Vanden Bout DA, Stevenson KJ (2009) Synthesis and catalytic evaluation of dendrimer-encapsulated Cu nanoparticles. an undergraduate expirement catalytic nanomaterials. J Chem Educ 86:368–372. https://doi.org/10.1021/ed086p368
Fierro JLG (1990) Structure and composition of perovskite surface in relation to adsorption and catalytic properties. Catal Today 8:153–174. https://doi.org/10.1016/0920-5861(90)87016-V
Fillafer N, Seewald T, Schmidt-Mende L, Polarz S (2020) Interfacial charge transfer processes in 2D and 3D semiconducting hybrid perovskites: azobenzene as photoswitchable ligand. Beilstein J Nanotechnol 11:466–479. https://doi.org/10.3762/bjnano.11.38
Fischer H, Schulz-Ekloff G, Wöhrle D (1997) Oxidation of aqueous sulfide solutions by dioxygen part II: catalysis by soluble and immobilized cobalt(II) phthalocyanines. Chem Eng Technol 20:624–632. https://doi.org/10.1002/ceat.270200909
Foo GS, Polo-Garzon F, Fung V, Jiang DE, Overbury SH, Wu Z (2017) Acid-base reactivity of perovskite catalysts probed via conversion of 2-propanol over titanates and zirconates. ACS Catal 7:4423–4434. https://doi.org/10.1021/ACSCATAL.7B00783/SUPPL_FILE/CS7B00783_SI_001.PDF
Fujii H, Mizuno N, Misono M (1987) Pronounced catalytic activity of La1–xSrxCoO3 highly dispersed on ZrO2 for complete oxidation of propane. Chem Lett 16:2147–2150. https://doi.org/10.1246/cl.1987.2147
Futai M, Yonghua C, Louhui (1986) Characterization of perovskite-type oxide catalysts RECoO3 by TPR. React Kinet Catal Lett 31:47–54. https://doi.org/10.1007/BF02062510
Gallezot P, Blanc B, Barthomeuf D, Païs da Silva MI (1994) Selective hydrogenation of cinnamaldehyde controlled by host/guest interactions in beta zeolite. Stud Surf Sci Catal 84:1433–1439. https://doi.org/10.1016/S0167-2991(08)63685-X
Gilkey MJ, Xu B (2016) Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal 6:1420–1436. https://doi.org/10.1021/acscatal.5b02171
Gliński M, Ulkowska U (2011) Reactivity of alcohols in chemoselective transfer hydrogenation of acrolein over magnesium oxide as the catalyst. Catal Lett 141:293–299. https://doi.org/10.1007/s10562-010-0497-7
Go YB, Jacobson AJ (2007) Solid solution precursors to gadolinia-doped ceria prepared via a low-temperature solution route. Chem Mater 19:4702–4709. https://doi.org/10.1021/cm071310k
Goldschmidt VM (1926) Die Gesetze der Krystallochemie. Naturwissenschaften 14:477–485. https://doi.org/10.1007/BF01507527
Gross BH, Mebane RC, Armstrong DL (2001) Transfer hydrogenolysis of aromatic alcohols using Raney catalysts and 2-propanol. Appl Catal A Gen 219:281–289. https://doi.org/10.1016/S0926-860X(01)00700-1
Hájek J, Kumar N, Mäki-Arvela P, Salmi T, Murzin DY, Paseka I, Heikkilä T, Laine E, Laukkanen P, Väyrynen J (2003) Ruthenium-modified MCM-41 mesoporous molecular sieve and Y zeolite catalysts for selective hydrogenation of cinnamaldehyde. Appl Catal A Gen 251:385–396. https://doi.org/10.1016/S0926-860X(03)00345-4
Hájek J, Kumar N, Salmi T, Murzin DY, Karhu H, Väyrynen J, Červený L, Paseka I (2003) Impact of catalyst reduction mode on selective hydrogenation of cinnamaldehyde over Ru–Sn sol–gel catalysts. Ind Eng Chem Res 42:295–305. https://doi.org/10.1021/ie020419t
Hájek J, Mäki-Arvela P, Toukoniitty E, Kumar N, Salmi T, Murzin DY, Červený L, Paseka I, Laine E (2004) The effect of chemical reducing agents in the synthesis of sol–gel Ru–Sn catalysts: selective hydrogenation of cinnamaldehyde. J Sol Gel Sci Technol 30:187–195. https://doi.org/10.1023/B:JSST.0000039504.89972.ee
Hájek J, Murzin DY (2004) Liquid-phase hydrogenation of cinnamaldehyde over a Ru–Sn sol–gel catalyst. 1. Evaluation of mass transfer via a combined experimental/theoretical approach. Ind Eng Chem Res 43:2030–2038. https://doi.org/10.1021/ie0340802
Haldar P, Mahajani VV (2004) Catalytic transfer hydrogenation: O-nitro anisole to o-anisidine, some process development aspects. Chem Eng J 104:27–33. https://doi.org/10.1016/j.cej.2004.08.002
Hammami R, Batis H, Minot C (2009) Combined experimental and theoretical investigation of the CO2 adsorption on LaMnO3+y perovskite oxide. Surf Sci 603:3057–3067. https://doi.org/10.1016/j.susc.2009.08.016
He X, Antonelli D (2002) Recent advances in synthesis and applications of transition metal containing mesoporous molecular sieves. Angew Chemie Int Ed 41:214–229. https://doi.org/10.1002/1521-3773(20020118)41:2%3c214::AID-ANIE214%3e3.0.CO;2-D
Helal MA, Kojima S (2022) Brillouin scattering and first-principles studies of BaMO3 (M = Ti, Zr, and Cu) perovskites. Materials 15:6747. https://doi.org/10.3390/MA15196747
Hicks RF, Qi H, Young ML, Lee RG (1990) Structure sensitivity of methane oxidation over platinum and palladium. J Catal 122:280–294. https://doi.org/10.1016/0021-9517(90)90282-O
Huang TL, MacInnes JM, Cliffe KR (2001) Nitrogen removal from wastewater by a catalytic oxidation method. Water Res 35:2113–2120. https://doi.org/10.1016/S0043-1354(00)00492-9
Huang YB, Zhang X, Zhang J, Chen H, Wang T, Lu Q (2022) Catalytic transfer hydrogenation of 5-hydroxymethylfurfural with primary alcohols over skeletal CuZnAl catalysts. Chemsuschem 15:e202200237. https://doi.org/10.1002/CSSC.202200237
Hwang J, Rao RR, Giordano L, Katayama Y, Yu Y, Shao-Horn Y (2017) Perovskites in catalysis and electrocatalysis. Science 358:751–756
Hyodo T (1996) Catalytic activities of rare-earth manganites for cathodic reduction of oxygen in alkaline solution. J Electrochem Soc 143:L266. https://doi.org/10.1149/1.1837229
Imai H, Takami K, Naito M (1984) Preparation of CoLaO3 catalyst fine particles by mist decomposition method II effect of additives for the increase of surface area. Mater Res Bull 19:1293–1300. https://doi.org/10.1016/0025-5408(84)90191-0
Irusta S, Pina MP, Menéndez M, Santamaría J (1998) Catalytic combustion of volatile organic compounds over La-based perovskites. J Catal 179:400–412. https://doi.org/10.1006/jcat.1998.2244
Jae J, Mahmoud E, Lobo RF, Vlachos DG (2014) Cascade of liquid-phase catalytic transfer hydrogenation and etherification of 5-hydroxymethylfurfural to potential biodiesel components over Lewis acid zeolites. ChemCatChem 6:508–513. https://doi.org/10.1002/cctc.201300978
Jae J, Zheng W, Lobo RF, Vlachos DG (2013) Production of dimethylfuran from hydroxymethylfurfural through catalytic transfer hydrogenation with ruthenium supported on carbon. Chemsuschem 6:1158–1162. https://doi.org/10.1002/cssc.201300288
Jagadeesh RV, Wienhöfer G, Westerhaus FA, Surkus AE, Pohl MM, Junge H, Junge K, Beller M (2011) Efficient and highly selective iron-catalyzed reduction of nitroarenes. Chem Commun 47:10972–10974. https://doi.org/10.1039/c1cc13728j
Jin S (2021) Can we find the perfect A-cations for halide perovskites? ACS Energy Lett 6:3386–3389. https://doi.org/10.1021/ACSENERGYLETT.1C01806/ASSET/IMAGES/MEDIUM/NZ1C01806_M001.GIF
Jin X, Yin B, Xia Q, Fang T, Shen J, Kuang L, Yang C (2019) Catalytic transfer hydrogenation of biomass-derived substrates to value-added chemicals on dual-function catalysts: opportunities and challenges. Chemsuschem 12:71–92. https://doi.org/10.1002/cssc.201801620
Johnstone RAW, Wilby AH (1981) Metal-assisted reactions-part 101 1 Part 9. I.D. Entwistle, B.J. Hussey and R.A.W. Johnstone, Tetrahedron Letters 4747 (1980). Rapid, stereoselective and specific catalytic transfer reduction of alkynes to cis-alkenes. Tetrahedron 37:3667–3670. https://doi.org/10.1016/S0040-4020(01)98896-9
Johnstone RAWW, Wilby AH, Entwistle ID (1985) Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. American Chemical Society
Kagan CR, Mitzi DB, Dimitrakopoulos CD (1999) Organic-inorganic hybrid materials as semiconducting channels in thin- film field-effect transistors. Science 286:945–947. https://doi.org/10.1126/science.286.5441.945
Kalbasi RJ, Nourbakhsh AA, Babaknezhad F (2011) Synthesis and characterization of Ni nanoparticles-polyvinylamine/SBA-15 catalyst for simple reduction of aromatic nitro compounds. Catal Commun 12:955–960. https://doi.org/10.1016/j.catcom.2011.02.019
Keav S, Matam SK, Ferri D, Weidenkaff A (2014) Structured perovskite-based catalysts and their application as three-way catalytic converters—a review. Catalysts 4:226–255. https://doi.org/10.3390/catal4030226
Keil FJ (1999) Diffusion and reaction in porous networks. Catal Today 53:245–258. https://doi.org/10.1016/S0920-5861(99)00119-4
Khalesi A, Arandiyan HR, Parvari M (2008) Effects of lanthanum substitution by strontium and calcium in La–Ni–Al perovskite oxides in dry reforming of methane. Chin J Catal 29:960–968. https://doi.org/10.1016/S1872-2067(08)60079-0
Khalesi A, Arandiyan HR, Parvari M (2008) Production of syngas by CO2 reforming on MxLa1−xNi0.3Al0.7O3−d (M = Li, Na, K) catalysts. Ind Eng Chem Res 47:5892–5898. https://doi.org/10.1021/ie800111e
Kijeński J, Winiarek P, Paryjczak T, Lewicki A, Mikolajska A (2002) Platinum deposited on monolayer supports in selective hydrogenation of furfural to furfuryl alcohol. Appl Catal A Gen 233:171–182. https://doi.org/10.1016/S0926-860X(02)00140-0
Kuhn JN, Ozkan US (2008) Surface properties of Sr- and Co-doped LaFeO3. J Catal 253:200–211. https://doi.org/10.1016/j.jcat.2007.10.005
Kulkarni AS, Jayaram RV (2004) Liquid phase catalytic transfer hydrogenation of aromatic nitro compounds on La1–xSrxFeO3 perovskites prepared by microwave irradiation. J Mol Catal A Chem 223:107–110. https://doi.org/10.1016/j.molcata.2003.12.042
Kun I, Szöllösi G, Bartók M (2001) Crotonaldehyde hydrogenation over clay-supported platinum catalysts. J Mol Catal A Chem 169:235–246. https://doi.org/10.1016/S1381-1169(00)00566-5
Kundra M, Grall T, Ng D, Xie Z, Hornung CH (2021) Continuous flow hydrogenation of flavorings and fragrances using 3D-printed catalytic static mixers. Ind Eng Chem Res 60:1989–2002. https://doi.org/10.1021/ACS.IECR.0C05671
Lafaye G, Ekou T, Micheaud-Especel C, Montassier C, Marecot P (2004) Citral hydrogenation over alumina supported Rh–Ge catalysts: effects of the reduction temperature. Appl Catal A Gen 257:107–117. https://doi.org/10.1016/S0926-860X(03)00634-3
Lashdaf M, Tiitta M, Venäläinen T, Österholm H, Krause AOI (2004) Ruthenium on beta zeolite in cinnamaldehyde hydrogenation. Catal Lett 94:7–14. https://doi.org/10.1023/b:catl.0000019323.61742.c4
Lee SH, Lee JY, Park YM, Wee JH, Lee KY (2006) Complete oxidation of methane and CO at low temperature over LaCoO3 prepared by spray-freezing/freeze-drying method. Catal Today 117:376–381. https://doi.org/10.1016/j.cattod.2006.05.035
Lee YN, Lago RM, Fierro JLG, González J (2001) Hydrogen peroxide decomposition over Ln1−xAxMnO3 (Ln = La or Nd and A = K or Sr) perovskites. Appl Catal A Gen 215:245–256. https://doi.org/10.1016/S0926-860X(01)00536-1
Leng F, Gerber IC, Axet MR, Serp P (2018) Selectivity shifts in hydrogenation of cinnamaldehyde on electron-deficient ruthenium nanoparticles. Comptes Rendus Chim 21:346–353. https://doi.org/10.1016/J.CRCI.2017.04.001
Lewis JD, Van De Vyver S, Crisci AJ, Gunther WR, Michaelis VK, Griffin RG, Romµn-Leshkov Y (2014) A continuous flow strategy for the coupled transfer hydrogenation and etherification of 5-(hydroxymethyl)furfural using Lewis acid zeolites. Chemsuschem 7:2255–2265. https://doi.org/10.1002/cssc.201402100
Li H, Zhang J, Huang G, Fu S, Ma C, Wang B, Huang Q, Liao H (2017) Hydrothermal synthesis and enhanced photocatalytic activity of hierarchical flower-like Fe-doped BiVO4. Trans Nonferrous Met Soc China (Engl Ed) 27:868–875. https://doi.org/10.1016/S1003-6326(17)60102-X
Li X, Liu Y, Deng J, Xie S, Zhao X, Zhang Y, Zhang K, Arandiyan H, Guo G, Dai H (2017) Enhanced catalytic performance for methane combustion of 3DOM CoFe2O4 by co-loading MnOx and Pd–Pt alloy nanoparticles. Appl Surf Sci 403:590–600. https://doi.org/10.1016/j.apsusc.2017.01.237
Liang H-W, Cao X, Zhou F, Cui C-H, Zhang W-J, Yu S-H (2011) A free-standing Pt-nanowire membrane as a highly stable electrocatalyst for the oxygen reduction reaction. Adv Mater 23:1467–1471. https://doi.org/10.1002/adma.201004377
Lin KS, Wang HP (2000) Supercritical water oxidation of 2-chlorophenol catalyzed by Cu2+ cations and copper oxide clusters. Environ Sci Technol 34:4849–4854. https://doi.org/10.1021/es001062s
López E, Ordóñez S, Díez FV, Sastre H (2001) Hydrodechlorination of chlorobenzene-tetrachloroethylene mixtures over a Pd/Al2O3 catalyst. In: Studies in surface science and catalysis. Elsevier Inc., pp 521–526
Lu L, Eychmüller A (2008) Ordered macroporous bimetallic nanostructures: design, characterization, and applications. Acc Chem Res 41:244–253. https://doi.org/10.1021/ar700143w
Mabate TP, Meijboom R, Bingwa N (2022) The inorganic perovskite-catalyzed transfer hydrogenation of cinnamaldehyde using glycerol as a hydrogen donor. Catalysts 12:241. https://doi.org/10.3390/CATAL12020241/S1
Mabate TP, Potgieter K, Molokoane PP, Meijboom R, Bingwa N (2022) Rapid kinetic evaluation of inorganic-perovskite-catalysed redox conversion of p-nitrophenol and morin aided by an opentrons robotic system. J Mater Sci 57:11590–11611. https://doi.org/10.1007/S10853-022-07393-4/SCHEMES/3
Mäki-Arvela P, Hájek J, Salmi T, Murzin DY (2005) Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts. Appl Catal A Gen. https://doi.org/10.1016/j.apcata.2005.05.045
Makshina EV, Sirotin SV, van den Berg MWE, Klementiev KV, Yushchenko VV, Mazo GN, Grünert W, Romanovsky BV (2006) Characterization and catalytic properties of nanosized cobaltate particles prepared by in situ synthesis inside mesoporous molecular sieves. Appl Catal A Gen 312:59–66. https://doi.org/10.1016/j.apcata.2006.06.021
Mandal PK, McMurray JS (2007) Pd-C-induced catalytic transfer hydrogenation with triethylsilane. J Org Chem 72:6599–6601. https://doi.org/10.1021/JO0706123/SUPPL_FILE/JO0706123SI20070604_010512.PDF
Matatov-Meytal YI, Sheintuch M (1998) Catalytic abatement of water pollutants. Ind Eng Chem Res 37:309–326
Mhlwatika Z, Meijboom R, Bingwa N (2021) Nanocasted perovskites as potential catalysts for acetalization of glycerol. Inorg Chem Commun 133:108962. https://doi.org/10.1016/J.INOCHE.2021.108962
Milone C, Ingoglia R, Tropeano ML, Neri G, Galvagno S (2003) First example of selective hydrogenation of unconstrained α, β-unsaturated ketone to α, β-unsaturated alcohol by molecular hydrogen. Chem Commun 3:868–869. https://doi.org/10.1039/b212441f
Misono M (2009) Recent progress in the practical applications of heteropolyacid and perovskite catalysts: Catalytic technology for the sustainable society. Catal Today 144:285–291. https://doi.org/10.1016/j.cattod.2008.10.054
Mizuno N (1990) Supported perovskites. Catal Today 8:221–230. https://doi.org/10.1016/0920-5861(90)87019-Y
Mogudi BM, Ncube P, Meijboom R (2016) Catalytic activity of mesoporous cobalt oxides with controlled porosity and crystallite sizes: evaluation using the reduction of 4-nitrophenol. Appl Catal B Environ 198:74–82. https://doi.org/10.1016/j.apcatb.2016.05.051
Moure C, Peña O (2015) Recent advances in perovskites: processing and properties. Prog Solid State Chem. https://doi.org/10.1016/j.progsolidstchem.2015.09.001
Nair MM, Kleitz F, Kaliaguine S (2012) Kinetics of methanol oxidation over mesoporous perovskite catalysts. ChemCatChem 4:387–394. https://doi.org/10.1002/cctc.201100356
Nair MM, Kleitz F, Kaliaguine S (2016) Pore structure effects on the kinetics of methanol oxidation over nanocast mesoporous perovskites. Cuihua Xuebao/Chin J Catal 37:32–42. https://doi.org/10.1016/S1872-2067(15)60909-3
Natile MM, Ugel E, Maccato C, Glisenti A (2007) LaCoO3: effect of synthesis conditions on properties and reactivity. Appl Catal B Environ 72:351–362. https://doi.org/10.1016/j.apcatb.2006.11.011
Nguyen SV, Szabo V, Trong On D, Kaliaguine S (2002) Mesoporous silica supported LaCoO3 perovskites as catalysts for methane oxidation. Microporous Mesoporous Mater 54:51–61. https://doi.org/10.1016/S1387-1811(02)00340-2
Nijhuis TA, Kreutzer MT, Romijn ACJ, Kapteijn F, Moulijn JA (2001) Monolithic catalysts as more efficient three-phase reactors. In: Catalysis today. Elsevier, pp 157–165
Nzuzo Y, Adeyinka A, Carleschi E, Doyle BP, Bingwa N (2021) Effect of d z2 orbital electron-distribution of La-based inorganic perovskites on surface kinetics of a model reaction. Inorg Chem Front 8:3037–3048. https://doi.org/10.1039/D1QI00297J
Orge CA, Órfão JJMM, Pereira MFRR, Barbero BP, Cadús LE (2013) Lanthanum-based perovskites as catalysts for the ozonation of selected organic compounds. Appl Catal B Environ 140–141:426–432. https://doi.org/10.1016/j.apcatb.2013.04.045
Panagiotopoulou P, Martin N, Vlachos DG (2014) Effect of hydrogen donor on liquid phase catalytic transfer hydrogenation of furfural over a Ru/RuO2/C catalyst. J Mol Catal A Chem 392:223–228. https://doi.org/10.1016/j.molcata.2014.05.016
Park BW, Philippe B, Zhang X, Rensmo H, Boschloo G, Johansson EMJ (2015) Bismuth based hybrid perovskites A3Bi2I9 (A: methylammonium or cesium) for solar cell application. Adv Mater 27:6806–6813. https://doi.org/10.1002/ADMA.201501978
Parlett CMA, Wilson K, Lee AF (2013) Hierarchical porous materials: catalytic applications. Chem Soc Rev 42:3876–3893
Parravano G (1952) Ferroelectric transitions and heterogenous catalysis. J Chem Phys. https://doi.org/10.1063/1.1700412
Pecchi G, Escalona N, Ghampson IT, Morales R (2016) Energy production, decontamination, and hydrogenation reactions over perovskite-type oxide catalyst. In: Perovskite materials—synthesis, characterisation, properties, and applications. InTech
Peña MA, Fierro JLG (2001) Chemical structures and performance of perovskite oxides. Chem Rev 101:1981–2017
Pérez-Ramírez J, Christensen CH, Egeblad K, Christensen CH, Groen JC (2008) Hierarchical zeolites: enhanced utilisation of microporous crystals in catalysis by advances in materials design. Chem Soc Rev 37:2530–2542
Pirkanniemi K, Sillanp M (2002) Heterogeneous water phase catalysis as an environmental application: a review. Chemosphere 48:1047–1060
Polo-Garzon F, Wu Z (2018) Acid-base catalysis over perovskites: a review. J Mater Chem A 6:2877–2894
Ponce S, Peña MA, Fierro JLG (2000) Surface properties and catalytic performance in methane combustion of SR-substituted lanthanum manganites. Appl Catal B Environ 24:193–205. https://doi.org/10.1016/S0926-3373(99)00111-3
Qiao ZA, Zhang P, Chai SH, Chi M, Veith GM, Gallego NC, Kidder M, Dai S (2014) Lab-in-a-shell: encapsulating metal clusters for size sieving catalysis. J Am Chem Soc 136:11260–11263. https://doi.org/10.1021/ja505903r
Rajmohan S, Manikandan A, Jeseentharani V, Antony SA, Pragasam J (2016) Simple co-precipitation synthesis and characterization studies of La1−xNixVO3 perovskites nanostructures for humidity sensing applications. J Nanosci Nanotechnol 16:1650–1655. https://doi.org/10.1166/jnn.2016.10745
Raveau B (2007) The perovskite history: more than 60 years of research from the discovery of ferroelectricity to colossal magnetoresistance via high TC superconductivity. Prog Solid State Chem 35:171–173
Raybaud P, Chizallet C, Mager-Maury C, Digne M, Toulhoat H, Sautet P (2013) From γ-alumina to supported platinum nanoclusters in reforming conditions: 10 years of DFT modeling and beyond. J Catal 308:328–340. https://doi.org/10.1016/j.jcat.2013.08.015
Roark RD, Kohler SD, Ekerdt JG (1992) Role of silanol groups in dispersing Mo(VI) on silica. Catal Lett 16:71–76. https://doi.org/10.1007/BF00764356
Royer S, Duprez D, Can F, Courtois X, Batiot-Dupeyrat C, Laassiri S, Alamdari H (2014) Perovskites as substitutes of noble metals for heterogeneous catalysis: dream or reality. Chem Rev 114:10292–10368. https://doi.org/10.1021/cr500032a
Royer S, Bérubé F, Kaliaguine S (2005) Effect of the synthesis conditions on the redox and catalytic properties in oxidation reactions of LaCo1−xFexO3. Appl Catal A Gen 282:273–284. https://doi.org/10.1016/j.apcata.2004.12.018
Sato S, Takahashi R, Sodesawa T, Nozaki F, Jin XZ, Suzuki S, Nakayama T (2000) Mass-transfer limitation in mesopores of Ni–MgO catalyst in liquid-phase hydrogenation. J Catal 191:261–270. https://doi.org/10.1006/jcat.1999.2797
Sayagués MJ, Vallet-Regí M, Caneiro A, González-Calbet JM (1994) Microstructural characterization of the LaNiO3−y system. J Solid State Chem 110:295–304. https://doi.org/10.1006/jssc.1994.1172
Scholz D, Aellig C, Hermans I (2014) Catalytic transfer hydrogenation/hydrogenolysis for reductive upgrading of furfural and 5-(hydroxymethyl)furfural. Chemsuschem 7:268–275. https://doi.org/10.1002/cssc.201300774
Selvam P, Mohapatra SK, Sonavane SU, Jayaram RV (2004) Chemo- and regioselective reduction of nitroarenes, carbonyls and azo dyes over nickel-incorporated hexagonal mesoporous aluminophosphate molecular sieves. Tetrahedron Lett 45:2003–2007. https://doi.org/10.1016/j.tetlet.2003.12.091
Shen W, Dong X, Zhu Y, Chen H, Shi J (2005) Mesoporous CeO2 and CuO-loaded mesoporous CeO2: synthesis, characterization, and CO catalytic oxidation property. Microporous Mesoporous Mater 85:157–162. https://doi.org/10.1016/j.micromeso.2005.06.006
Shu J, Kaliaguine S (1998) Well-dispersed perovskite-type oxidation catalysts. Appl Catal B Environ 16:L303. https://doi.org/10.1016/S0926-3373(97)00097-0
Singh D, Tabari T, Trochowski M, LastNameYağcı MB, Singh D, Tabari T, Baris Yagci M, Macyk W (2018) Efficient synthesis of BiFeO3 by the microwave-assisted sol-gel method: “A” site influence on the photoelectrochemical activity of perovskites. Appl Surf Sci 471:1017–1027. https://doi.org/10.1016/j.apsusc.2018.12.082
Singh UK, Vannice MA (2001) Kinetics of liquid-phase hydrogenation reactions over supported metal catalysts—a review. Appl Catal A Gen 213:1–24
Smyth DM, Tejuca LG, Fierro JLG (1993) Properties and applications of perovskite-type oxides. In: Deker M (ed) Google books, p 47
Taguchi A, Schüth F (2005) Ordered mesoporous materials in catalysis. Microporous Mesoporous Mater 77:1–45
Tejuca LG, Fierro JLG, Tascón JMD (1989) Structure and reactivity of perovskite-type oxides. Adv Catal 36:237–328. https://doi.org/10.1016/S0360-0564(08)60019-X
Thananatthanachon T, Rauchfuss TB (2010) Efficient production of the liquid fuel 2,5-dimethylfuran from fructose using formic acid as a reagent. Angew Chemie 122:6766–6768. https://doi.org/10.1002/ange.201002267
Thomas JM, Raja R (2008) Exploiting nanospace for asymmetric catalysis: confinement of immobilized, single-site chiral catalysts enhances enantioselectivity. Acc Chem Res 41:708–720
Tian B, Liu X, Yang H, Xie S, Yu C, Tu B, Zhao D (2003) General synthesis of ordered crystallized metal oxide nanoarrays replicated by microwave-digested mesoporous silica. Adv Mater 15:1370–1374. https://doi.org/10.1002/adma.200305211
Toro RD, Hernández P, Díaz Y, Brito JL (2013) Synthesis of La0.8Sr0.2FeO3 perovskites nanocrystals by Pechini sol-gel method. Mater Lett 107:231–234. https://doi.org/10.1016/j.matlet.2013.05.139
Tundo P, Anastas P, Black DSC, Breen J, Collins T, Memoli S, Miyamoto J, Polyakoff M, Tumas W (2000) Synthetic pathways and processes in green chemistry. Introductory overview. Pure Appl Chem 72:1207–1228. https://doi.org/10.1351/pac200072071207
Veitía MSI, Ferroud C (2015) New activation methods used in green chemistry for the synthesis of high added value molecules. Int J Energy Environ Eng 6:37–46. https://doi.org/10.1007/s40095-014-0148-7
Venezia AM, La Parola V, Nicoli V, Deganello G (2002) Effect of gold on the HDS activity of supported palladium catalysts. J Catal 212:56–62. https://doi.org/10.1006/jcat.2002.3778
Vijayakumar M, Inaguma Y, Mashiko W, Crosnier-Lopez MP, Bohnke C (2004) Synthesis of fine powders of Li3xLa2/3−xTiO3 perovskite by a polymerizable precursor method. Chem Mater 16:2719–2724. https://doi.org/10.1021/cm049869x
Villoria J-A, Mota N, Al-Sayari S, Alvarez-Galvan M-C (2012) Perovskites as catalysts in the reforming of hydrocarbons: a review. Micro Nanosyst 4:231–252
Voorhoeve RJH, Johnson DW, Remeika JP, Gallagher PK (1977) Perovskite oxides: materials science in catalysis. Science 195:827–833. https://doi.org/10.1126/science.195.4281.827
Voorhoeve RJH, Remeika JP, Johnson DW (1973) Rare-earth manganites: Catalysts with low ammonia yield in the reduction of nitrogen oxides. Science 180:62–64. https://doi.org/10.1126/science.180.4081.62
Voorhoeve RJH, Remeika JP, Trimble LE, Cooper AS, Disalvo FJ, Gallagher PK (1975) Perovskite-like La1−xKxMnO3 and related compounds: solid state chemistry and the catalysis of the reduction of NO by CO and H2. J Solid State Chem 14:395–406. https://doi.org/10.1016/0022-4596(75)90061-4
Wachs IE (2005) Recent conceptual advances in the catalysis science of mixed metal oxide catalytic materials. Catal Today 100:79–94. https://doi.org/10.1016/J.CATTOD.2004.12.019
Walcarius A (2013) Mesoporous materials and electrochemistry. Chem Soc Rev 42:4098–4140
Wan Y, Zhao D, Zhang FQ, Meng Y, Gu D, Yan Y, Yu CZ, Tu B, Zhao DY, Shi YF, Yang HF, Li Z, Liu RL, Wan Y, Chen ZX, Am Chem Soc J, Huang Y, Cai HQ, Yu T, Zhang F, Deng YH, Zhang LJ, Liu C, Wu XJ (2007) Ordered mesoporous polymers and carbon molecular sieves
Wang F, Jiang Y, Wen X, Xia J, Sha G, Amal R (2013) Confined Au–Pd ensembles in mesoporous TiO2 spheres for the photocatalytic oxidation of acetaldehyde. ChemCatChem 5:3557–3561. https://doi.org/10.1002/cctc.201300584
Wang W, Shao Z (2015) Research progress of perovskite materials in photocatalysis-and photovoltaics-related energy conversion and environmental treatment. Chem Soc Rev 00:1–3. https://doi.org/10.1039/x0xx00000x
Wang Y, Arandiyan H, Scott J, Bagheri A, Dai H, Amal R (2017) Recent advances in ordered meso/macroporous metal oxides for heterogeneous catalysis: a review. J Mater Chem A 5:8825–8846. https://doi.org/10.1039/c6ta10896b
Wang Y, Cui X, Li Y, Chen L, Shu Z, Chen H, Shi J (2013) High surface area mesoporous LaFexCo1−xO3 oxides: synthesis and electrocatalytic property for oxygen reduction. J Chem Soc Dalt Trans 42:9448–9452. https://doi.org/10.1039/c3dt50151e
Wang Y, Cui X, Li Y, Shu Z, Chen H, Shi J (2013) A simple co-nanocasting method to synthesize high surface area mesoporous LaCoO3 oxides for CO and NO oxidations. Microporous Mesoporous Mater 176:8–15. https://doi.org/10.1016/j.micromeso.2013.03.033
Wang Y, Dai H, Deng J, Liu Y, Zhao Z, Li X, Arandiyan H (2013) Three-dimensionally ordered macroporous InVO4: fabrication and excellent visible-light-driven photocatalytic performance for methylene blue degradation. Chem Eng J 226:87–94. https://doi.org/10.1016/j.cej.2013.04.032
Wang Y, Xie S, Deng J, Deng S, Wang H, Yan H, Dai H (2014) Morphologically controlled synthesis of porous spherical and cubic LaMnO3 with high activity for the catalytic removal of toluene. ACS Appl Mater Interfaces 6:17394–17401. https://doi.org/10.1021/am500489x
Wang YY, Ren J, Wang YY, Zhang F, Liu X, Guo Y, Lu G (2008) Nanocasted synthesis of mesoporous LaCoO3 perovskite with extremely high surface area and excellent activity in methane combustion. J Phys Chem C 112:15293–15298. https://doi.org/10.1021/jp8048394
Weidenkaff A (2004) Preparation and application of nanostructured perovskite phases. Adv Eng Mater 6:709–714. https://doi.org/10.1002/adem.200400098
Weidenkaff A, Ebbinghaus SG, Lippert T, Montenegro MJ, Soltmann C, Wessicken R (2002) Phase formation and phase transition of Ln1−xCaxCoO3-δ (Ln= La, Er) applied for bifunctional air electrodes. Cryst Eng 5:449–457. https://doi.org/10.1016/S1463-0184(02)00056-4
Wilson Bradley (2020) Perovskites—the best material you’ve never heard of—theGIST. https://the-gist.org/2020/01/perovskites-best-material-youve-never-heard-of/. Accessed 15 May 2023
Xiao P, Xu X, Zhu J, Zhu Y (2020) In situ generation of perovskite oxides and carbon composites: a facile, effective and generalized route to prepare catalysts with improved performance. J Catal 383:88–96. https://doi.org/10.1016/j.jcat.2020.01.007
Xiao P, Zhu J, Zhao D, Zhao Z, Zaera F, Zhu Y (2019) Porous LaFeO3 prepared by an in situ carbon templating method for catalytic transfer hydrogenation reactions. ACS Appl Mater Interfaces 11:15517–15527. https://doi.org/10.1021/acsami.9b00506
Yang Q, Liu G, Liu Y (2015) Perovskite-type oxides as the catalyst precursors for preparing supported metallic nanocatalysts: a review the contents include ABO3, partially substituted ABO3 (A1−yA′). https://doi.org/10.1021/acs.iecr.7b03251
Ye H, Zhao H, Jiang Y, Liu H, Hou Z (2020) Catalytic transfer hydrogenation of the CO bond in unsaturated aldehydes over Pt nanoparticles embedded in porous UiO-66 nanoparticles. ACS Appl Nano Mater 3:12260–12268. https://doi.org/10.1021/ACSANM.0C02735/ASSET/IMAGES/LARGE/AN0C02735_0009.JPEG
Yoon S, Maegli AE, Karvonen L, Matam SK, Shkabko A, Riegg S, Großmann T, Ebbinghaus SG, Pokrant S, Weidenkaff A (2013) Bandgap tuning in SrTi(N, O, F)3 by anionic-lattice variation. J Solid State Chem 206:226–232. https://doi.org/10.1016/j.jssc.2013.08.001
Yue W, Zhou W (2007) Porous crystals of cubic metal oxides templated by cage-containing mesoporous silica. J Mater Chem 17:4947–4952. https://doi.org/10.1039/b709076e
Zanella R, Louis C, Giorgio S, Touroude R (2004) Crotonaldehyde hydrogenation by gold supported on TiO2: structure sensitivity and mechanism. J Catal 223:328–339. https://doi.org/10.1016/j.jcat.2004.01.033
Zhang L, Xu Z, Cao L, Yao X (2007) Synthesis of BF-PT perovskite powders by high-energy ball milling. Mater Lett 61:1130–1133. https://doi.org/10.1016/j.matlet.2006.06.069
Zhang P, Zhu H, Dai S (2015) Porous carbon supports: recent advances with various morphologies and compositions. ChemCatChem 7:2788–2805. https://doi.org/10.1002/cctc.201500368
Zhang Q, Saito F (2000) Mechanochemical synthesis of LaMnO3 from La2O3 and Mn2O3 powders. J Alloys Compd 297:99–103. https://doi.org/10.1016/S0925-8388(99)00606-4
Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD (1998) Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 279:548–552. https://doi.org/10.1126/science.279.5350.548
Zhou W, Profile S, Shao Z, Zhu Y (2017) Electrocatalysis perovskite/carbon composites: applications in oxygen electrocatalysis. Small. https://doi.org/10.1002/smll.201603793
Zhu H, Zhang P, Dai S (2015) Recent advances of lanthanum-based perovskite oxides for catalysis. American Chemical Society
Zhu J, Thomas A (2009) Perovskite-type mixed oxides as catalytic material for NO removal. Appl Catal B Environ 92:225–233. https://doi.org/10.1016/j.apcatb.2009.08.008
Zou H, Ge X, Shen J (2003) Surface acidity and basicity of γ-Al2O3 doped with K+ and La3+ and calcined at elevated temperatures. Thermochim Acta 397:81–86. https://doi.org/10.1016/S0040-6031(02)00329-5
Zuo L, Wang J, Mei D, Adu-Mensah D, Gao Y (2022) Ultrasonic-assisted catalytic transfer hydrogenation of cottonseed biodiesel using Raney-Ni catalyst in aqueous environment. Chem Eng J 437:135193. https://doi.org/10.1016/J.CEJ.2022.135193
Acknowledgements
This is work is supported by the National Research Foundation of South Africa (Grant UID: 138186) and the Research Center for Synthesis and Catalysis hosted by the University of Johannesburg.
Author information
Authors and Affiliations
Contributions
N.B. and M.M. did conceptualization T.P.M., N.P.M., and S.N. gathered the data T.PM. wrote and formatted the manuscript. All authors proof-read the manuscript. N.B. secured funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Mabate, T.P., Maqunga, N.P., Ntshibongo, S. et al. Metal oxides and their roles in heterogeneous catalysis: special emphasis on synthesis protocols, intrinsic properties, and their influence in transfer hydrogenation reactions. SN Appl. Sci. 5, 196 (2023). https://doi.org/10.1007/s42452-023-05416-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-023-05416-6